The Role of TNFR2 and DR3 in the In Vivo Expansion of Tregs in T Cell Depleting Transplantation Regimens

 , ,
, , 
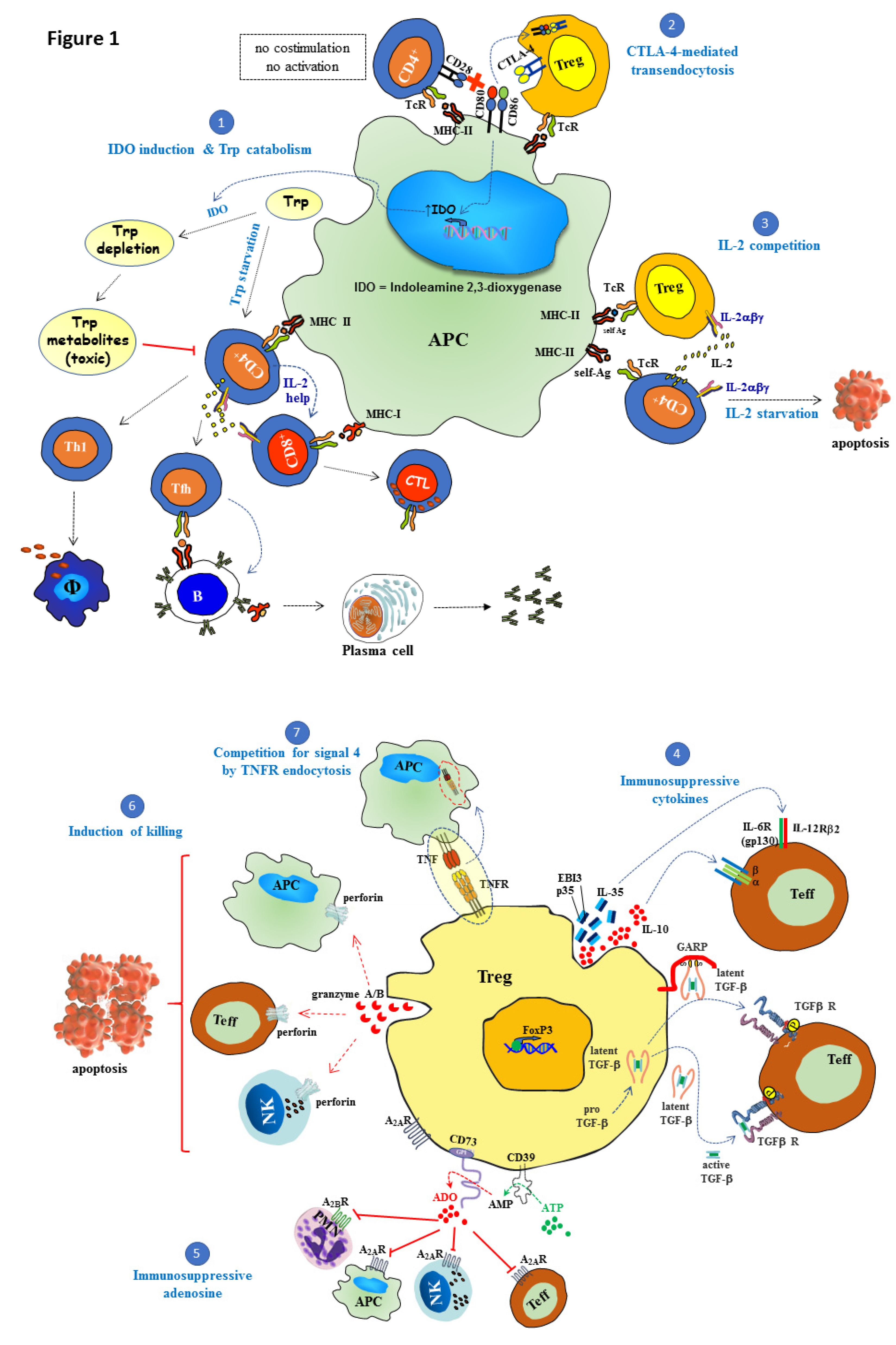{kind=link}
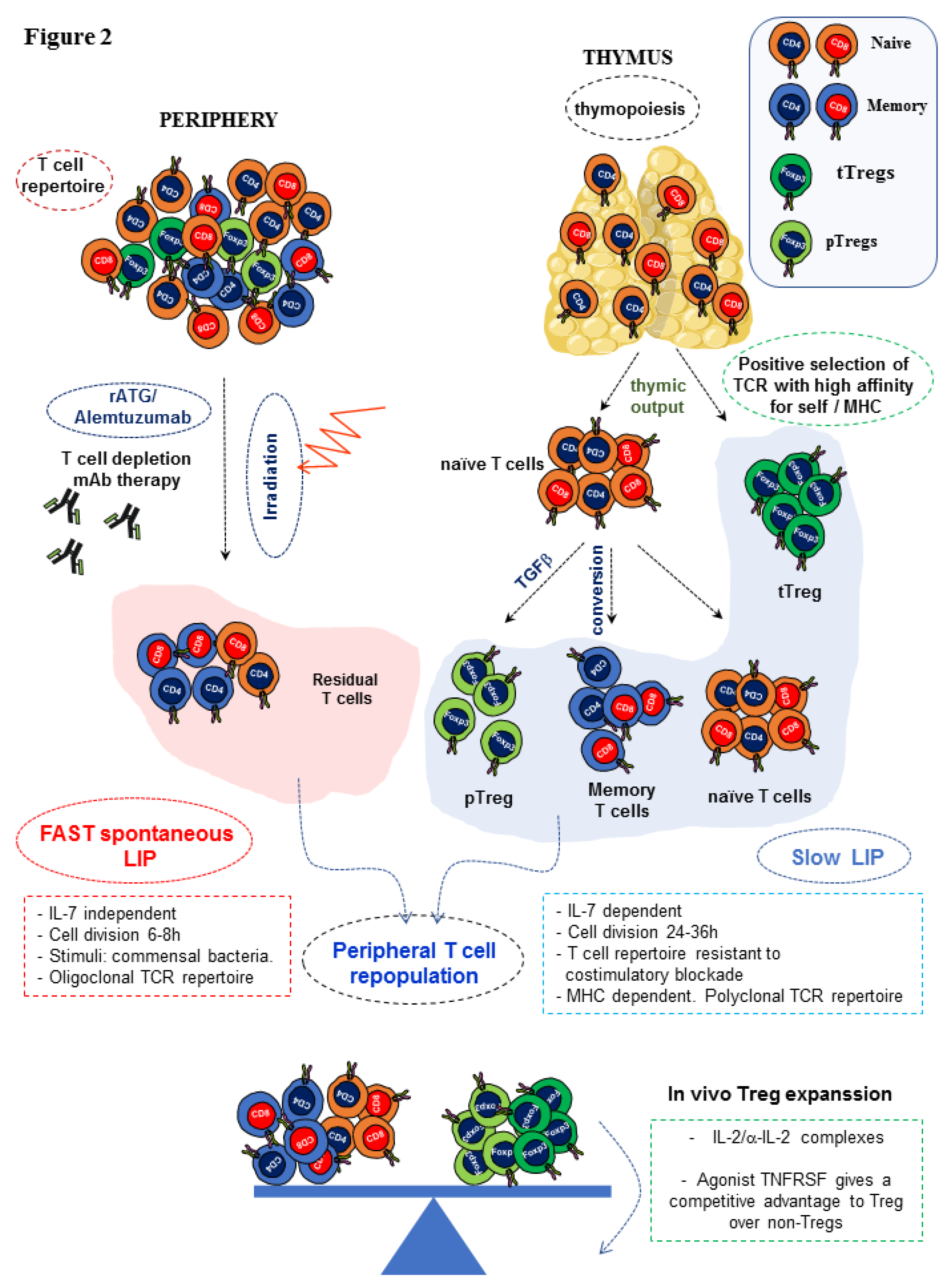{kind=link}
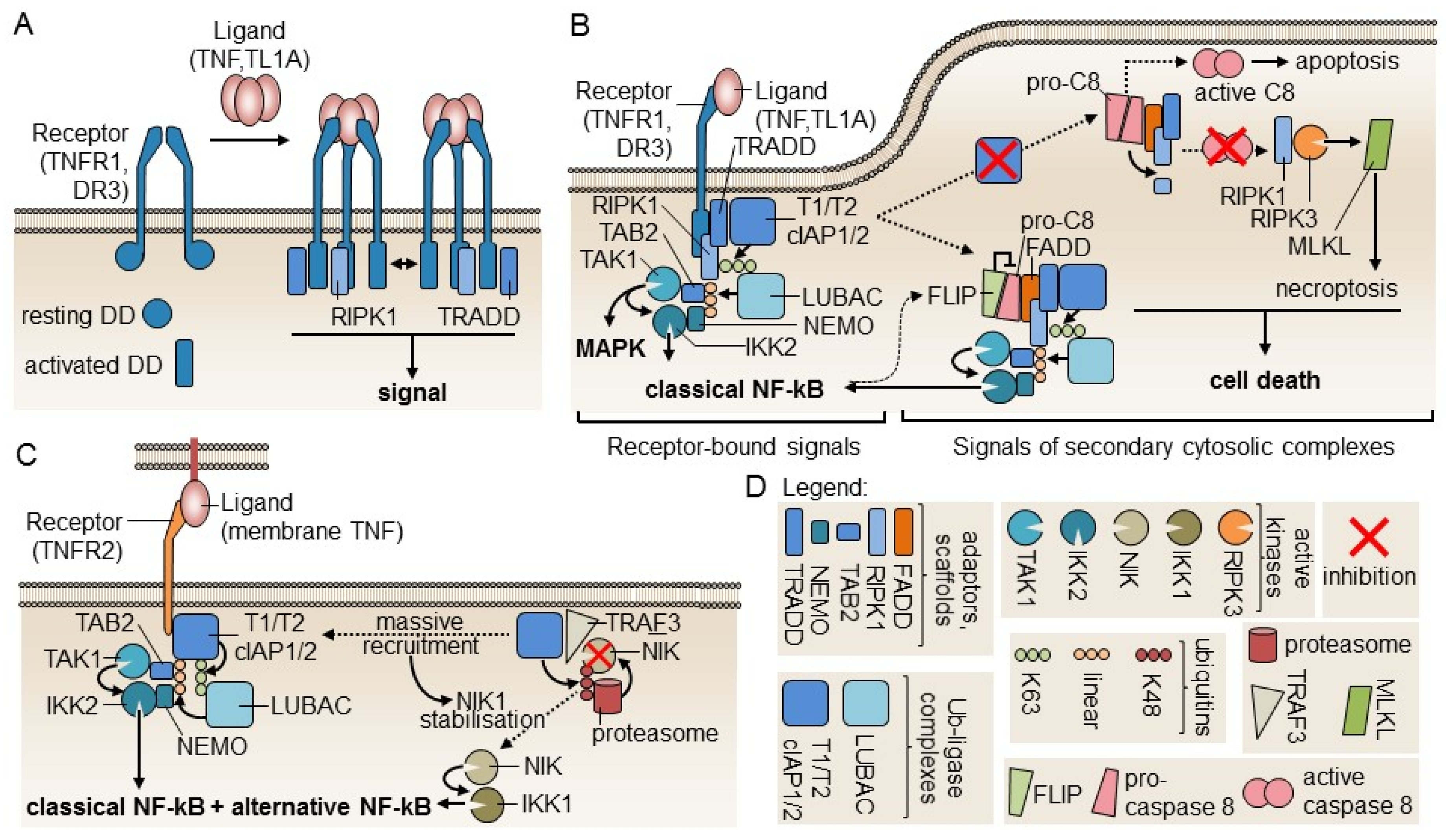{kind=link}
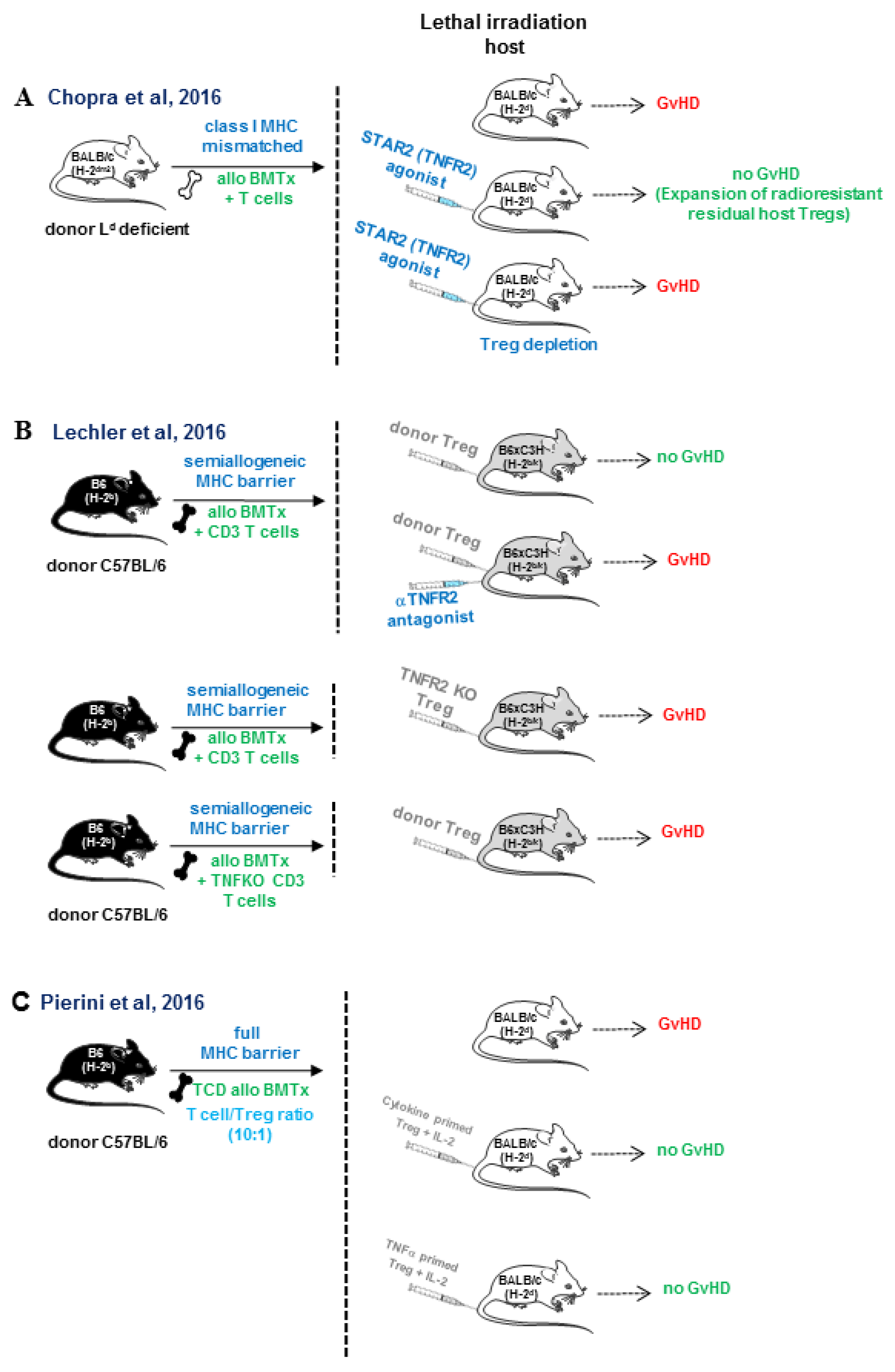{kind=link}
Abstract
1. Introduction
1.1. The Role of Regulatory T Cells in Transplantation
1.2. T Cell Depletion in Peri-Transplant Regimens and Allogeneic Bone Marrow Transplantation to Promote Long-Term Survival of Allografts
1.3. In Vivo versus in Vitro Tregs Expansion to Modulate Graft Rejection
2. Molecules of the Tumor Necrosis Factor (TNF)/TNFR Superfamily Involved in Tregs Expansion
3. Redundant Functional Activity of TNFRSF Members in Tregs Differentiation and Survival
4. The Role of TNFR1 and TNFR2 in In Vivo Tregs Expansion
5. Evidence of the Anti-Inflammatory Functions of TNF and Its Role in Tregs Function
6. Role of TNFR2 in Tregs Expansion in Allogeneic Bone Marrow Transplantation and Cardiac Transplantation
7. TL1A (TNFSF15)/DR3 (TNFRSF25) Pathway
8. Concluding Remarks on the Therapeutic Implication of Targeting TNFR in Inflammatory Diseases
Funding
Conflicts of Interest
Abbreviations
| FLIP | FLICE/Caspase 8 inhibitory protein |
| LIP | Lymphopenia-induced proliferation |
| tTregs | thymic regulatory T cells |
| pTregs | peripheral regulatory T cells |
| TCR | T cell receptor |
| rATG | rabbit polyclonal anti-thymocyte globulin |
| MHC | major histocompatibility complex |
| TNFR | tumor necrosis factor receptor |
| Alemtuzumab | chimeric recombinant antibody against CD52 |
| IL-2 | interleukin 2 |
| IL-7 | interleukin 7 |
| LIP | Lymphopenia-induced proliferation |
| THD | TNF homology domain |
| Tregs | regulatory T cells |
| TCD | T-cell depleted |
| TNF | Tumor necrosis factor |
| TGF-β | Tumor growth factor |
| TNFRSF | TNF receptor superfamily |
| Foxp3 | Forkhead box P3 transcription factor |
| AIRE | Autoimmune regulator |
| TCR | T cell receptor |
| MHC | Major histocompatibility complex |
| GvHD | Graft versus Host Disease |
| tTregs | thymic Tregs |
| pTregs | Peripheral Treg |
| CTLA-4 (CD152) | Cytotoxic T-Lymphocyte-Associated protein 4 |
| IDO | Indoleamine 2,3-dioxygenase |
| LAP | Latency-Associated Protein |
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| AMP | Adenosine monophosphate |
| rATG | rabbit anti-thymocyte globulin |
| MAPK | Mitogen-activated protein kinase |
| FADD | Fas-associated death domain adaptor proteins |
| TRADD | TNFR1-associated death domain |
| MDSC | Myeloid-derived suppressive cells |
| IL-2 | Interleukin 2 |
| DR3 | Death receptor 3 |
| HSCT | Hematopoietic stem cell transplantation |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| GM-CSF | Granulocyte/monocyte-colony stimulating factor |
| IPEX | Immunodysregulation polyendocrinopathy and enteropathy X-linked |
| MHC | major histocompatibility complex |
| BMTx | bone marrow transplantation |
| TNFKO | tumor necrosis factor knock-out |
| STAR2 | Multimeric TNFR2 agonist |
| GvHD | graft versus-host reaction |
| TNFR2 | tumor necrosis factor 2 |
| IL-2 | interleukin 2 |
| TCD | T cell-depleted |
| Tregs | regulatory T cells |
References
- Nishizuka, Y.; Sakakura, T. Thymus and reproduction: Sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 1969, 166, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.J.; Waldmann, H. Induction of tolerance by monoclonal antibody therapy. Nature 1986, 320, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.X.; Cobbold, S.; Benjamin, R.; Waldmann, H. Induction of classical transplantation tolerance in the adult. J. Exp. Med. 1989, 169, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Cobbold, S.P.; Pope, H.; Elliott, J.; Kioussis, D.; Davies, J.; Waldmann, H. “Infectious” transplantation tolerance. Science 1993, 259, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Graca, L.; Honey, K.; Adams, E.; Cobbold, S.P.; Waldmann, H. Cutting edge: Anti-CD154 therapeutic antibodies induce infectious transplantation tolerance. J. Immunol. 2000, 165, 4783–4786. [Google Scholar] [CrossRef]
- Kendal, A.R.; Chen, Y.; Regateiro, F.S.; Ma, J.; Adams, E.; Cobbold, S.P.; Hori, S.; Waldmann, H. Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J. Exp. Med. 2011, 208, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Asano, M.; Toda, M.; Sakaguchi, N.; Sakaguchi, S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 1996, 184, 387–396. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Graca, L.; Le, M.A.; Lin, C.Y.; Fairchild, P.J.; Cobbold, S.P.; Waldmann, H. Donor-specific transplantation tolerance: The paradoxical behavior of CD4+CD25+ T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10122–10126. [Google Scholar] [CrossRef]
- Graca, L.; Cobbold, S.P.; Waldmann, H. Identification of regulatory T cells in tolerated allografts. J. Exp. Med. 2002, 195, 1641–1646. [Google Scholar] [CrossRef] [PubMed]
- Golshayan, D.; Jiang, S.; Tsang, J.; Garin, M.I.; Mottet, C.; Lechler, R.I. In vitro-expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood 2007, 109, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Dhamne, C.; Chung, Y.; Alousi, A.M.; Cooper, L.J.; Tran, D.Q. Peripheral and thymic foxp3(+) regulatory T cells in search of origin, distinction, and function. Front. Immunol. 2013, 4, 253. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Ann. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Richert-Spuhler, L.E.; Lund, J.M. The Immune Fulcrum: Regulatory T Cells Tip the Balance Between Pro- and Anti-inflammatory Outcomes upon Infection. Prog. Mol. Biol. Transl. Sci. 2015, 136, 217–243. [Google Scholar]
- Mills, K.H. Regulatory T cells: Friend or foe in immunity to infection? Nat. Rev. Immunol. 2004, 4, 841–855. [Google Scholar] [CrossRef]
- Wood, K.J.; Sakaguchi, S. Regulatory T cells in transplantation tolerance. Nat. Rev. Immunol. 2003, 3, 199–210. [Google Scholar] [CrossRef]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef]
- Ohkura, N.; Kitagawa, Y.; Sakaguchi, S. Development and maintenance of regulatory T cells. Immunity 2013, 38, 414–423. [Google Scholar] [CrossRef]
- Wing, J.B.; Sakaguchi, S. Multiple treg suppressive modules and their adaptability. Front. Immunol. 2012, 3, 178. [Google Scholar] [CrossRef]
- Fonseca, V.R.; Ribeiro, F.; Graca, L. T follicular regulatory (Tfr) cells: Dissecting the complexity of Tfr-cell compartments. Immunol. Rev. 2019, 288, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; DiPaolo, R.A.; Andersson, J.; Zhao, D.M.; Stephens, G.L.; Thornton, A.M. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol. Rev. 2006, 212, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Colvin, R.B.; Smith, R.N. Antibody-mediated organ-allograft rejection. Nat. Rev. Immunol. 2005, 5, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Nankivell, B.J.; Borrows, R.J.; Fung, C.L.; O’Connell, P.J.; Allen, R.D.; Chapman, J.R. The natural history of chronic allograft nephropathy. N. Engl. J. Med. 2003, 349, 2326–2333. [Google Scholar] [CrossRef]
- Buhler, L.H.; Spitzer, T.R.; Sykes, M.; Sachs, D.H.; Delmonico, F.L.; Tolkoff-Rubin, N.; Saidman, S.L.; Sackstein, R.; McAfee, S.; Dey, B.; et al. Induction of kidney allograft tolerance after transient lymphohematopoietic chimerism in patients with multiple myeloma and end-stage renal disease. Transplantation 2002, 74, 1405–1409. [Google Scholar] [CrossRef]
- Sykes, M. Bone marrow and solid organ transplantation start to converge. Curr. Opin. Immunol. 1999, 11, 493–496. [Google Scholar] [CrossRef]
- Tchao, N.K.; Turka, L.A. Lymphodepletion and homeostatic proliferation: Implications for transplantation. Am. J. Transplant. 2012, 12, 1079–1090. [Google Scholar] [CrossRef]
- Williams, K.M.; Hakim, F.T.; Gress, R.E. T cell immune reconstitution following lymphodepletion. Semin. Immunol. 2007, 19, 318–330. [Google Scholar] [CrossRef]
- Min, B. Spontaneous T Cell Proliferation: A Physiologic Process to Create and Maintain Homeostatic Balance and Diversity of the Immune System. Front. Immunol. 2018, 9, 547. [Google Scholar] [CrossRef]
- Wu, Z.; Bensinger, S.J.; Zhang, J.; Chen, C.; Yuan, X.; Huang, X.; Markmann, J.F.; Kassaee, A.; Rosengard, B.R.; Hancock, W.W.; et al. Homeostatic proliferation is a barrier to transplantation tolerance. Nat.Med. 2004, 10, 87–92. [Google Scholar] [CrossRef]
- Stockinger, B.; Barthlott, T.; Kassiotis, G. The concept of space and competition in immune regulation. Immunology 2004, 111, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.A.; Rocha, B. Population biology of lymphocytes: The flight for survival. Ann. Rev. Immunol. 2000, 18, 83–111. [Google Scholar] [CrossRef] [PubMed]
- Tanchot, C.; Rosado, M.M.; Agenes, F.; Freitas, A.A.; Rocha, B. Lymphocyte homeostasis. Semin. Immunol. 1997, 9, 331–337. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Ilic, A.; Koelsch, K.; Sarvetnick, N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell 2004, 117, 265–277. [Google Scholar] [CrossRef]
- Datta, S.; Sarvetnick, N. Lymphocyte proliferation in immune-mediated diseases. Trends Immunol. 2009, 30, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, H.; Meadors, J.; Poon, R.; Guimond, M.; Mackall, C.L. Harnessing the physiology of lymphopenia to support adoptive immunotherapy in lymphoreplete hosts. Blood 2009, 114, 3831–3840. [Google Scholar] [CrossRef]
- Mackall, C.L.; Fry, T.J.; Bare, C.; Morgan, P.; Galbraith, A.; Gress, R.E. IL-7 increases both thymic-dependent and thymic-independent T-cell regeneration after bone marrow transplantation. Blood 2001, 97, 1491–1497. [Google Scholar] [CrossRef]
- Surh, C.D.; Boyman, O.; Purton, J.F.; Sprent, J. Homeostasis of memory T cells. Immunol. Rev. 2006, 211, 154–163. [Google Scholar] [CrossRef]
- Goldrath, A.W.; Sivakumar, P.V.; Glaccum, M.; Kennedy, M.K.; Bevan, M.J.; Benoist, C.; Mathis, D.; Butz, E.A. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J. Exp. Med. 2002, 195, 1515–1522. [Google Scholar] [CrossRef]
- Berzins, S.P.; Uldrich, A.P.; Sutherland, J.S.; Gill, J.; Miller, J.F.; Godfrey, D.I.; Boyd, R.L. Thymic regeneration: Teaching an old immune system new tricks. Trends Mol. Med. 2002, 8, 469–476. [Google Scholar] [CrossRef]
- Mackall, C.L.; Bare, C.V.; Granger, L.A.; Sharrow, S.O.; Titus, J.A.; Gress, R.E. Thymic-independent T cell regeneration occurs via antigen-driven expansion of peripheral T cells resulting in a repertoire that is limited in diversity and prone to skewing. J. Immunol. 1996, 156, 4609–4616. [Google Scholar] [PubMed]
- Salomon, B.; Lenschow, D.J.; Rhee, L.; Ashourian, N.; Singh, B.; Sharpe, A.; Bluestone, J.A. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000, 12, 431–440. [Google Scholar] [CrossRef]
- Boden, E.; Tang, Q.; Bour-Jordan, H.; Bluestone, J.A. The role of CD28 and CTLA4 in the function and homeostasis of CD4+CD25+ regulatory T cells. Novartis Found. Symp. 2003, 252, 55–63. [Google Scholar] [PubMed]
- Tang, Q.; Henriksen, K.J.; Boden, E.K.; Tooley, A.J.; Ye, J.; Subudhi, S.K.; Zheng, X.X.; Strom, T.B.; Bluestone, J.A. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J. Immunol. 2003, 171, 3348–3352. [Google Scholar] [CrossRef]
- Riella, L.V.; Liu, T.; Yang, J.; Chock, S.; Shimizu, T.; Mfarrej, B.; Batal, I.; Xiao, X.; Sayegh, M.H.; Chandraker, A. Deleterious effect of CTLA4-Ig on a Treg-dependent transplant model. Am. J. Transplant. 2012, 12, 846–855. [Google Scholar] [CrossRef]
- Verbinnen, B.; Billiau, A.D.; Vermeiren, J.; Galicia, G.; Bullens, D.M.; Boon, L.; Cadot, P.; Hens, G.; Dewolf-Peeters, C.; Van Gool, S.W.; et al. Contribution of regulatory T cells and effector T cell deletion in tolerance induction by costimulation blockade. J. Immunol. 2008, 181, 1034–1042. [Google Scholar] [CrossRef]
- Schwarz, C.; Unger, L.; Mahr, B.; Aumayr, K.; Regele, H.; Farkas, A.M.; Hock, K.; Pilat, N.; Wekerle, T. The Immunosuppressive Effect of CTLA4 Immunoglobulin Is Dependent on Regulatory T Cells at Low But Not High Doses. Am. J. Transplant. 2016, 16, 3404–3415. [Google Scholar] [CrossRef]
- Vincenti, F.; Rostaing, L.; Grinyo, J.; Rice, K.; Steinberg, S.; Gaite, L.; Moal, M.C.; Mondragon-Ramirez, G.A.; Kothari, J.; Polinsky, M.S.; et al. Belatacept and Long-Term Outcomes in Kidney Transplantation. N. Engl. J. Med. 2016, 374, 333–343. [Google Scholar] [CrossRef]
- Levitsky, J.; Miller, J.; Huang, X.; Chandrasekaran, D.; Chen, L.; Mathew, J.M. Inhibitory effects of belatacept on allospecific regulatory T-cell generation in humans. Transplantation 2013, 96, 689–696. [Google Scholar] [CrossRef]
- Sanchez-Fueyo, A.; Domenig, C.; Strom, T.B.; Zheng, X.X. The complement dependent cytotoxicity (CDC) immune effector mechanism contributes to anti-CD154 induced immunosuppression. Transplantation 2002, 74, 898–900. [Google Scholar] [CrossRef]
- Hancock, W.W.; Sayegh, M.H.; Zheng, X.G.; Peach, R.; Linsley, P.S.; Turka, L.A. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc. Natl. Acad. Sci. USA 1996, 93, 13967–13972. [Google Scholar] [CrossRef] [PubMed]
- Vogel, I.; Verbinnen, B.; Van Gool, S.; Ceuppens, J.L. Regulatory T Cell-Dependent and -Independent Mechanisms of Immune Suppression by CD28/B7 and CD40/CD40L Costimulation Blockade. J. Immunol. 2016, 197, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Koyama, I.; Kawai, T.; Andrews, D.; Boskovic, S.; Nadazdin, O.; Wee, S.L.; Sogawa, H.; Wu, D.L.; Smith, R.N.; Colvin, R.B.; et al. Thrombophilia associated with anti-CD154 monoclonal antibody treatment and its prophylaxis in nonhuman primates. Transplantation 2004, 77, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.M.; Shevach, E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998, 188, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Gregori, S.; Bacchetta, R.; Passerini, L.; Levings, M.K.; Roncarolo, M.G. Isolation, expansion, and characterization of human natural and adaptive regulatory T cells. Methods Mol. Biol. 2007, 380, 83–105. [Google Scholar] [PubMed]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2019, 20, 158–172. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Huehn, J.; Polansky, J.K.; Hamann, A. Epigenetic control of FOXP3 expression: The key to a stable regulatory T-cell lineage? Nat. Rev. Immunol. 2009, 9, 83–89. [Google Scholar] [CrossRef]
- Sanchez-Abarca, L.I.; Gutierrez-Cosio, S.; Santamaria, C.; Caballero-Velazquez, T.; Blanco, B.; Herrero-Sanchez, C.; Garcia, J.L.; Carrancio, S.; Hernandez-Campo, P.; Gonzalez, F.J.; et al. Immunomodulatory effect of 5-azacytidine (5-azaC): Potential role in the transplantation setting. Blood 2010, 115, 107–121. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Del Rio, M.L.; Schneider, P.; Fernandez-Renedo, C.; Perez-Simon, J.A.; Rodriguez-Barbosa, J.I. LIGHT/HVEM/LTbetaR Interaction as a Target for the Modulation of the Allogeneic Immune Response in Transplantation. Am. J. Transplant. 2013, 13, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Ward-Kavanagh, L.K.; Lin, W.W.; Sedy, J.R.; Ware, C.F. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, T.H.; Wolf, D.; Tsai, M.S.; Chirinos, J.; Deyev, V.V.; Gonzalez, L.; Malek, T.R.; Levy, R.B.; Podack, E.R. Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J. Clin. Investig. 2010, 120, 3629–3640. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Baumel, M.; Mannel, D.N.; Howard, O.M.; Oppenheim, J.J. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 2007, 179, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Wing, J.B.; Tay, C.; Sakaguchi, S. Control of Regulatory T Cells by Co-signal Molecules. Adv. Exp. Med. Biol. 2019, 1189, 179–210. [Google Scholar]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef]
- Mahmud, S.A.; Manlove, L.S.; Schmitz, H.M.; Xing, Y.; Wang, Y.; Owen, D.L.; Schenkel, J.M.; Boomer, J.S.; Green, J.M.; Yagita, H.; et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat. Immunol. 2014, 15, 473–481. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Liao, Y.; Teh, P.; Pascutti, M.F.; Oja, A.E.; Garnham, A.L.; Gloury, R.; Tempany, J.C.; Sidwell, T.; Cuadrado, E.; et al. The TNF Receptor Superfamily-NF-kappaB Axis Is Critical to Maintain Effector Regulatory T Cells in Lymphoid and Non-lymphoid Tissues. Cell Rep. 2017, 20, 2906–2920. [Google Scholar] [CrossRef]
- Ronin, E.; Lubrano di Ricco, M.; Vallion, R.; Divoux, J.; Kwon, H.K.; Gregoire, S.; Collares, D.; Rouers, A.; Baud, V.; Benoist, C.; et al. The NF-kappaB RelA Transcription Factor Is Critical for Regulatory T Cell Activation and Stability. Front. Immunol. 2019, 10, 2487. [Google Scholar] [CrossRef]
- Kim, E.Y.; Teh, H.S. TNF type 2 receptor (p75) lowers the threshold of T cell activation. J. Immunol. 2001, 167, 6812–6820. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Milsark, I.W.; Cerami, A.C. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 1985, 229, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Adhesion receptors of the immune system. Nature 1990, 346, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; Chantry, D.; Jackson, A.; Maini, R.; Feldmann, M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet 1989, 2, 244–247. [Google Scholar] [CrossRef]
- Pfeffer, K. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. 2003, 14, 185–191. [Google Scholar] [CrossRef]
- Chen, X.; Subleski, J.J.; Hamano, R.; Howard, O.M.; Wiltrout, R.H.; Oppenheim, J.J. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur. J. Immunol. 2010, 40, 1099–1106. [Google Scholar] [CrossRef]
- Chen, X.; Subleski, J.J.; Kopf, H.; Howard, O.M.; Mannel, D.N.; Oppenheim, J.J. Cutting edge: Expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: Applicability to tumor-infiltrating T regulatory cells. J. Immunol. 2008, 180, 6467–6471. [Google Scholar] [CrossRef]
- Chen, X.; Wu, X.; Zhou, Q.; Howard, O.M.; Netea, M.G.; Oppenheim, J.J. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J. Immunol. 2013, 190, 1076–1084. [Google Scholar] [CrossRef]
- Cohen, J.L.; Wood, K.J. TNFR2: The new Treg switch? Oncoimmunology 2017, 7, e1373236. [Google Scholar] [CrossRef]
- Yin, X.; Li, W.; Ma, H.; Zeng, W.; Peng, C.; Li, Y.; Liu, M.; Chen, Q.; Zhou, R.; Jin, T. Crystal structure and activation mechanism of DR3 death domain. FEBS J. 2019, 286, 2593–2610. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Atretkhany, K.N.; Mufazalov, I.A.; Dunst, J.; Kuchmiy, A.; Gogoleva, V.S.; Andruszewski, D.; Drutskaya, M.S.; Faustman, D.L.; Schwabenland, M.; Prinz, M.; et al. Intrinsic TNFR2 signaling in T regulatory cells provides protection in CNS autoimmunity. Proc. Natl. Acad. Sci. USA 2018, 115, 13051–13056. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, M.; Ziegler, H.K. Paradoxical anti-inflammatory actions of TNF-alpha: Inhibition of IL-12 and IL-23 via TNF receptor 1 in macrophages and dendritic cells. J. Immunol. 2005, 175, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Erickson, S.L.; de Sauvage, F.J.; Kikly, K.; Carver-Moore, K.; Pitts-Meek, S.; Gillett, N.; Sheehan, K.C.; Schreiber, R.D.; Goeddel, D.V.; Moore, M.W. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 1994, 372, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Bossen, C.; Ingold, K.; Tardivel, A.; Bodmer, J.L.; Gaide, O.; Hertig, S.; Ambrose, C.; Tschopp, J.; Schneider, P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 2006, 281, 13964–13971. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Kashiwase, K.; Akaza, T.; Morishima, Y.; Inoko, H.; Sasazuki, T.; Kodera, Y.; Juji, T. Polymorphisms in TNFA and TNFR2 affect outcome of unrelated bone marrow transplantation. Bone Marrow Transplant. 2002, 29, 569–575. [Google Scholar] [CrossRef]
- Johnson, K.J.; Sanchez, H.N.; Schoenbrunner, N. Defining response to TNF-inhibitors in rheumatoid arthritis: The negative impact of anti-TNF cycling and the need for a personalized medicine approach to identify primary non-responders. Clin. Rheumatol. 2019, 38, 2967–2976. [Google Scholar] [CrossRef]
- Nie, H.; Zheng, Y.; Li, R.; Zhang, J. Reply to Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat. Med. 2016, 22, 18–19. [Google Scholar] [CrossRef]
- Zaragoza, B.; Chen, X.; Oppenheim, J.J.; Baeyens, A.; Gregoire, S.; Chader, D.; Gorochov, G.; Miyara, M.; Salomon, B.L. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat. Med. 2016, 22, 16–17. [Google Scholar] [CrossRef]
- Okubo, Y.; Mera, T.; Wang, L.; Faustman, D.L. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci. Rep. 2013, 3, 3153. [Google Scholar] [CrossRef]
- Chen, X.; Hamano, R.; Subleski, J.J.; Hurwitz, A.A.; Howard, O.M.; Oppenheim, J.J. Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3- conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J. Immunol. 2010, 185, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Ferrara, J.L. Immunobiology of acute graft-versus-host disease. Blood Rev. 2003, 17, 187–194. [Google Scholar] [CrossRef]
- Blazar, B.R.; MacDonald, K.P.A.; Hill, G.R. Immune regulatory cell infusion for graft-versus-host disease prevention and therapy. Blood 2018, 131, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.; Santolaria, T.; Calise, D.; Al, S.T.; Hudrisier, D.; Romagnoli, P.; van Meerwijk, J.P. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat. Med. 2008, 14, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Chopra, M.; Biehl, M.; Steinfatt, T.; Brandl, A.; Kums, J.; Amich, J.; Vaeth, M.; Kuen, J.; Holtappels, R.; Podlech, J.; et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med. 2016, 213, 1881–1900. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, M.; Naserian, S.; Pilon, C.; Thiolat, A.; Martin, G.H.; Pouchy, C.; Dominique, C.; Belkacemi, Y.; Charlotte, F.; Maury, S.; et al. Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells. Blood 2016, 128, 1651–1659. [Google Scholar] [CrossRef]
- Pierini, A.; Strober, W.; Moffett, C.; Baker, J.; Nishikii, H.; Alvarez, M.; Pan, Y.; Schneidawind, D.; Meyer, E.; Negrin, R.S. TNF-alpha priming enhances CD4+FoxP3+ regulatory T-cell suppressive function in murine GVHD prevention and treatment. Blood 2016, 128, 866–871. [Google Scholar] [CrossRef]
- Suzuki, J.; Cole, S.E.; Batirel, S.; Kosuge, H.; Shimizu, K.; Isobe, M.; Libby, P.; Mitchell, R.N. Tumor necrosis factor receptor -1 and -2 double deficiency reduces graft arterial disease in murine cardiac allografts. Am. J. Transplant. 2003, 3, 968–976. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M.; O’Rourke, K.; Yu, G.L.; Lyons, R.H.; Garg, M.; Duan, D.R.; Xing, L.; Gentz, R.; Ni, J.; Dixit, V.M. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science 1996, 274, 990–992. [Google Scholar] [CrossRef]
- Migone, T.S.; Zhang, J.; Luo, X.; Zhuang, L.; Chen, C.; Hu, B.; Hong, J.S.; Perry, J.W.; Chen, S.F.; Zhou, J.X.; et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 2002, 16, 479–492. [Google Scholar] [CrossRef]
- Richard, A.C.; Ferdinand, J.R.; Meylan, F.; Hayes, E.T.; Gabay, O.; Siegel, R.M. The TNF-family cytokine TL1A: From lymphocyte costimulator to disease co-conspirator. J. Leukoc. Biol. 2015, 98, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Taraban, V.Y.; Ferdinand, J.R.; Al-Shamkhani, A. Expression of TNFRSF25 on conventional T cells and Tregs. J. Clin. Investig. 2011, 121, 463–464. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Nishikii, H.; Baker, J.; Pierini, A.; Schneidawind, D.; Pan, Y.; Beilhack, A.; Park, C.G.; Negrin, R.S. Treatment with agonistic DR3 antibody results in expansion of donor Tregs and reduced graft-versus-host disease. Blood 2015, 126, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Nishikii, H.; Kim, B.S.; Yokoyama, Y.; Chen, Y.; Baker, J.; Pierini, A.; Alvarez, M.; Mavers, M.; Maas-Bauer, K.; Pan, Y.; et al. DR3 signaling modulates the function of Foxp3+ regulatory T cells and the severity of acute graft-versus-host disease. Blood 2016, 128, 2846–2858. [Google Scholar] [CrossRef] [PubMed]
- Mavers, M.; Simonetta, F.; Nishikii, H.; Ribado, J.V.; Maas-Bauer, K.; Alvarez, M.; Hirai, T.; Turkoz, M.; Baker, J.; Negrin, R.S. Activation of the DR3-TL1A Axis in Donor Mice Leads to Regulatory T Cell Expansion and Activation With Reduction in Graft-Versus-Host Disease. Front. Immunol. 2019, 10, 1624. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Schreiber, T.H.; Tryphonopoulos, P.; Li, S.; Tzakis, A.G.; Ruiz, P.; Podack, E.R. Tregs expanded in vivo by TNFRSF25 agonists promote cardiac allograft survival. Transplantation 2012, 94, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Mancusi, A.; Alvarez, M.; Piccinelli, S.; Velardi, A.; Pierini, A. TNFR2 signaling modulates immunity after allogeneic hematopoietic cell transplantation. Cytokine Growth Factor Rev. 2019, 47, 54–61. [Google Scholar] [CrossRef]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Barbosa, J.-I.; Schneider, P.; Graca, L.; Bühler, L.; Perez-Simon, J.-A.; del Rio, M.-L. The Role of TNFR2 and DR3 in the In Vivo Expansion of Tregs in T Cell Depleting Transplantation Regimens. Int. J. Mol. Sci. 2020, 21, 3347. https://doi.org/10.3390/ijms21093347
Rodriguez-Barbosa J-I, Schneider P, Graca L, Bühler L, Perez-Simon J-A, del Rio M-L. The Role of TNFR2 and DR3 in the In Vivo Expansion of Tregs in T Cell Depleting Transplantation Regimens. International Journal of Molecular Sciences. 2020; 21(9):3347. https://doi.org/10.3390/ijms21093347
Chicago/Turabian StyleRodriguez-Barbosa, Jose-Ignacio, Pascal Schneider, Luis Graca, Leo Bühler, Jose-Antonio Perez-Simon, and Maria-Luisa del Rio. 2020. "The Role of TNFR2 and DR3 in the In Vivo Expansion of Tregs in T Cell Depleting Transplantation Regimens" International Journal of Molecular Sciences 21, no. 9: 3347. https://doi.org/10.3390/ijms21093347
APA StyleRodriguez-Barbosa, J.-I., Schneider, P., Graca, L., Bühler, L., Perez-Simon, J.-A., & del Rio, M.-L. (2020). The Role of TNFR2 and DR3 in the In Vivo Expansion of Tregs in T Cell Depleting Transplantation Regimens. International Journal of Molecular Sciences, 21(9), 3347. https://doi.org/10.3390/ijms21093347