Predictive Potential of Circulating Ube2h mRNA as an E2 Ubiquitin-Conjugating Enzyme for Diagnosis or Treatment of Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
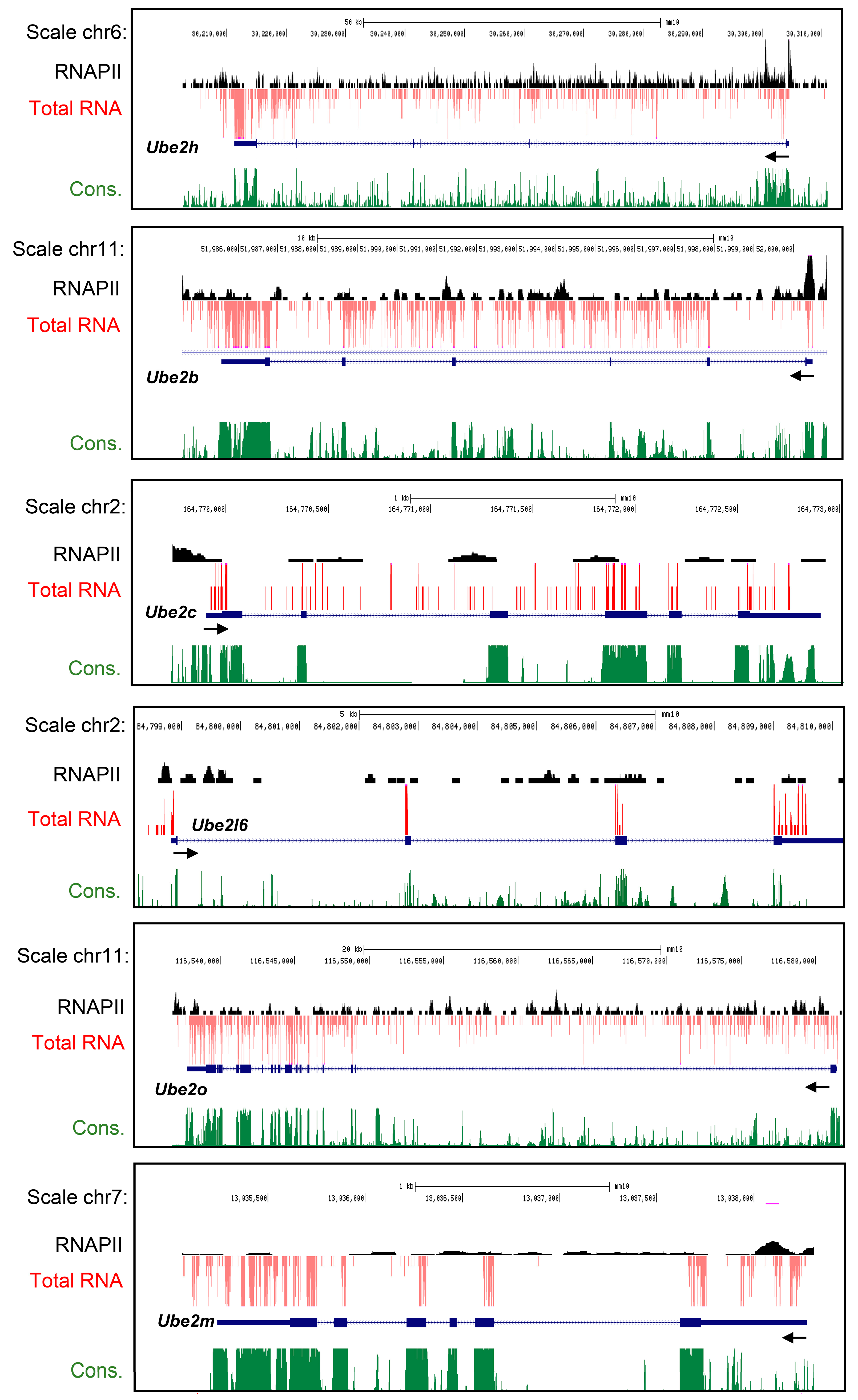
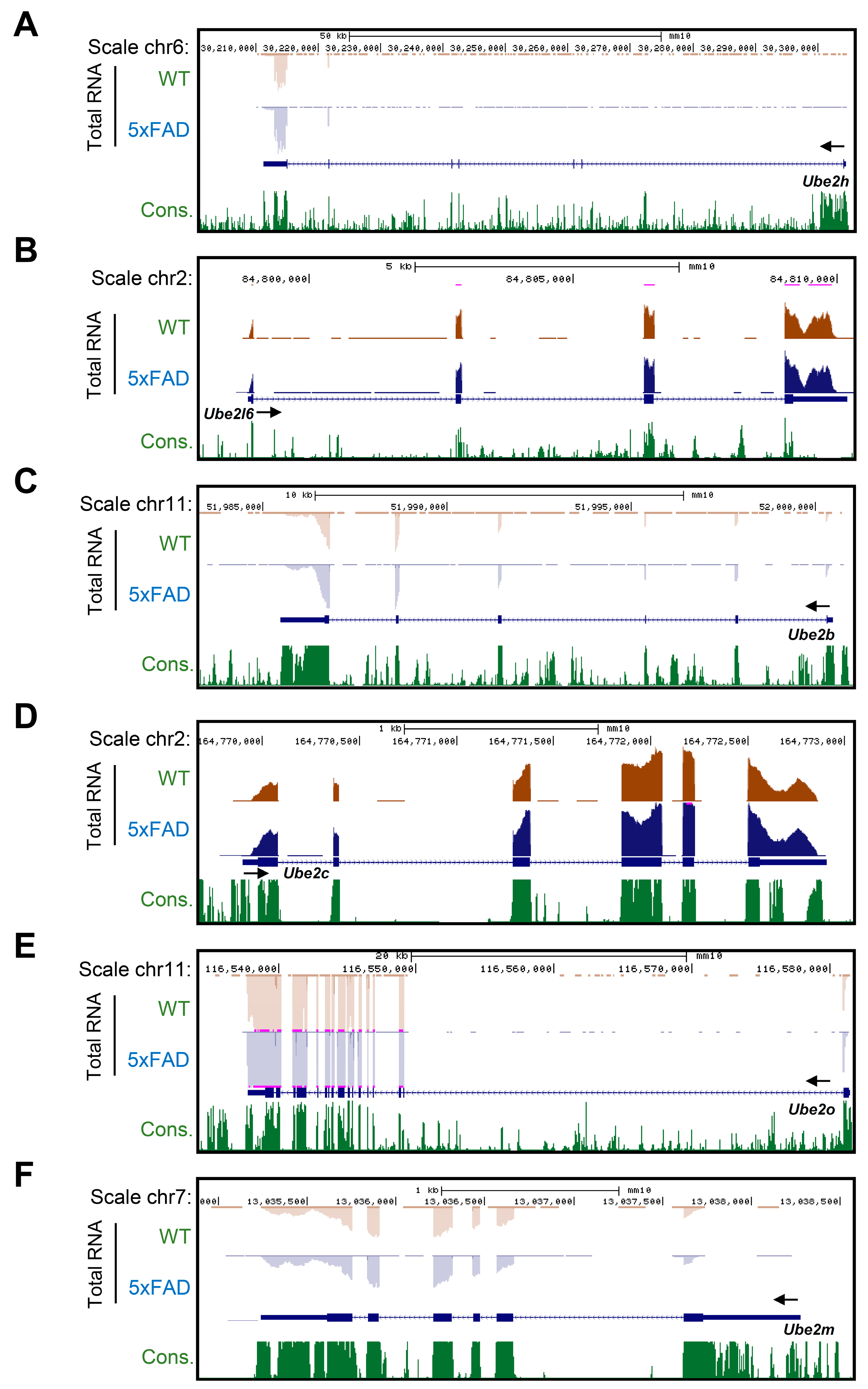
2.1. Characterization of Ube2 Subfamily Genes Expression in the Primary Cortical Neurons from RNA-Seq Data Base
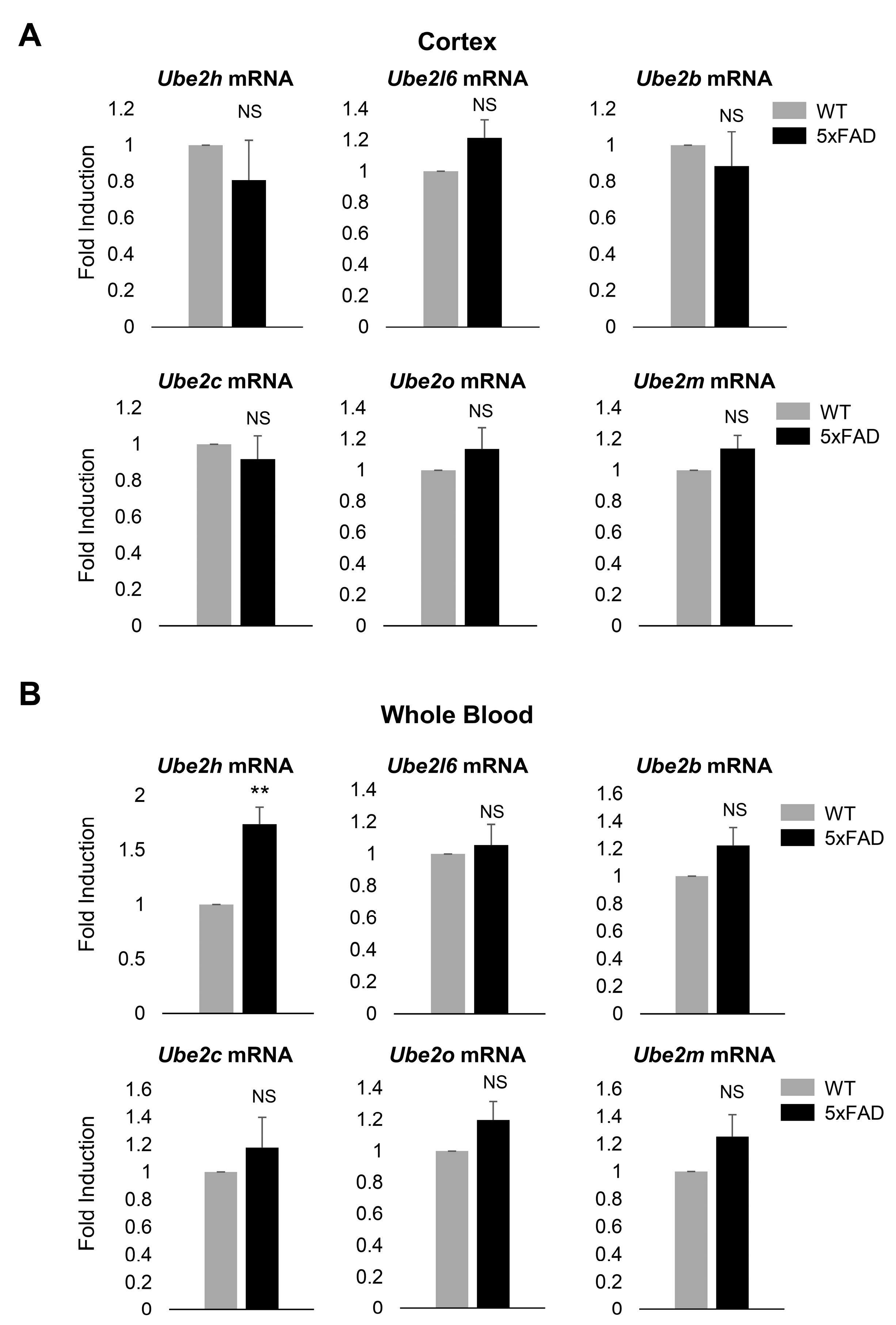
2.2. Ube2h mRNA is Abundantly Expressed in the Blood from AD
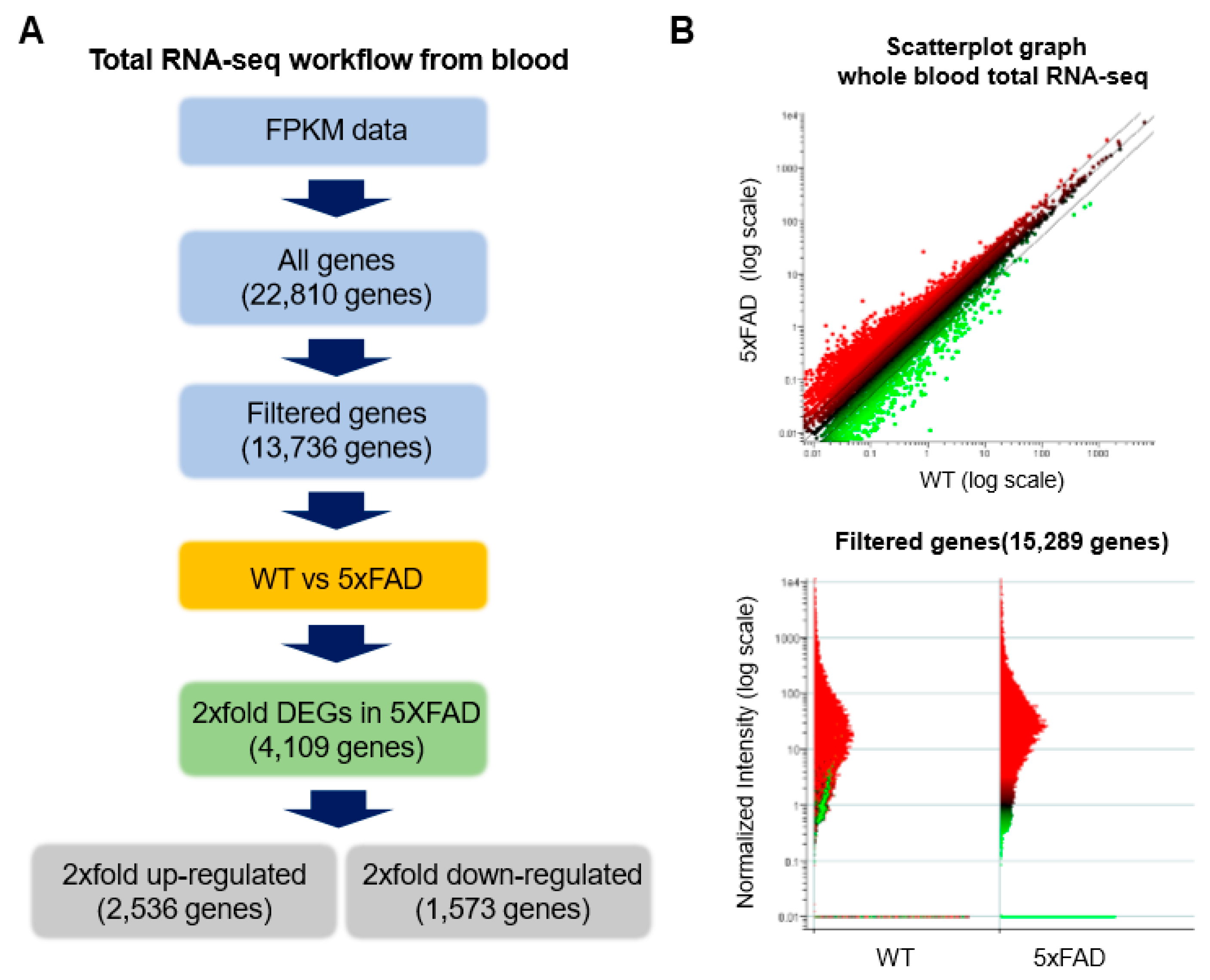
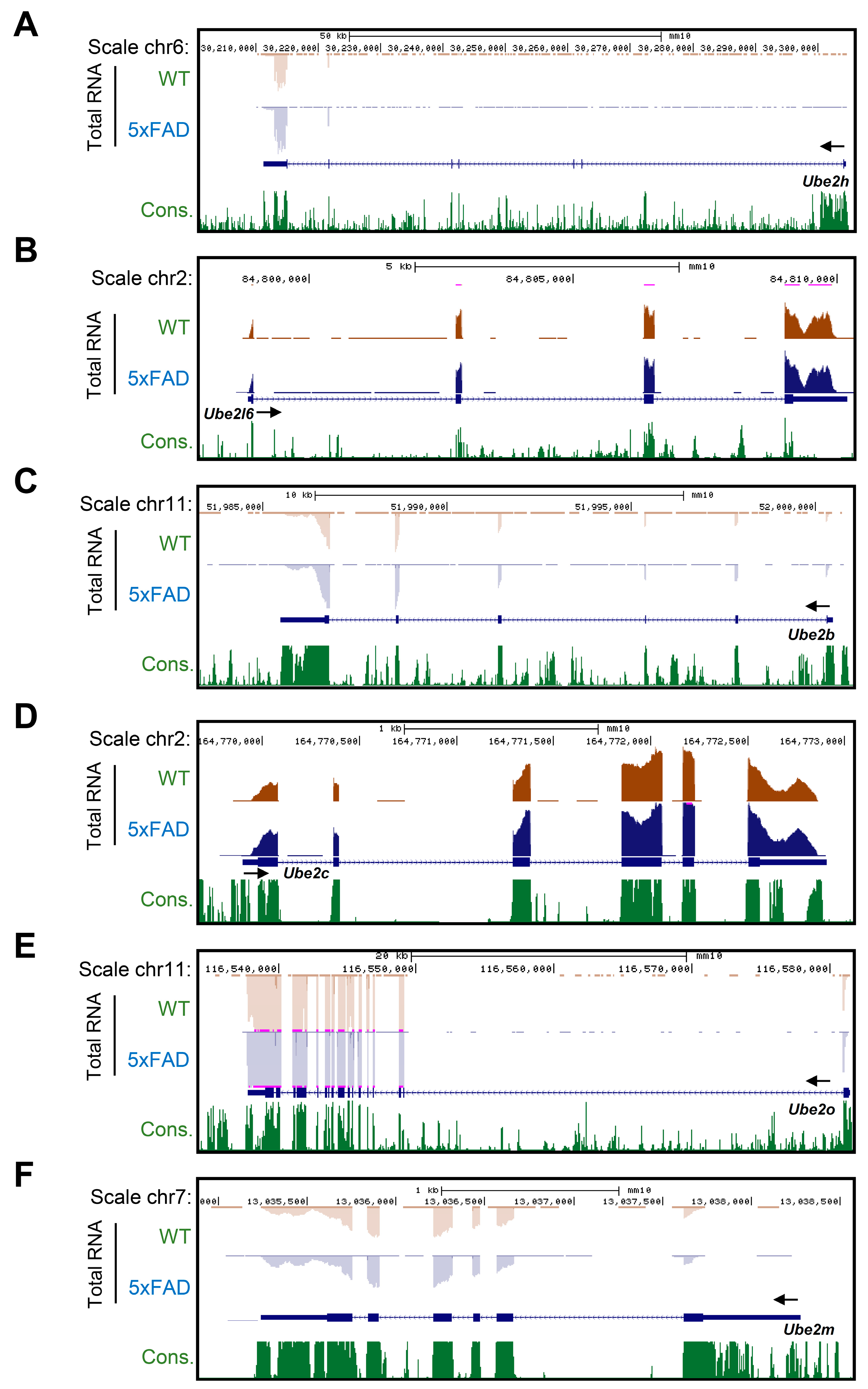
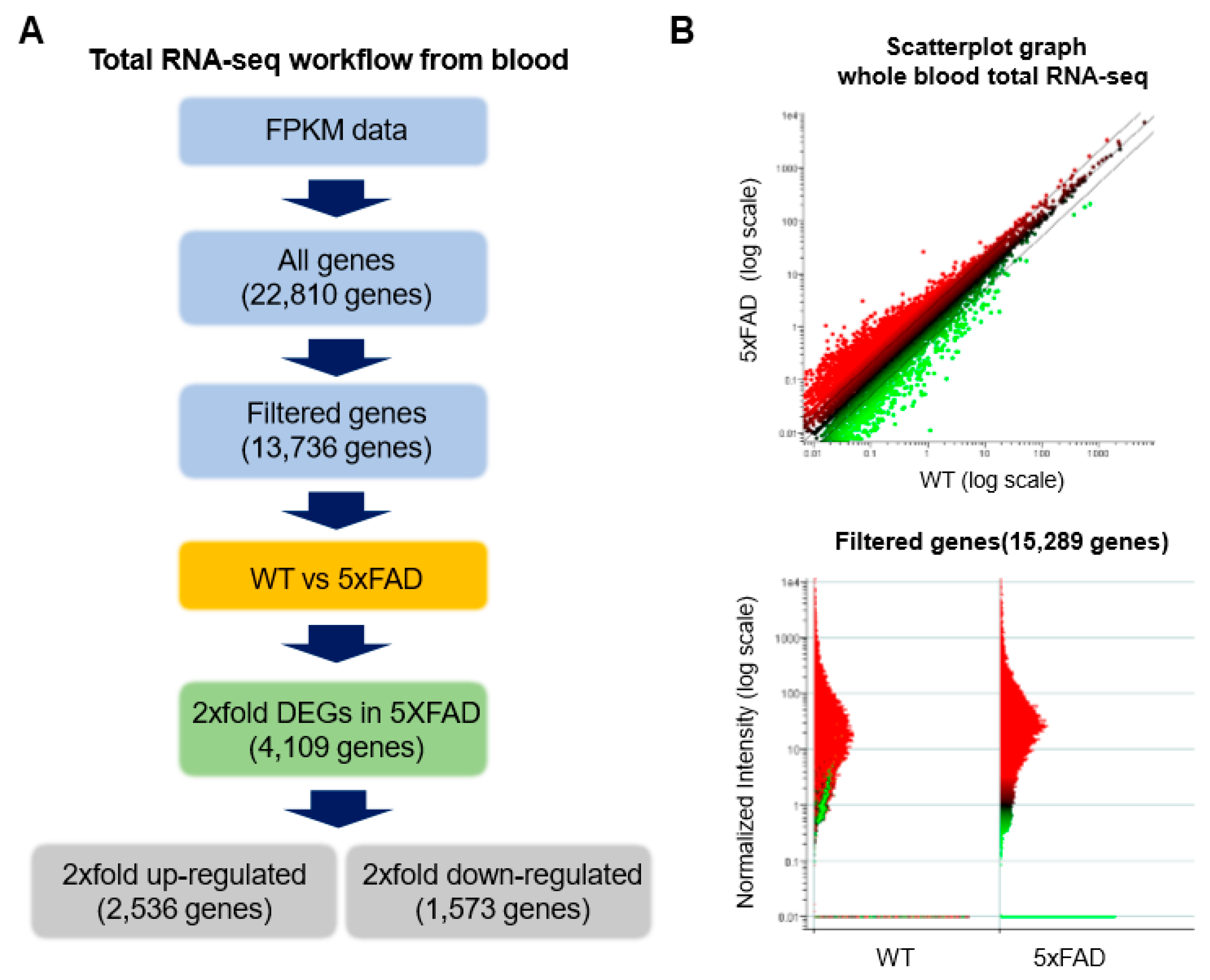
2.3. Accurate Prediction of Gene Expression Change through Total RNA-Seq for AD from Whole Blood
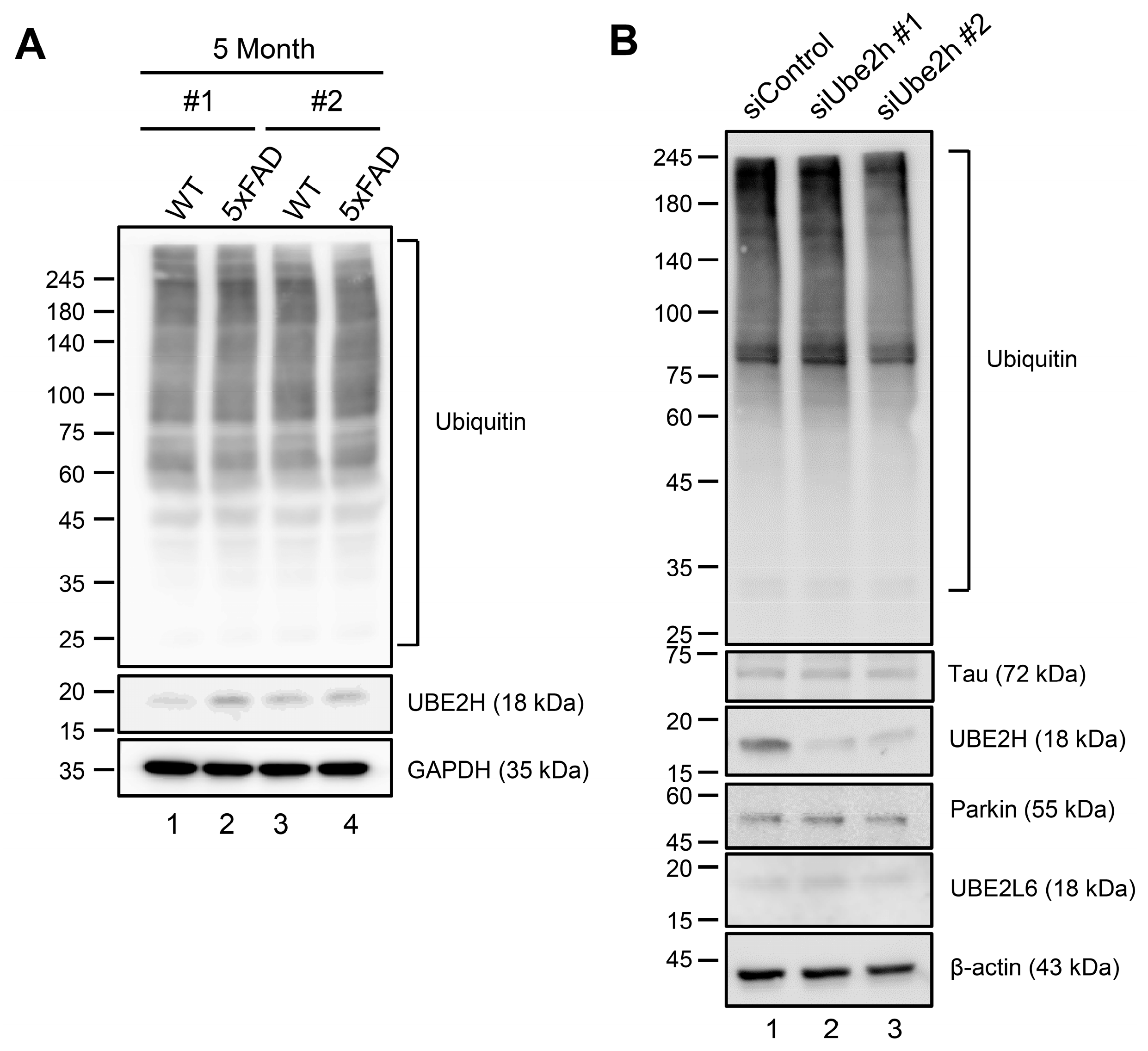
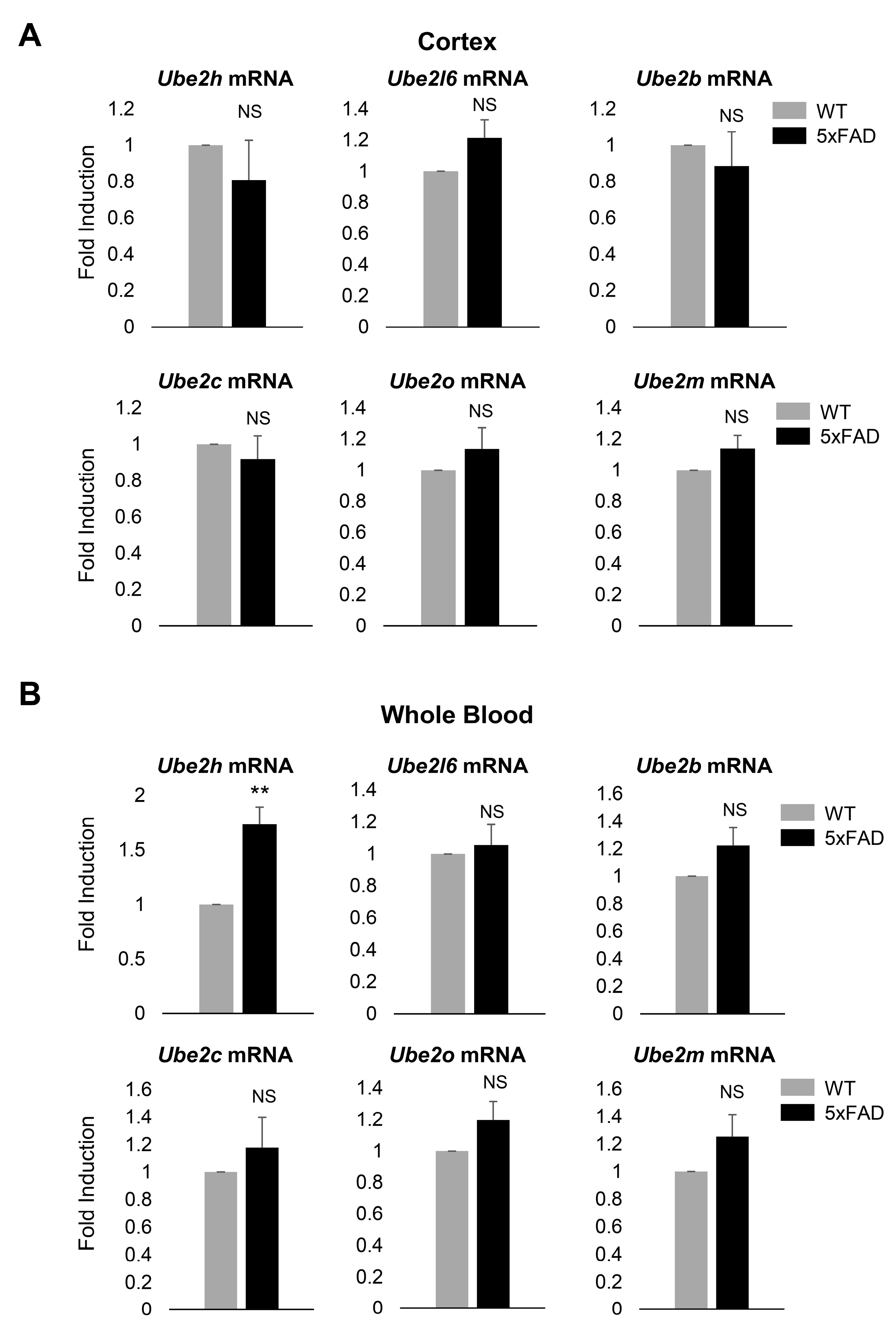
2.4. Biochemical Characterization of UBE2H on AD in Cortex
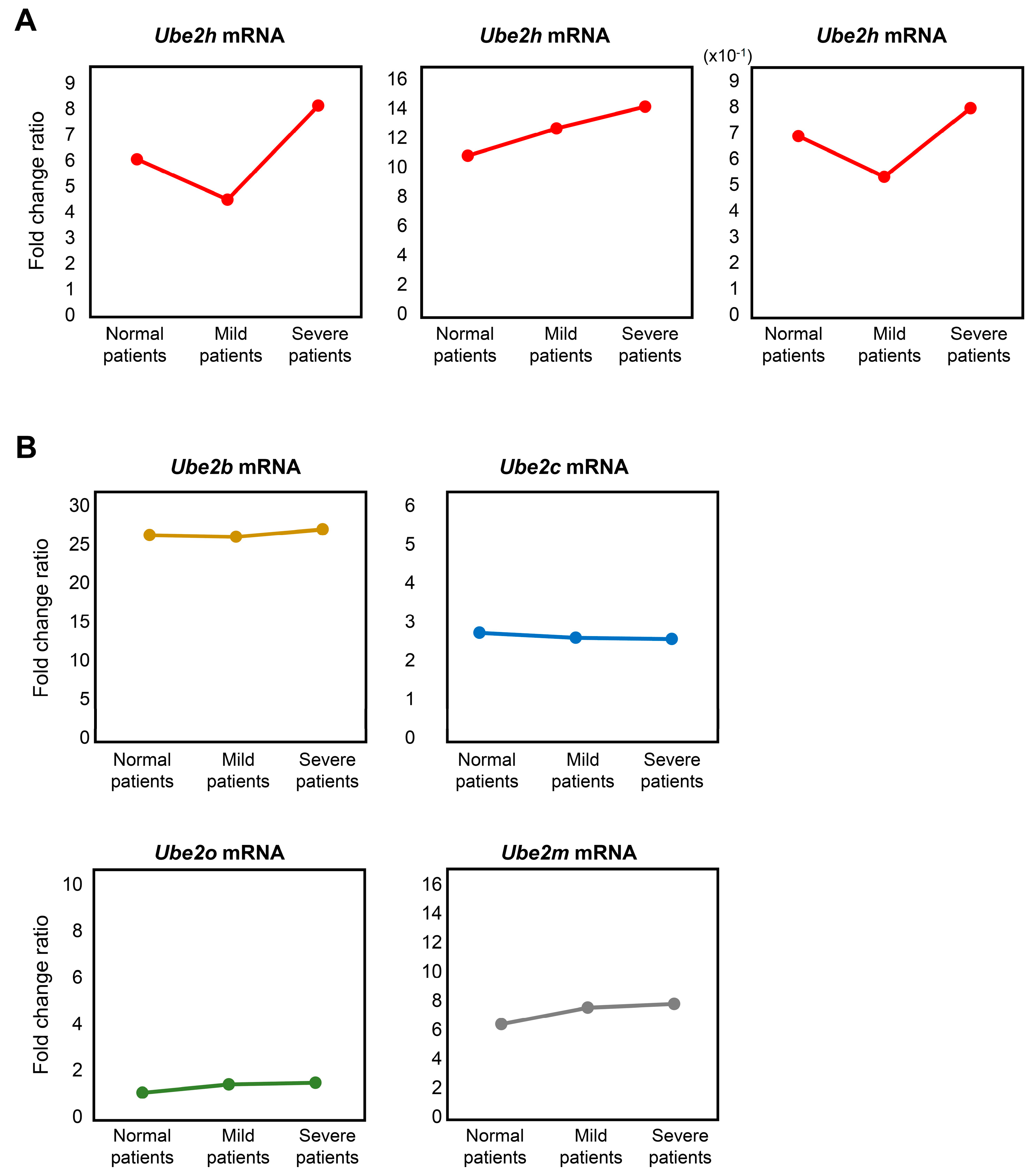
2.5. Analysis of Ube2 Subfamilies’ Gene Expression Profiles in AD Patients from Peripheral Blood Mononuclear Cells (PBMCs) Microarray Dataset
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. RNA-Seq
4.3. Sequencing Data Analysis
4.4. Microarray Data
4.5. Data Analysis
4.6. RT-qPCR
4.7. Cell Culture, siRNA Transfection and Immunoblotting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2019, 61, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.E.; Balendra, S.I.; Almonte, M.T.; Cordeiro, M.F. Retinal correlates of neurological disorders. Ther. Adv. Chronic Dis. 2019, 10, 2040622319882205. [Google Scholar] [CrossRef]
- Fu, P.; Gao, M.; Yung, K.K.L. Association of intestinal disorders with Parkinson’s disease and Alzheimer’s disease: A systematic review and meta-analysis. ACS Chem. Neurosci. 2019, 11, 395–405. [Google Scholar] [CrossRef]
- Davis, L.; Chin, J.W. Designer proteins: Applications of genetic code expansion in cell biology. Nat. Rev. Mol. Cell Biol. 2012, 13, 168–182. [Google Scholar] [CrossRef]
- Lim, K.H.; Song, M.H.; Baek, K.H. Decision for cell fate: Deubiquitinating enzymes in cell cycle checkpoint. Cell Mol. Life Sci. 2016, 73, 1439–1455. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Bertrand, M.J. The role of the IAP E3 ubiquitin ligases in regulating pattern-recognition receptor signalling. Nat. Rev. Immunol. 2012, 12, 833–844. [Google Scholar] [CrossRef]
- Haakonsen, D.L.; Rape, M. Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 2019, 29, 704–716. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Nakajima, B.; Nomura, M.; Arfin, S.M. Cloning and characterization of a Saccharomyces cerevisiae gene encoding a new member of the ubiquitin-conjugating protein family. J. Biol. Chem. 1991, 266, 15549–15554. [Google Scholar]
- Kaiser, P.; Seufert, W.; Hofferer, L.; Kofler, B.; Sachsenmaier, C.; Herzog, H.; Jentsch, S.; Schweiger, M.; Schneider, R. A human ubiquitin-conjugating enzyme homologous to yeast UBC8. J. Biol. Chem. 1994, 269, 8797–8802. [Google Scholar] [PubMed]
- Kaiser, P.; Mandl, S.; Schweiger, M.; Schneider, R. Characterization of functionally independent domains in the human ubiquitin conjugating enzyme UbcH2. FEBS Lett. 1995, 377, 193–196. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.P.; Lecker, S.H.; Chen, Y.; Waddell, I.D.; Goldberg, A.L.; Reid, M.B. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J. 2003, 17, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Vourc’h, P.; Martin, I.; Bonnet-Brilhault, F.; Marouillat, S.; Barthelemy, C.; Pierre Muh, J.; Andres, C. Mutation screening and association study of the UBE2H gene on chromosome 7q32 in autistic disorder. Psychiatr. Genet. 2003, 13, 221–225. [Google Scholar] [CrossRef]
- Martin, I.; Vourc’h, P.; Mahe, M.; Thepault, R.A.; Antar, C.; Vedrine, S.; Praline, J.; Camu, W.; Andres, C.R.; Corcia, P.; et al. Association study of the ubiquitin conjugating enzyme gene UBE2H in sporadic ALS. Amyotroph. Lateral. Scler. 2009, 10, 432–435. [Google Scholar] [CrossRef]
- Sokolowski, M.; Wasserman, J.; Wasserman, D. Gene-level associations in suicide attempter families show overrepresentation of synaptic genes and genes differentially expressed in brain development. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 774–784. [Google Scholar] [CrossRef]
- Kim, K.M.; Abdelmohsen, K.; Mustapic, M.; Kapogiannis, D.; Gorospe, M. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8, e1413. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiklejohn, H.; Mostaid, M.S.; Luza, S.; Mancuso, S.G.; Kang, D.; Atherton, S.; Rothmond, D.A.; Weickert, C.S.; Opazo, C.M.; Pantelis, C.; et al. Blood and brain protein levels of ubiquitin-conjugating enzyme E2K (UBE2K) are elevated in individuals with schizophrenia. J. Psychiatr. Res. 2019, 113, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Liu, M.; Du, K.; Zhong, X.; Gong, S.; Jiao, L.; Wei, M. Differential Expression of mRNAs in the Brain Tissues of Patients with Alzheimer’s Disease Based on GEO Expression Profile and Its Clinical Significance. Biomed. Res. Int. 2019, 2019, 8179145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Kim, Y.S.; Jung, H.M.; Yoon, B.E. Exploring glia to better understand Alzheimer’s disease. Anim. Cells Syst. 2018, 22, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, Y.; Zhang, L.; Dong, Y.; Ji, H.; Shen, L. The Potential Markers of Circulating microRNAs and long non-coding RNAs in Alzheimer’s Disease. Aging Dis. 2019, 10, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Arning, A.; Hiersche, M.; Witten, A.; Kurlemann, G.; Kurnik, K.; Manner, D.; Stoll, M.; Nowak-Gottl, U. A genome-wide association study identifies a gene network of ADAMTS genes in the predisposition to pediatric stroke. Blood 2012, 120, 5231–5236. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.E.; Lee, T.; Lee, I. From sequencing data to gene functions: Co-functional network approaches. Anim. Cells Syst. 2017, 21, 77–83. [Google Scholar] [CrossRef]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.C.; Ponting, C.P. Catalogues of mammalian long noncoding RNAs: Modest conservation and incompleteness. Genome Biol. 2009, 10, R124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, J.Y.; Schaukowitch, K.; Farbiak, L.; Kilaru, G.; Kim, T.K. Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat. Neurosci. 2016, 19, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Schaukowitch, K.; Joo, J.Y.; Liu, X.; Watts, J.K.; Martinez, C.; Kim, T.K. Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 2014, 56, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Siddeek, B.; Inoubli, L.; Lakhdari, N.; Rachel, P.B.; Fussell, K.C.; Schneider, S.; Mauduit, C.; Benahmed, M. MicroRNAs as potential biomarkers in diseases and toxicology. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 764–765, 46–57. [Google Scholar] [CrossRef]
- Yu, H.W.; Cho, W.C. The role of microRNAs in toxicology. Arch. Toxicol. 2015, 89, 319–325. [Google Scholar] [CrossRef]
- Kopreski, M.S.; Benko, F.A.; Kwak, L.W.; Gocke, C.D. Detection of tumor messenger RNA in the serum of patients with malignant melanoma. Clin. Cancer Res. 1999, 5, 1961–1965. [Google Scholar]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Song, Y.; Liu, Y.; Wu, P.; Zhang, F.; Wang, G. Genome-wide mRNA expression analysis of peripheral blood from patients with obsessive-compulsive disorder. Sci. Rep. 2018, 8, 12583. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Yu, D.R.; Huang, J.; Liu, Q.; Wang, D.X.; Luo, N.; Jia, H.; Fan, H.F.; Liu, Q.B. Multimodal Rehabilitation Program Promotes Motor Function Recovery of Rats After Ischemic Stroke by Upregulating Expressions of GAP-43, SYN, HSP70, and C-MYC. J. Stroke Cerebrovasc. Dis. 2018, 27, 2829–2839. [Google Scholar] [CrossRef] [PubMed]
- Gliddon, H.D.; Herberg, J.A.; Levin, M.; Kaforou, M. Genome-wide host RNA signatures of infectious diseases: Discovery and clinical translation. Immunology 2018, 153, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, M.F.; Kuasne, H.; Barros-Filho, M.C.; Ciliao, H.L.; Marchi, F.A.; Fuganti, P.E.; Paschoal, A.R.; Rogatto, S.R.; Colus, I.M.S. Circulating mRNAs and miRNAs as candidate markers for the diagnosis and prognosis of prostate cancer. PLoS ONE 2017, 12, e0184094. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Nakamura, H.; Sato, R.; Tsukamoto, T.; Harada, T.; Takahashi, S.; Adachi, Y.; Shomori, K.; Sano, A.; Kishimoto, Y.; et al. Clinical usefulness of serum telomerase reverse transcriptase (hTERT) mRNA and epidermal growth factor receptor (EGFR) mRNA as a novel tumor marker for lung cancer. Cancer Sci. 2006, 97, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Boscaro, M.; Pacenti, M.; Taccaliti, A.; Palu, G. Evaluation of circulating thyroid-specific transcripts as markers of thyroid cancer relapse. Int. J. Cancer 2004, 110, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Viereck, J.; Thum, T. Circulating Noncoding RNAs as Biomarkers of Cardiovascular Disease and Injury. Circ. Res. 2017, 120, 381–399. [Google Scholar] [CrossRef]
- Fenouil, T.; Fourier, A.; Quadrio, I.; Streichenberger, N.; Bernardini, S.; Zima, T.; Perret-Liaudet, A.; Meyronet, D. The standardization of cerebrospinal fluid markers and neuropathological diagnoses brings to light the frequent complexity of concomitant pathology in Alzheimer’s disease: The next challenge for biochemical markers? Clin. Biochem. 2019, 72, 15–23. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Pulliam, L.; Sun, B.; Mustapic, M.; Chawla, S.; Kapogiannis, D. Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’s disease. J. Neurovirol. 2019, 25, 702–709. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Vollmer, S.; Golombek, S.; Kahle, P.J. The ubiquitin-conjugating enzymes UBE2N, UBE2L3 and UBE2D2/3 are essential for Parkin-dependent mitophagy. J. Cell Sci. 2014, 127, 3280–3293. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.-H.; Joo, J.-Y. Predictive Potential of Circulating Ube2h mRNA as an E2 Ubiquitin-Conjugating Enzyme for Diagnosis or Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3398. https://doi.org/10.3390/ijms21093398
Lim K-H, Joo J-Y. Predictive Potential of Circulating Ube2h mRNA as an E2 Ubiquitin-Conjugating Enzyme for Diagnosis or Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(9):3398. https://doi.org/10.3390/ijms21093398
Chicago/Turabian StyleLim, Key-Hwan, and Jae-Yeol Joo. 2020. "Predictive Potential of Circulating Ube2h mRNA as an E2 Ubiquitin-Conjugating Enzyme for Diagnosis or Treatment of Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 9: 3398. https://doi.org/10.3390/ijms21093398
APA StyleLim, K.-H., & Joo, J.-Y. (2020). Predictive Potential of Circulating Ube2h mRNA as an E2 Ubiquitin-Conjugating Enzyme for Diagnosis or Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences, 21(9), 3398. https://doi.org/10.3390/ijms21093398