AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
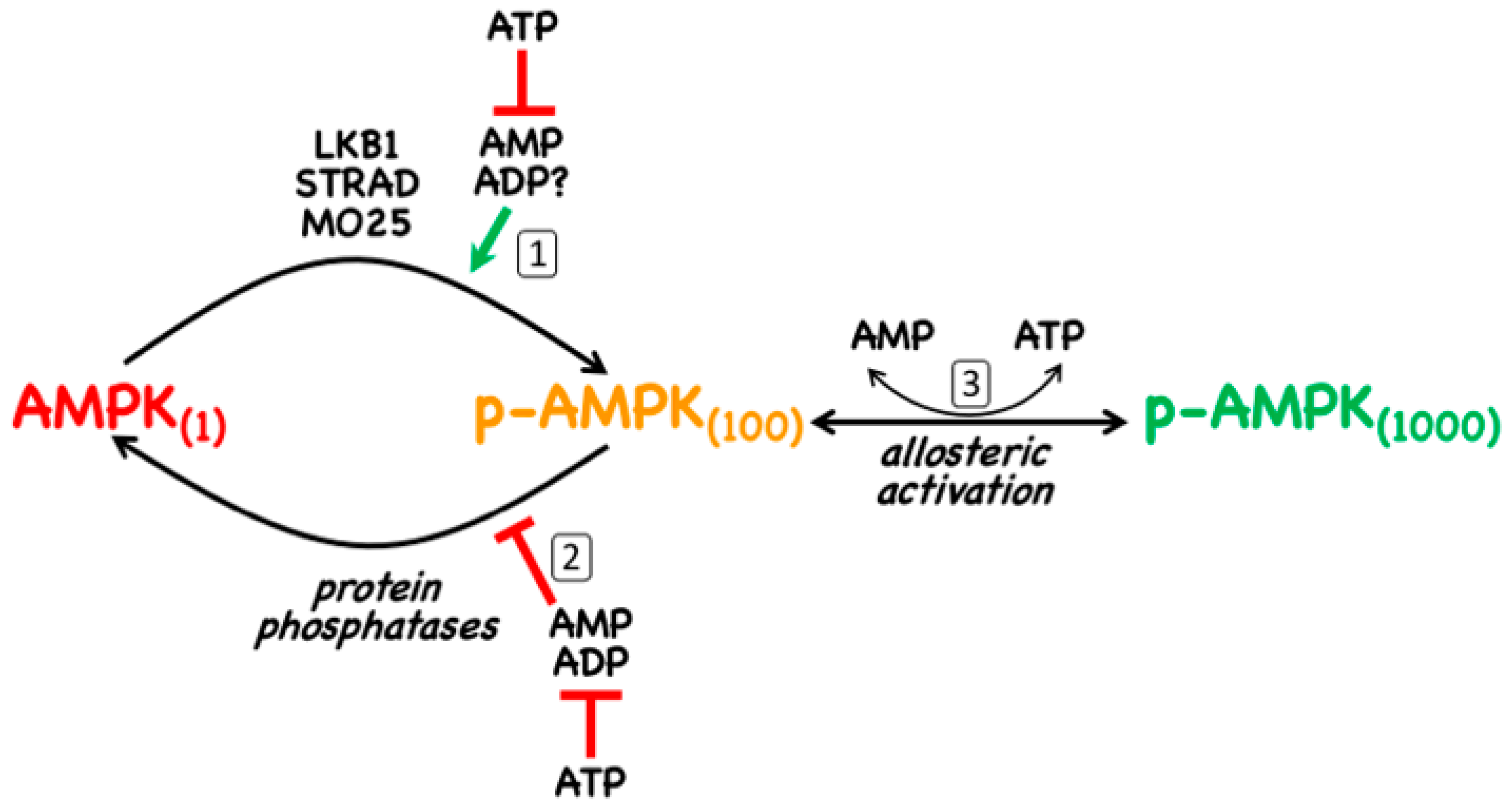
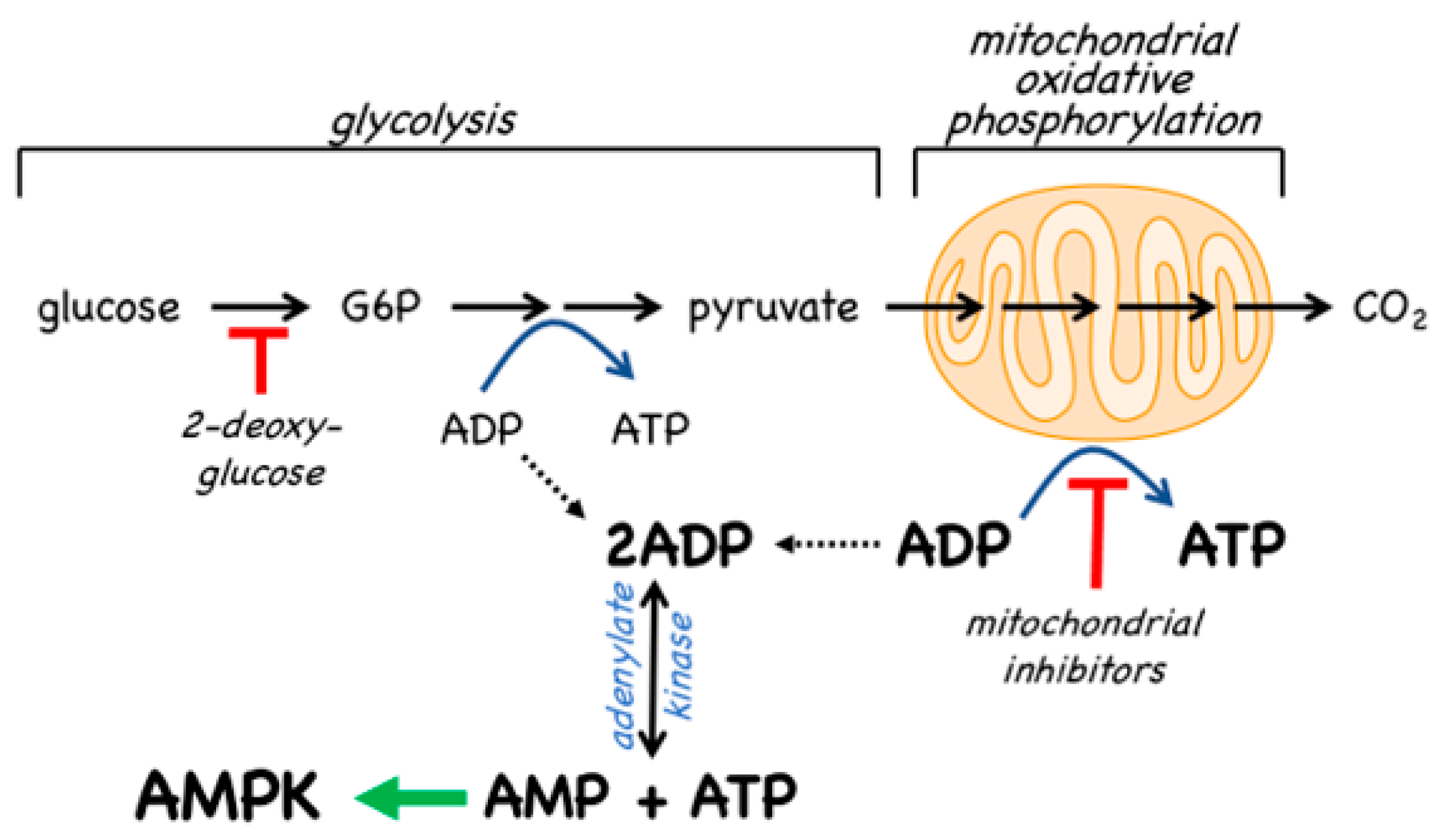
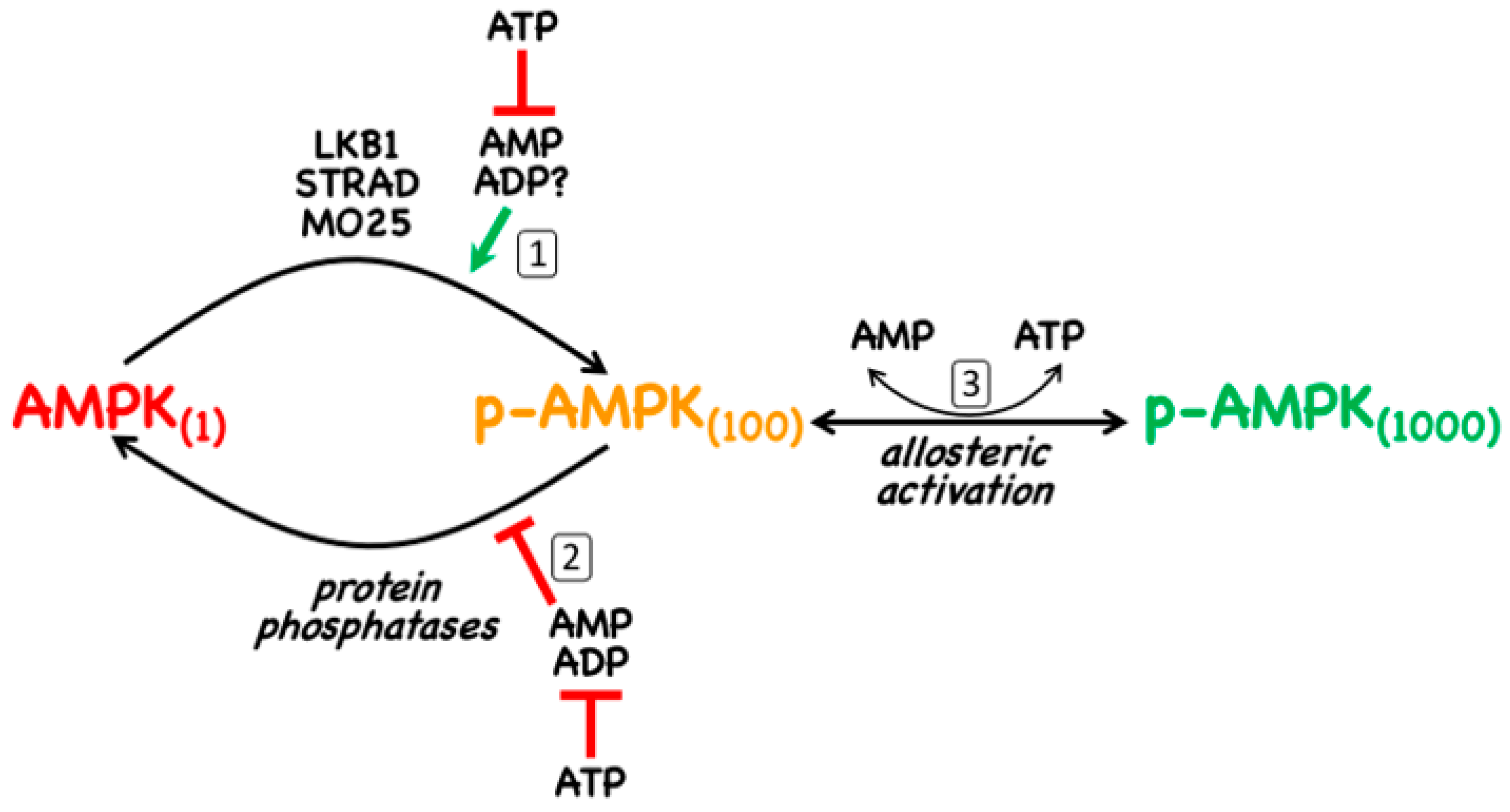
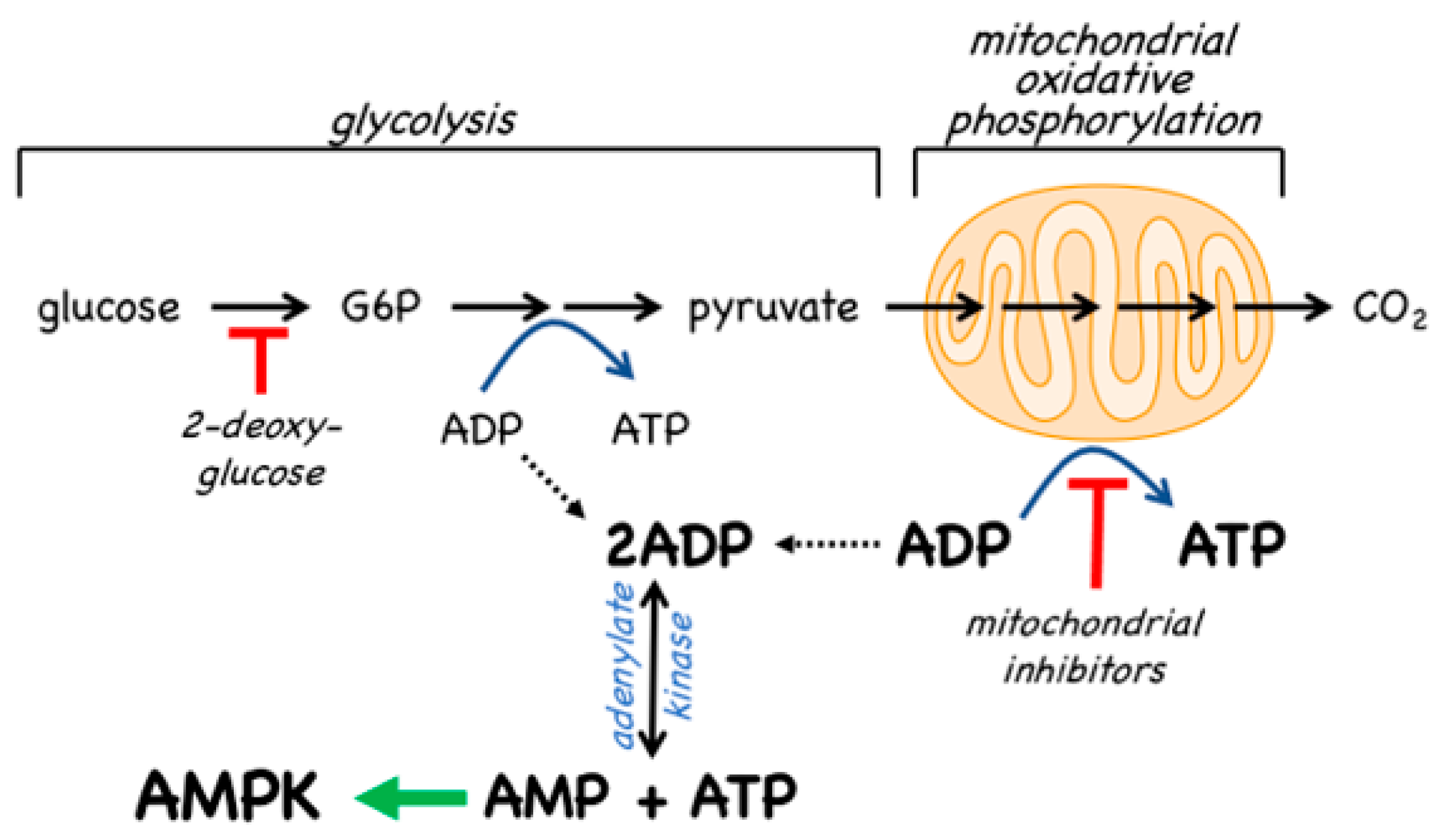
- The canonical pathway is triggered by increases in the cellular ratios of AMP:ATP and/or ADP:ATP. In most eukaryotes (apart from plants) ATP is generated from ADP mainly by mitochondrial oxidative metabolism, with a smaller contribution from glycolysis (although the latter can become more important in proliferating cells such as tumour cells). The resultant high ratios of ATP:ADP generated by these catabolic pathways represent a store of energy (analogous to a fully charged battery) that can be used to drive energy-requiring cellular processes including anabolic pathways (leading to cell growth) and progress through the cell cycle (leading to cell proliferation). An increase in the ratio of ADP:ATP indicates that the energy status of the cell is becoming compromised, and the reaction catalysed by adenylate kinases (2ADP ↔ ATP + AMP) amplifies this into even larger increases in AMP:ATP ratios [4]. Binding of AMP or ADP to a crucial site on the AMPK γ subunit (where they displace ATP) then activates AMPK by a complex mechanism (discussed in Section 2) involving phosphorylation at a conserved threonine residue on the α subunit (Thr172) by the upstream kinase and tumour suppressor, LKB1.
- A non-canonical pathway in which AMPK is activated by glucose starvation (see Section 3.1). This activation takes place on the surface of the lysosome where AMPK is regulated in a reciprocal manner with the mammalian target-of-rapamycin complex-1 (mTORC1), a key pathway that promotes cell growth [5]. Although glucose deprivation can cause increases in AMP:ATP and ADP:ATP ratios in cells that are dependent upon glycolysis, in other cells this lysosomal pathway of AMPK activation can occur without any changes in these ratios [6].
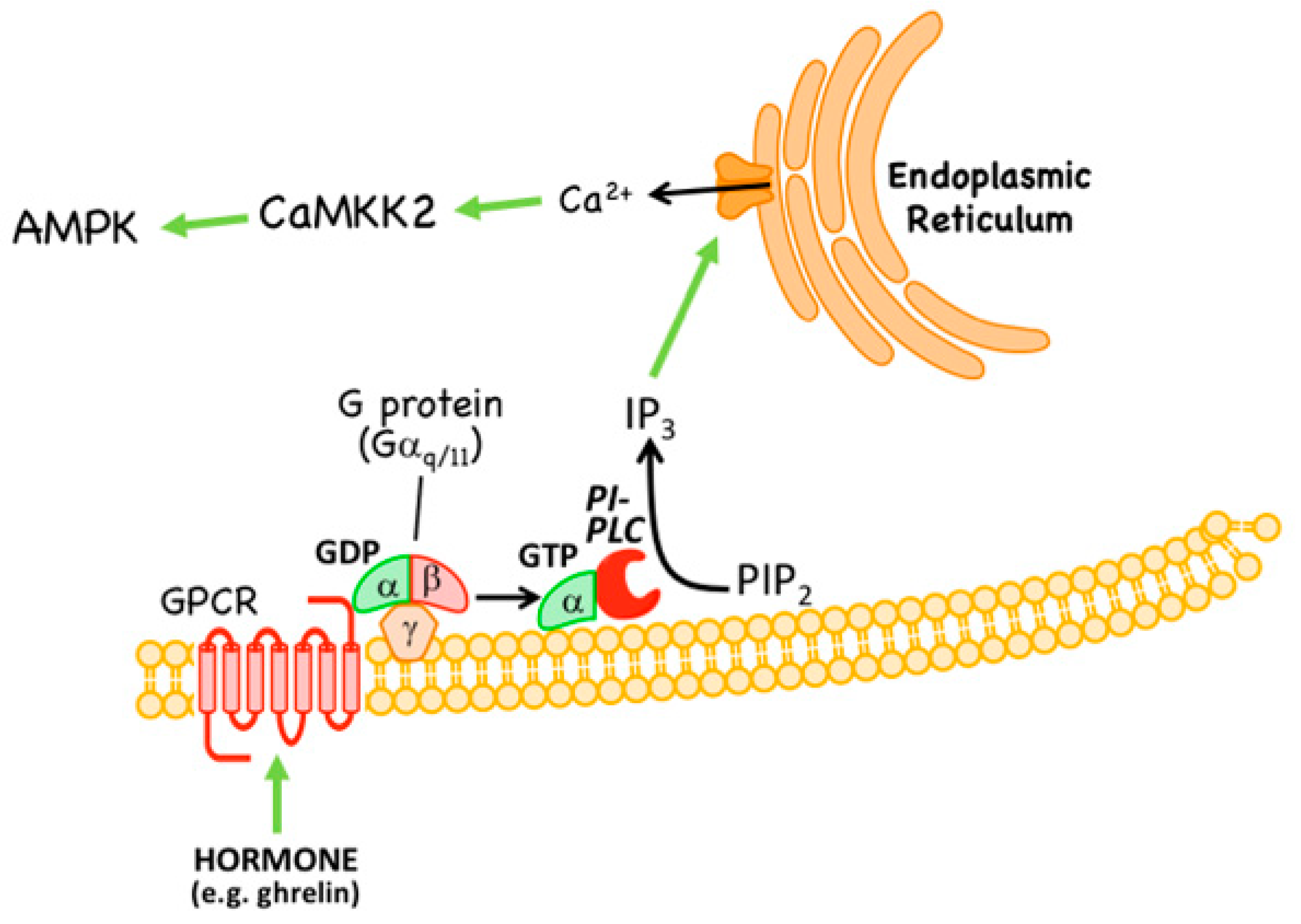
- A non-canonical pathway involving activation, by rising intracellular Ca2+, of the calcium/calmodulin-dependent kinase CaMKK2 which, like LKB1, phosphorylates Thr172 (see Section 3.2). This pathway is utilized by many hormones that switch on AMPK.
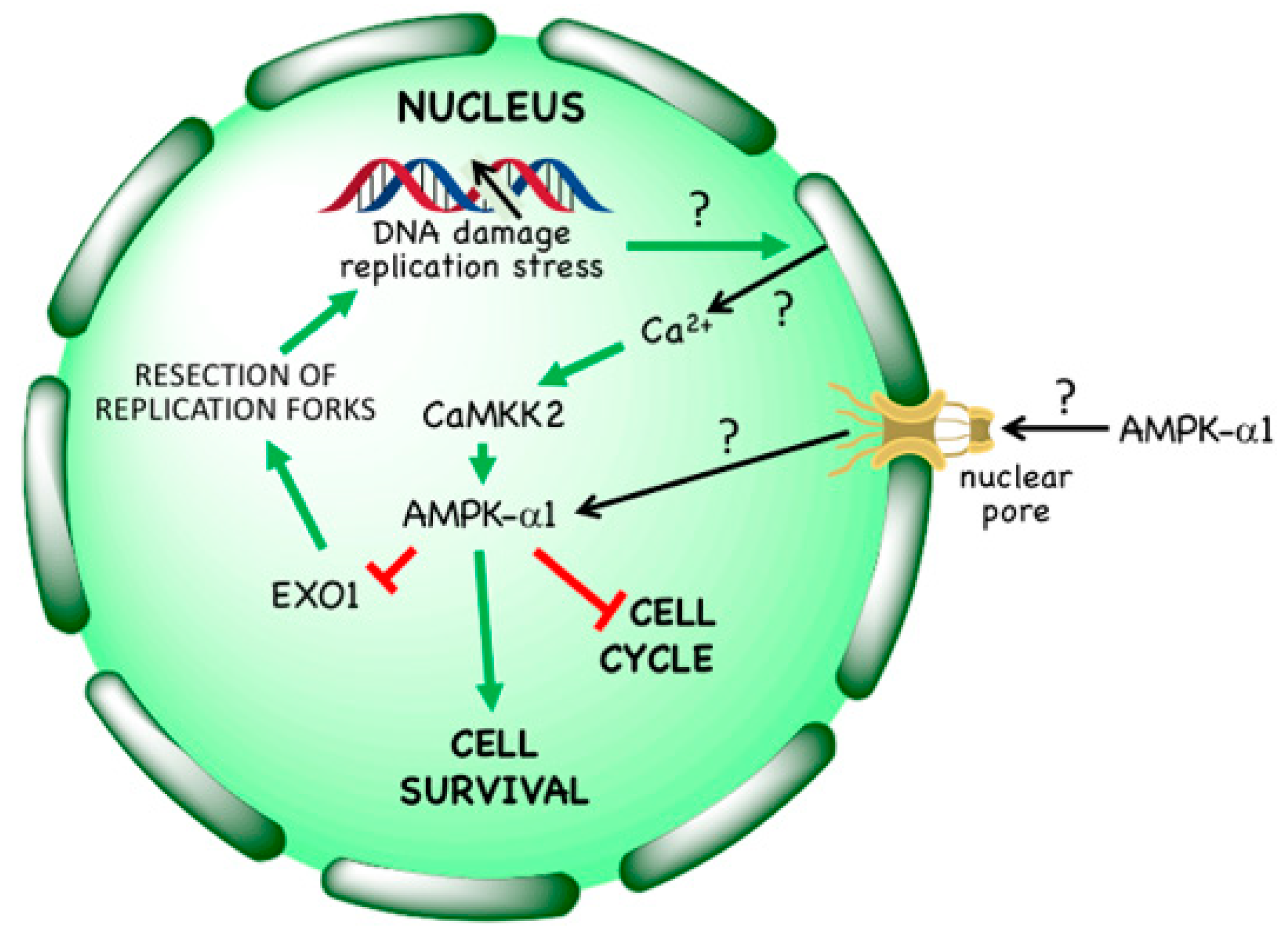
- A non-canonical pathway in which AMPK is activated by DNA-damaging treatments often used in cancer therapy, such as etoposide [7,8] (see Section 3.3). Although the mechanisms involved in this pathway are not yet completely understood, it seems possible that it may eventually have a major impact on treatment of cancer using cytotoxic, DNA-damaging agents (see Section 5.3).
- A non-canonical pathway (recently reported) in which AMPK is activated by direct binding of long-chain fatty acyl-CoA esters (see Section 4.3).
2. AMPK—Structure and Canonical Regulation by Adenine Nucleotides
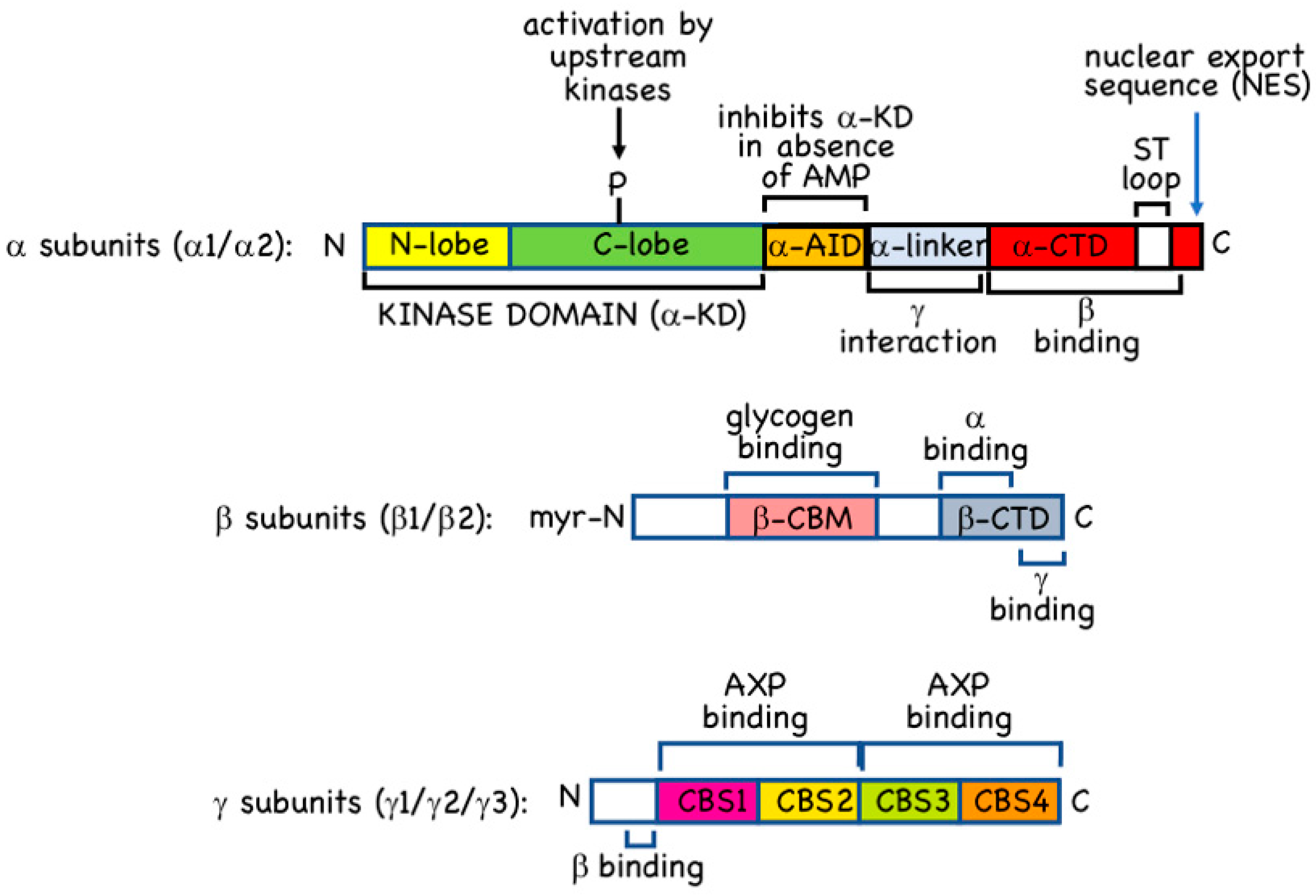
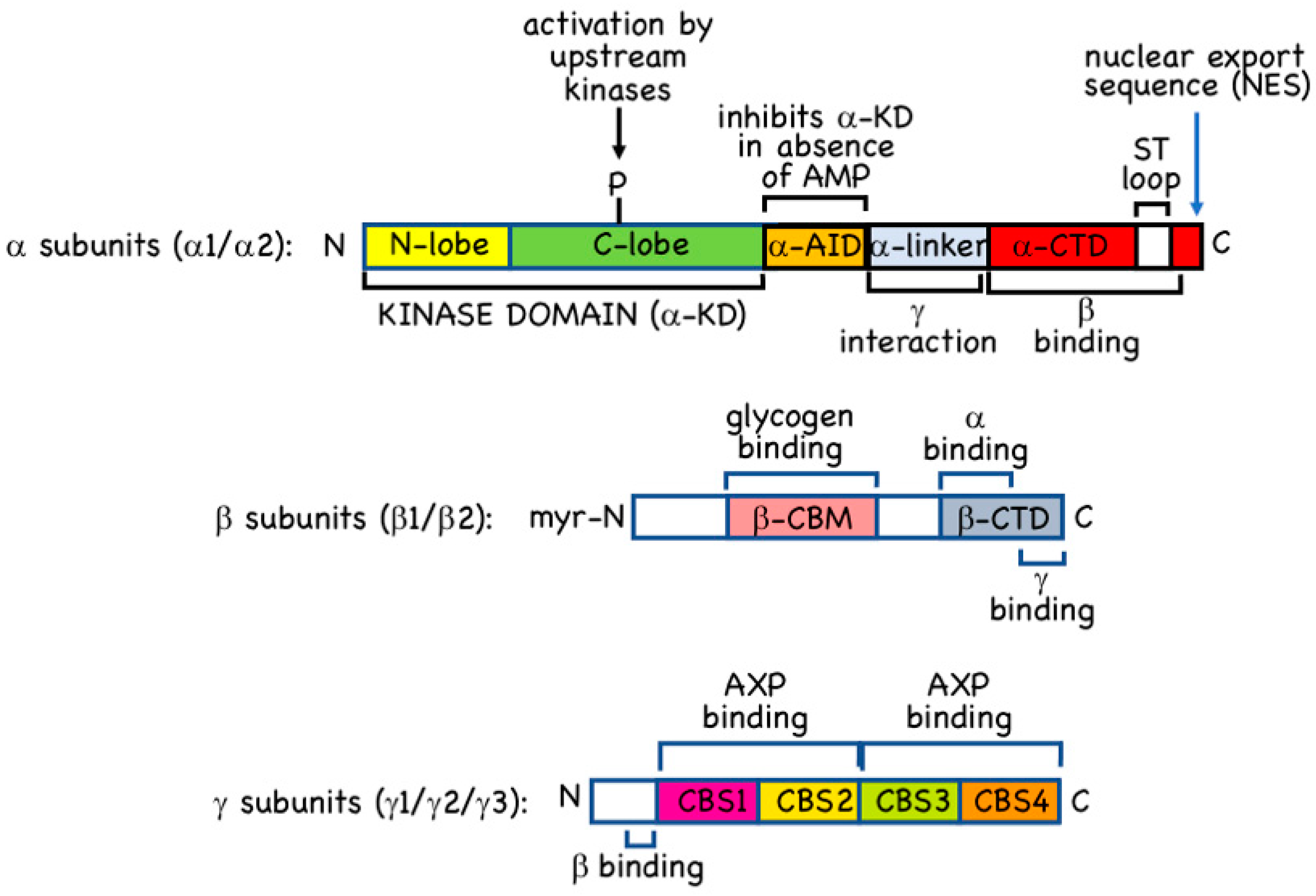
2.1. α Subunits
2.2. β Subunits
2.3. γ Subunits
3. Non-Canonical Regulation of AMPK
3.1. Non-Canonical Regulation by Glucose Starvation
3.2. Non-Canonical Regulation by CaMKK2
3.3. Non-Canonical Regulation by DNA Damage
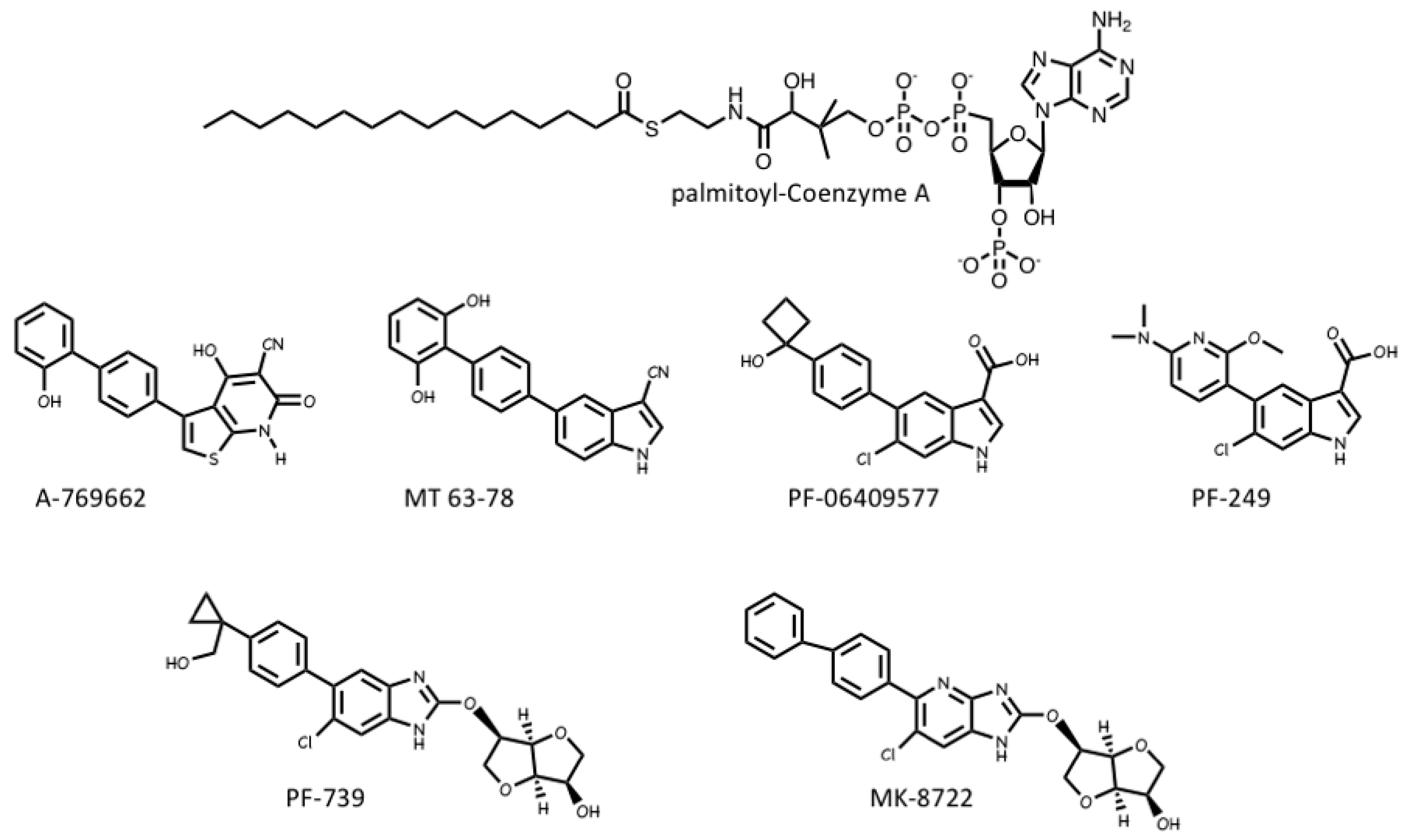
4. Pharmacological Activation and Inhibition of AMPK
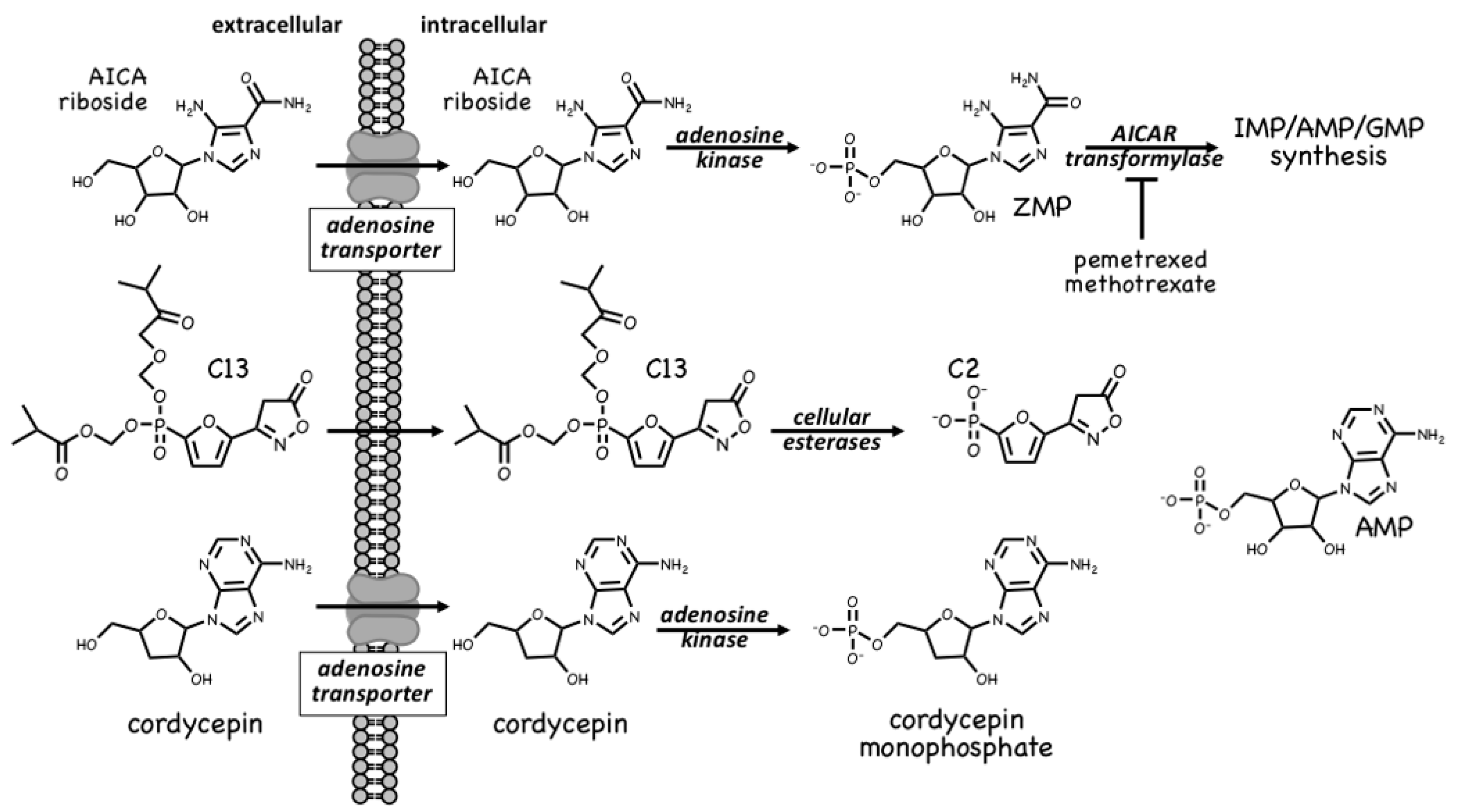
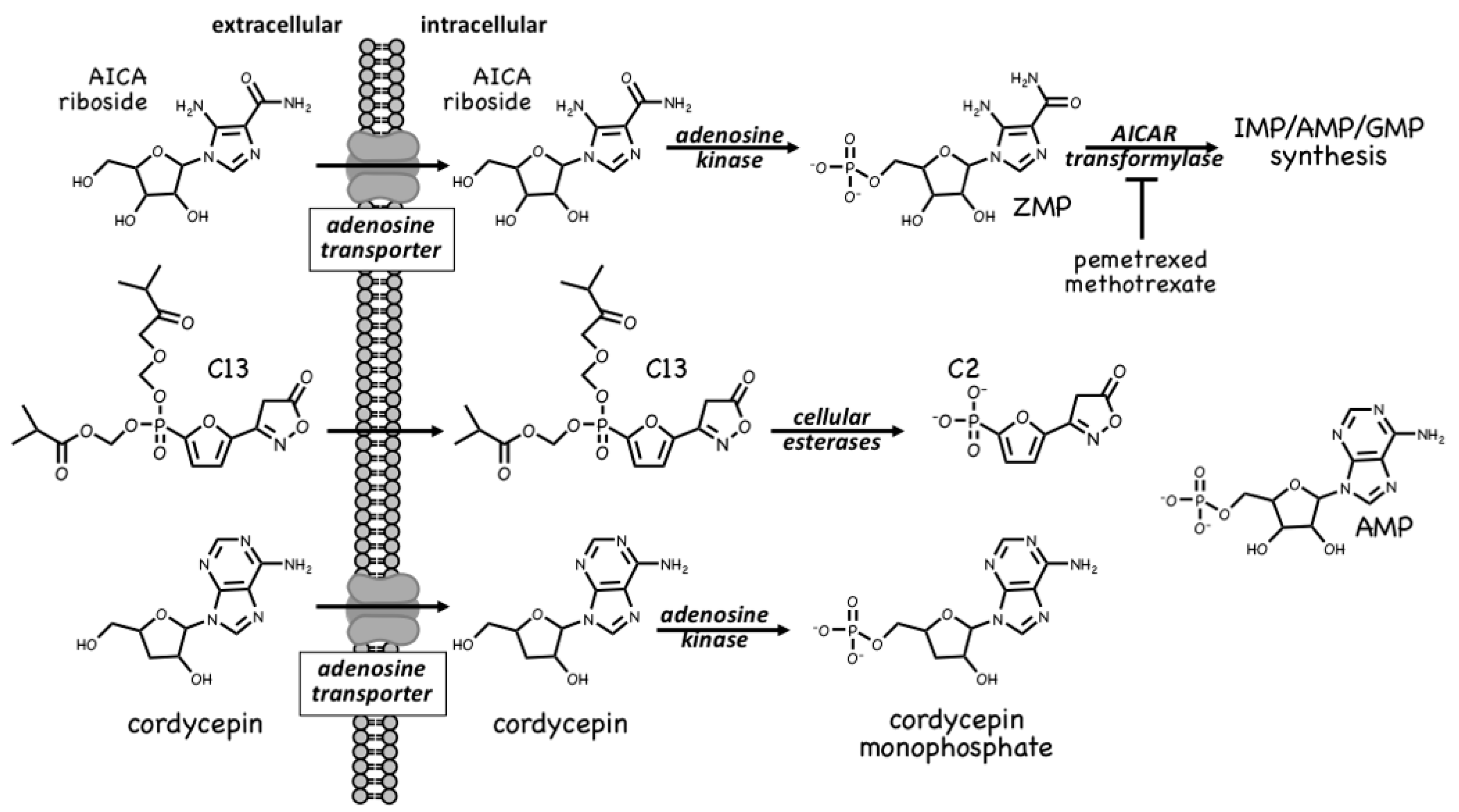
4.1. AMPK Activators: Pro-Drugs That Are Converted to AMP Analogues
4.2. AMPK Activators: Indirect Activation via Inhibition of ATP Synthesis
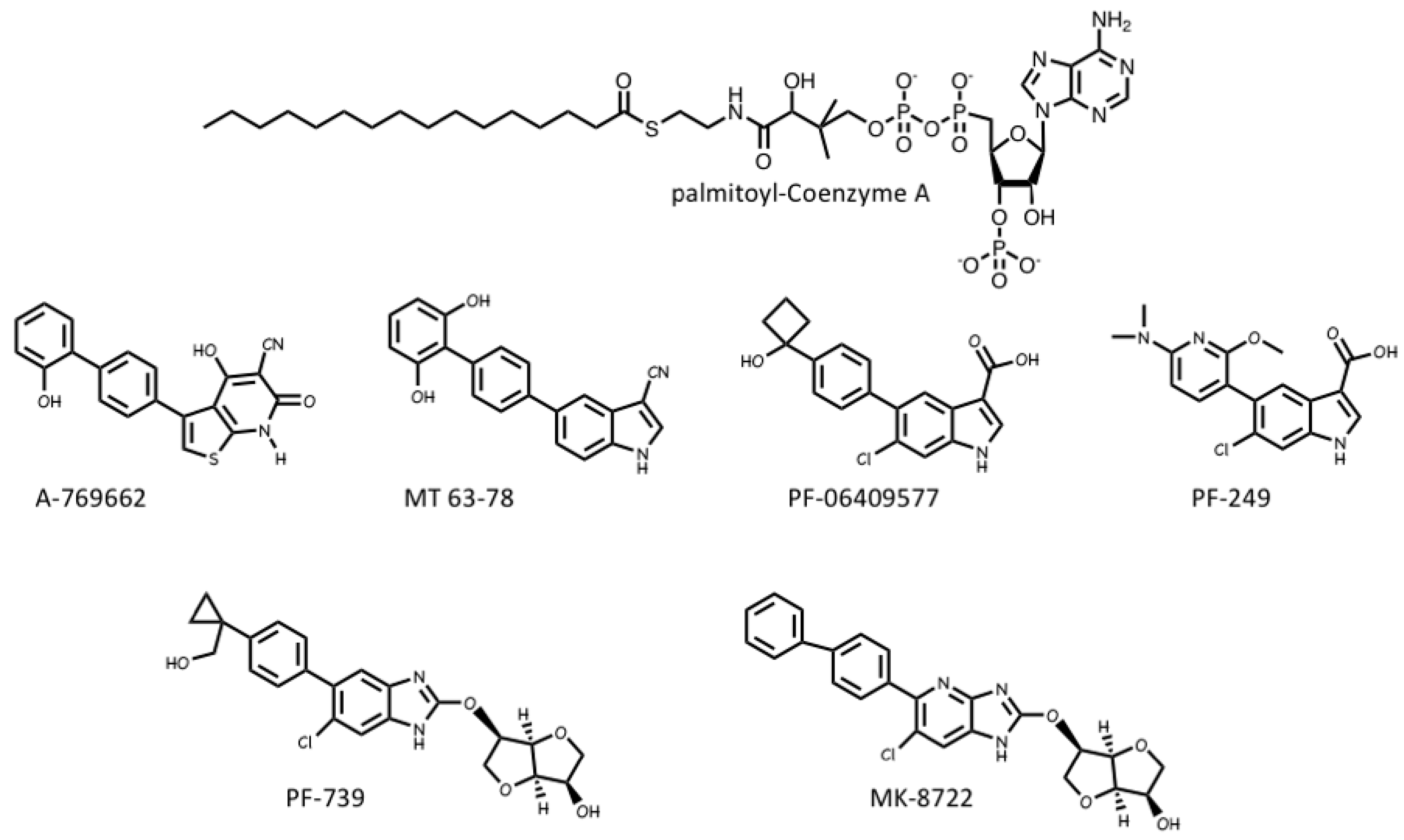
4.3. AMPK Activators: Drugs and Metabolites Binding at the AdaM Site
4.4. AMPK Inhibitors
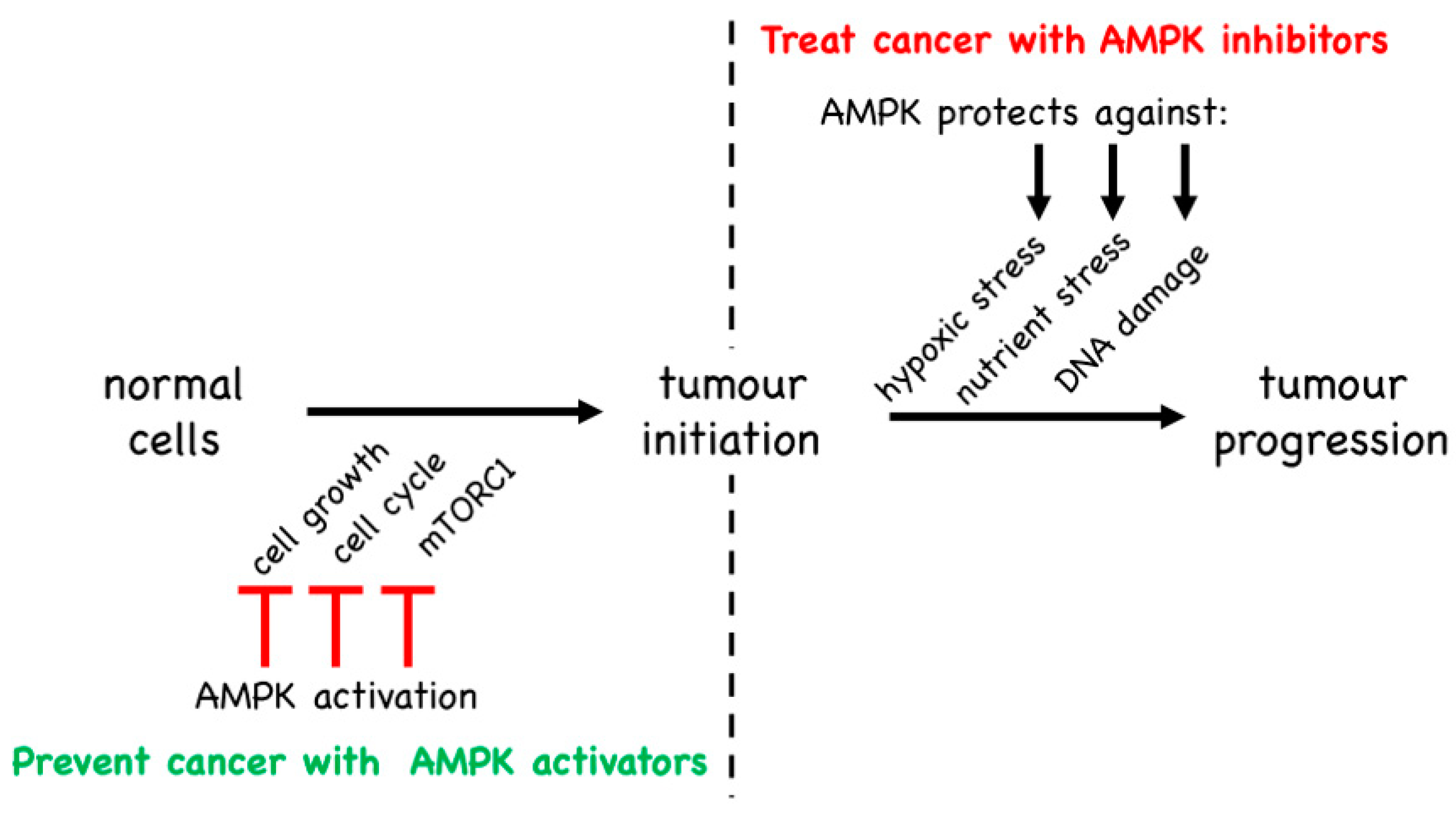
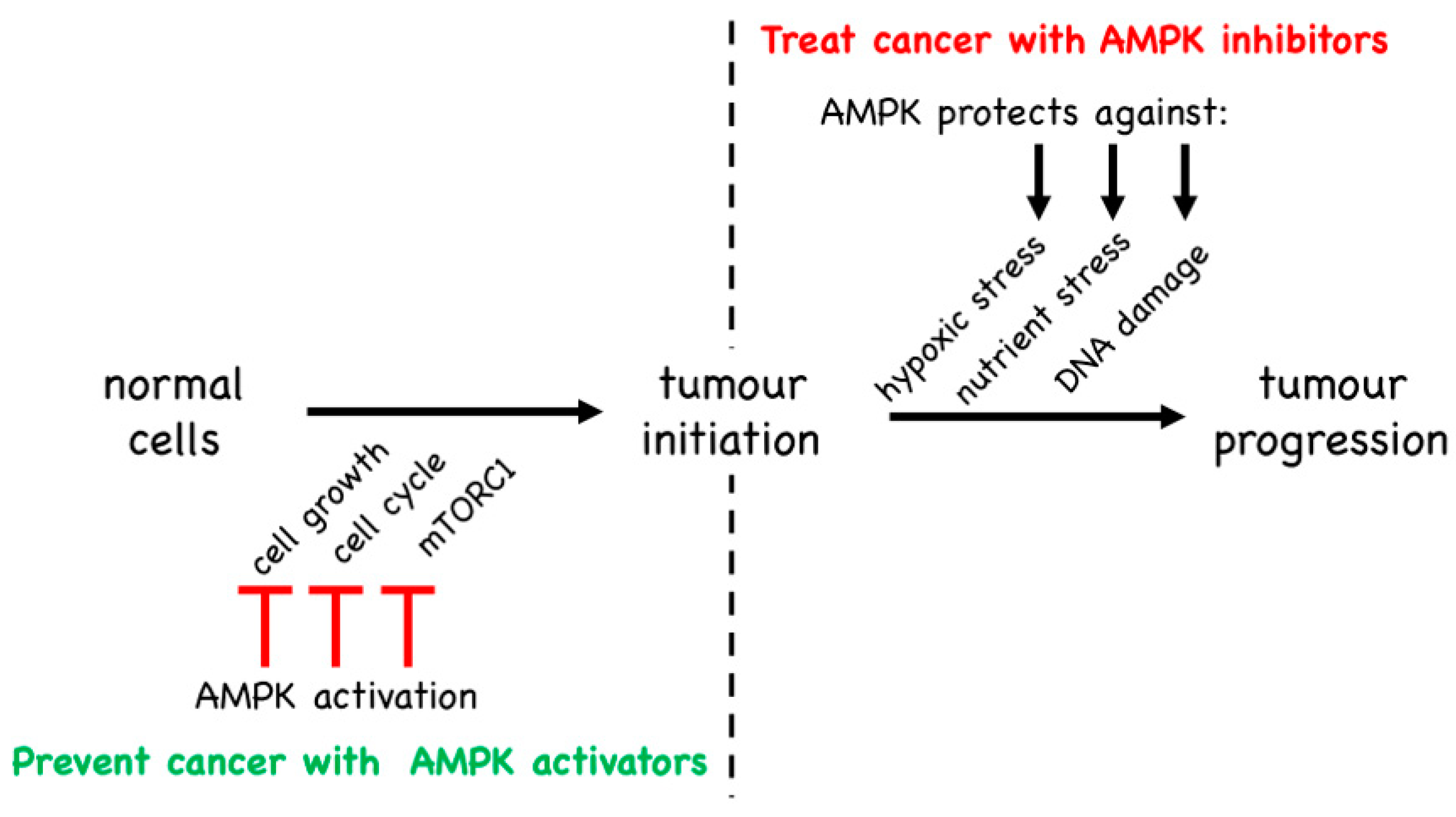
5. Is AMPK a Tumour Suppressor or a Tumour Promoter?
5.1. Evidence from Mouse Models That AMPK Is a Tumour Suppressor
5.2. Evidence from Mouse Models That AMPK Is a Tumour Promoter
5.3. Evidence from Genetic Changes in AMPK Genes in Human Cancer
5.4. Role of AMPK in Cancer Stem Cells
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| ADaM | Allosteric drug and metabolite (binding site on AMPK heterotrimer) |
| AICA | 5-aminoimidazole-4-carboxamide |
| α-AID | Autoinhibitory domain of the AMPK-α subunit |
| AMPK | AMP-activated protein kinase |
| ARK | AMPK-related kinase |
| ASA | Acetyl salicylic acid |
| ATIC | Human gene encoding both AICAR transformylase and IMP cyclohydrolase |
| ATM | Serine-protein kinase ATM (Ataxia telangiectasia mutated) |
| CaMKI | Calmodulin-dependent kinase-I |
| CaMKK2 | Ca2+/calmodulin-dependent kinase kinase-2 (-β) |
| β-CBM | Carbohydrate-binding module of the AMPK-β subunit |
| CBS | Cystathionine β-synthase (refers to CBS domain or CBS repeat) |
| CBS1 | CBS repeat 1; refers to an adenine nucleotide-binding site on the AMPK-γ subunit |
| CBS3 | CBS repeat 3; refers to an adenine nucleotide-binding site on the AMPK-γ subunit |
| CBS4 | CBS repeat 4; refers to an adenine nucleotide-binding site on the AMPK-γ subunit |
| C-lobe | C-terminal lobe of a kinase domain |
| CSCs | Cancer stem cells |
| α-CTD | C-terminal domain of the AMPK-α subunit |
| β-CTD | C-terminal domain of the AMPK-β subunit |
| EXO1 | Exonuclease 1 |
| FBP | Fructose-1,6-bisphosphate |
| GHSR1 | Growth hormone secretagogue receptor-1 |
| IGF-1 | Insulin-like growth factor-1 |
| IP3 | Inositol-1,4,5-triphosphate |
| iPSCs | Inducible pluripotent stem cells |
| α-KD | Kinase domain of the AMPK-α subunit |
| KO | Knockout |
| LCFA | Long chain fatty acid |
| α-linker | Linker region of the AMPK-α subunit |
| LKB1 | Liver kinase B-1 |
| MAGE | Melanoma antigen gene (a gene family) |
| MEFs | Mouse embryonic fibroblasts |
| MO25 | Mouse protein 25 |
| mTORC1 | mammalian (or mechanistic) target-of-rapamycin complex-1 |
| NES | Nuclear exclusion sequence |
| N-lobe | N-terminal lobe of a kinase domain |
| NLS | Nuclear localization sequence |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| OCT1 | Organic cation transporter 1 |
| PPP1R3C | Human gene encoding a glycogen-targetting regulatory subunit of protein phosphatase-1 |
| PRKAA1 | Human gene encoding AMPK-α1 |
| PRKAA2 | Human gene encoding AMPK-α2 |
| PRKAB1 | Human gene encoding AMPK-β1 |
| PRKAG1 | Human gene encoding AMPK-γ1 |
| PRKAG2 | Human gene encoding AMPK-γ2 |
| PRKAG3 | Human gene encoding AMPK-γ3 |
| PTEN | Phosphatase and tensin homolog protein |
| shRNA | Short hairpin RNA |
| siRNA | Small interfering RNA |
| STRAD | Ste20-related adaptor |
| STK11 | Human gene encoding LKB1 |
| ST loop | Serine/threonine-rich regulatory loop near the C-terminus of the AMPK-α subunit |
| T-ALL | T cell acute lymphoblastic leukameia/lymphoma |
| TCGA | The Cancer Genome Atlas |
| TRIM-28 | Tripartite motif containing 28 (ubiquitin E3 ligase) |
| UBE2O | E2/E3 hybrid ubiquitin-protein ligase |
| VEGF | Vascular endothelial cell growth factor |
| ZMP | 5-aminoimidazole-4-carboxamide ribonucleoside monophosphate |
References
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing glucose as well as cellular energy status. Cell Metab. 2017, 27, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase: The energy charge hypothesis revisited. BioEssays 2001, 23, 1112–1119. [Google Scholar] [CrossRef]
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Dandapani, M.; Gray, A.; Egbani, E.O.; Evans, A.M.; Hardie, D.G. Genotoxic damage activates the AMPK-alpha1 isoform in the nucleus via Ca2+/CaMKK2 signaling to enhance tumor cell survival. Mol. Cancer Res. 2018, 16, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Lavagnino, Z.; Lemacon, D.; Kong, L.; Ustione, A.; Ng, X.; Zhang, Y.; Wang, Y.; Zheng, B.; Piwnica-Worms, H.; et al. Ca(2+)-stimulated AMPK-dependent phosphorylation of Exo1 protects stressed replication forks from aberrant resection. Mol. Cell 2019, 74, 1123–1137. [Google Scholar] [CrossRef]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Munday, M.R.; Campbell, D.G.; Carling, D.; Hardie, D.G. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur. J. Biochem. 1988, 175, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.W.; Guan, H.P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Trotter, D.G.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different gamma subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef]
- Oakhill, J.S.; Chen, Z.P.; Scott, J.W.; Steel, R.; Castelli, L.A.; Ling, N.; Macaulay, S.L.; Kemp, B.E. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. USA 2010, 107, 19237–19241. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Makela, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRADa/b and MO25a/b are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [Green Version]
- Zeqiraj, E.; Filippi, B.M.; Deak, M.; Alessi, D.R.; van Aalten, D.M. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science 2009, 326, 1707–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. Lkb1-dependent signaling pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef] [PubMed]
- Wingo, S.N.; Gallardo, T.D.; Akbay, E.A.; Liang, M.C.; Contreras, C.M.; Boren, T.; Shimamura, T.; Miller, D.S.; Sharpless, N.E.; Bardeesy, N.; et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS ONE 2009, 4, e5137. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5’-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [PubMed] [Green Version]
- Wang, L.; Brautigan, D.L. alpha-SNAP inhibits AMPK signaling to reduce mitochondrial biogenesis and dephosphorylates Thr172 in AMPKalpha in vitro. Nature Commun. 2013, 4, 1559. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.; Huang, P.; Wu, J.; Song, H. Drosophila protein phosphatase V regulates lipid homeostasis via the AMPK pathway. J. Mol. Cell. Biol. 2014, 6, 100–102. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Jiao, Z.H.; Zheng, L.S.; Zhang, Y.Y.; Xie, S.T.; Wang, Z.X.; Wu, J.W. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature 2009, 459, 1146–1149. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nature Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [Green Version]
- Horman, S.; Vertommen, D.; Heath, R.; Neumann, D.; Mouton, V.; Woods, A.; Schlattner, U.; Wallimann, T.; Carling, D.; Hue, L.; et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J. Biol. Chem. 2006, 281, 5335–5340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, R.L.; Barre, L.K.; Wood, S.D.; Anderson, K.A.; Kemp, B.E.; Means, A.R.; Witters, L.A. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 2006, 281, 36662–36672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heathcote, H.R.; Mancini, S.J.; Strembitska, A.; Jamal, K.; Reihill, J.A.; Palmer, T.M.; Gould, G.W.; Salt, I.P. Protein kinase C phosphorylates AMP-activated protein kinase alpha1 Ser487. Biochem. J. 2016, 473, 4681–4697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Ross, F.A.; Gowans, G.J.; Tibarewal, P.; Leslie, N.R.; Hardie, D.G. Phosphorylation by Akt within the ST loop of AMPK-α1 down-regulates its activation in tumour cells. Biochem. J. 2014, 459, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Kazgan, N.; Williams, T.; Forsberg, L.J.; Brenman, J.E. Identification of a nuclear export signal in the catalytic subunit of AMP-activated protein kinase. Mol. Biol. Cell 2010, 21, 3433–3442. [Google Scholar] [CrossRef] [Green Version]
- Salt, I.P.; Celler, J.W.; Hawley, S.A.; Prescott, A.; Woods, A.; Carling, D.; Hardie, D.G. AMP-activated protein kinase—Greater AMP dependence, and preferential nuclear localization, of complexes containing the a2 isoform. Biochem. J. 1998, 334, 177–187. [Google Scholar] [CrossRef]
- Turnley, A.M.; Stapleton, D.; Mann, R.J.; Witters, L.A.; Kemp, B.E.; Bartlett, P.F. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J. Neurochem. 1999, 72, 1707–1716. [Google Scholar] [CrossRef]
- McGee, S.L.; Howlett, K.F.; Starkie, R.L.; Cameron-Smith, D.; Kemp, B.E.; Hargreaves, M. Exercise increases nuclear AMPK alpha-2 in human skeletal muscle. Diabetes 2003, 52, 926–928. [Google Scholar] [CrossRef] [Green Version]
- Ai, H.; Ihlemann, J.; Hellsten, Y.; Lauritzen, H.P.; Hardie, D.G.; Galbo, H.; Ploug, T. Effect of fiber type and nutritional state on AICAR- and contraction-stimulated glucose transport in rat muscle. Am. J. Physiol. 2002, 282, E1291–E1300. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Okamoto, S.; Lee, S.; Saito, K.; Shiuchi, T.; Minokoshi, Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol. Cell. Biol. 2007, 27, 4317–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warden, S.M.; Richardson, C.; O’Donnell, J., Jr.; Stapleton, D.; Kemp, B.E.; Witters, L.A. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem. J. 2001, 354 Pt 2, 275–283. [Google Scholar] [CrossRef]
- McLaughlin, S.; Aderem, A. The myristoyl-electrostatic switch: A modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 1995, 20, 272–276. [Google Scholar] [CrossRef]
- Machovic, M.; Janecek, S.H. The evolution of putative starch-binding domains. FEBS Lett. 2006, 580, 6349–6356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, E.R.; Pan, D.A.; James, J.; Lucocq, J.M.; Hawley, S.A.; Green, K.A.; Baba, O.; Terashima, T.; Hardie, D.G. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr. Biol. 2003, 13, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Polekhina, G.; Gupta, A.; Michell, B.J.; van Denderen, B.; Murthy, S.; Feil, S.C.; Jennings, I.G.; Campbell, D.J.; Witters, L.A.; Parker, M.W.; et al. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr. Biol. 2003, 13, 867–871. [Google Scholar] [CrossRef] [Green Version]
- Polekhina, G.; Gupta, A.; van Denderen, B.J.; Feil, S.C.; Kemp, B.E.; Stapleton, D.; Parker, M.W. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure 2005, 13, 1453–1462. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; Nielsen, J.N.; Birk, J.B.; Olsen, G.S.; Viollet, B.; Andreelli, F.; Schjerling, P.; Vaulont, S.; Hardie, D.G.; Hansen, B.F.; et al. The a2-5′AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 2004, 53, 3074–3081. [Google Scholar] [CrossRef] [Green Version]
- Bultot, L.; Guigas, B.; Von Wilamowitz-Moellendorff, A.; Maisin, L.; Vertommen, D.; Hussain, N.; Beullens, M.; Guinovart, J.J.; Foretz, M.; Viollet, B.; et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem. J. 2012, 443, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Vernia, S.; Solaz-Fuster, M.C.; Gimeno-Alcaniz, J.V.; Rubio, T.; Garcia-Haro, L.; Foretz, M.; de Cordoba, S.R.; Sanz, P. AMP-activated protein kinase phosphorylates R5/PTG, the glycogen targeting subunit of the R5/PTG-protein phosphatase 1 holoenzyme, and accelerates its down-regulation by the laforin-malin complex. J. Biol. Chem. 2009, 284, 8247–8255. [Google Scholar] [CrossRef] [Green Version]
- Ducommun, S.; Deak, M.; Zeigerer, A.; Goransson, O.; Seitz, S.; Collodet, C.; Madsen, A.B.; Jensen, T.E.; Viollet, B.; Foretz, M.; et al. Chemical genetic screen identifies Gapex-5/GAPVD1 and STBD1 as novel AMPK substrates. Cell. Signal. 2019, 57, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J.; Whitfield, J.; Janzen, N.R.; Belhaj, M.R.; Galic, S.; Murray-Segal, L.; Smiles, W.J.; Ling, N.X.Y.; Dite, T.A.; Scott, J.W.; et al. Genetic loss of AMPK-glycogen binding destabilizes AMPK and disrupts metabolism. Mol. Metab. 2020, 41, 101048. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef]
- Ignoul, S.; Eggermont, J. CBS domains: Structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 2005, 289, C1369–C1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004, 113, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, B.E.; Oakhill, J.S.; Scott, J.W. AMPK structure and regulation from three angles. Structure 2007, 15, 1161–1163. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Yan, Y.; Novick, S.J.; Kovich, A.; Goswami, D.; Ke, J.; Tan, M.H.E.; Wang, L.; Li, X.; de Waal, P.; et al. Deconvoluting AMP-dependent kinase (AMPK) adenine nucleotide binding and sensing. J. Biol. Chem. 2017, 292, 12653–12666. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef]
- Salt, I.P.; Johnson, G.; Ashcroft, S.J.H.; Hardie, D.G. AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic b cells, and may regulate insulin release. Biochem. J. 1998, 335, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhang, C.S.; Zong, Y.; Feng, J.W.; Ma, T.; Hu, M.; Lin, Z.; Li, X.; Xie, C.; Wu, Y.; et al. Transient Receptor Potential V channels are essential for glucose sensing by aldolase and AMPK. Cell Metab. 2019, 30, 508–524. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Munday, M.R.; Scott, J.; Yang, X.; Carlson, M.; Carling, D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J. Biol. Chem. 1994, 269, 19509–19515. [Google Scholar] [PubMed]
- Wilson, W.A.; Hawley, S.A.; Hardie, D.G. Glucose repression/derepression in budding yeast: SNF1 protein kinase is activated by phosphorylation under derepressing conditions, and this correlates with a high AMP:ATP ratio. Curr. Biol. 1996, 6, 1426–1434. [Google Scholar] [CrossRef] [Green Version]
- Baena-Gonzalez, E.; Rolland, F.; Thevelein, J.M.; Sheen, J. A central integrator of transcription networks in plant stress and energy signalling. Nature 2007, 448, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Thelander, M.; Olsson, T.; Ronne, H. Snf1-related protein kinase 1 is needed for growth in a normal day-night light cycle. EMBO J. 2004, 23, 1900–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackintosh, R.W.; Davies, S.P.; Clarke, P.R.; Weekes, J.; Gillespie, J.G.; Gibb, B.J.; Hardie, D.G. Evidence for a protein kinase cascade in higher plants. 3-Hydroxy-3-methylglutaryl-CoA reductase kinase. Eur. J. Biochem. 1992, 209, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.V.; Heath, R.; Underwood, E.; Sanders, M.J.; Carmena, D.; McCartney, R.R.; Leiper, F.C.; Xiao, B.; Jing, C.; Walker, P.A.; et al. ADP regulates SNF1, the Saccharomyces cerevisiae homolog of AMP-activated protein kinase. Cell Metab. 2011, 14, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Sugden, C.; Crawford, R.M.; Halford, N.G.; Hardie, D.G. Regulation of spinach SNF1-related (SnRK1) kinases by protein kinases and phosphatases is associated with phosphorylation of the T loop and is regulated by 5′-AMP. Plant J. 1999, 19, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmoldulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahmann, N.; Woods, A.; Carling, D.; Heller, R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol. Cell. Biol. 2006, 26, 5933–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahmann, N.; Woods, A.; Spengler, K.; Heslegrave, A.; Bauer, R.; Krause, S.; Viollet, B.; Carling, D.; Heller, R. Activation of AMP-activated protein kinase by vascular endothelial growth factor mediates endothelial angiogenesis independently of nitric-oxide synthase. J. Biol. Chem. 2010, 285, 10638–10652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 2011, 146, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Wan, S.; Lyu, Y.L.; Liu, L.F.; Qi, H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS ONE 2008, 3, e2009. [Google Scholar] [CrossRef]
- Sanli, T.; Rashid, A.; Liu, C.; Harding, S.; Bristow, R.G.; Cutz, J.C.; Singh, G.; Wright, J.; Tsakiridis, T. Ionizing radiation activates AMP-activated kinase (AMPK): A target for radiosensitization of human cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 221–229. [Google Scholar] [CrossRef]
- Luo, L.; Huang, W.; Tao, R.; Hu, N.; Xiao, Z.X.; Luo, Z. ATM and LKB1 dependent activation of AMPK sensitizes cancer cells to etoposide-induced apoptosis. Cancer Lett. 2013, 328, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Sapkota, G.P.; Deak, M.; Kieloch, A.; Morrice, N.; Goodarzi, A.A.; Smythe, C.; Shiloh, Y.; Lees-Miller, S.P.; Alessi, D.R. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem. J. 2002, 368 Pt 2, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Boudeau, J.; Baas, A.F.; Deak, M.; Morrice, N.A.; Kieloch, A.; Schutkowski, M.; Prescott, A.R.; Clevers, H.C.; Alessi, D.R. MO25a/b interact with STRADa/b enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003, 22, 5102–5114. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Ogura, T.; Kishimoto, A.; Kaminishi, M.; Esumi, H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-d- ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 2001, 287, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Winder, W.W.; Hardie, D.G. The AMP-activated protein kinase, a metabolic master switch: Possible roles in Type 2 diabetes. Am. J. Physiol. 1999, 277, E1–E10. [Google Scholar] [CrossRef] [PubMed]
- Henin, N.; Vincent, M.F.; Gruber, H.E.; Van den Berghe, G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995, 9, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.E.; Brocklehurst, K.J.; Marley, A.E.; Carey, F.; Carling, D.; Beri, R.K. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994, 353, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-Aminoimidazole-4-carboxamide ribonucleoside: A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, A.E.; Pearson, T.; Currie, A.J.; Dale, N.; Hawley, S.A.; Sheehan, M.; Hirst, W.; Michel, A.D.; Randall, A.; Hardie, D.G.; et al. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J. Neurochem. 2004, 88, 1272–1282. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Ross, F.A.; Russell, F.M.; Atrih, A.; Lamont, D.J.; Hardie, D.G. Mechanism of activation of AMPK by cordycepin. Cell Chem. Biol. 2020, 27, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Longnus, S.L.; Wambolt, R.B.; Parsons, H.L.; Brownsey, R.W.; Allard, M.F. 5-Aminoimidazole-4-carboxamide 1-beta -D-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. Am. J. Physiol. 2003, 284, R936–R944. [Google Scholar]
- Vincent, M.F.; Marangos, P.J.; Gruber, H.E.; Van den Berghe, G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes 1991, 40, 1259–1266. [Google Scholar] [CrossRef]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racanelli, A.C.; Rothbart, S.B.; Heyer, C.L.; Moran, R.G. Therapeutics by cytotoxic metabolite accumulation: Pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009, 69, 5467–5474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirkmajer, S.; Kulkarni, S.S.; Tom, R.Z.; Ross, F.A.; Hawley, S.A.; Hardie, D.G.; Zierath, J.R.; Chibalin, A.V. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes 2015, 64, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Galeno, J.E.; Dang, Q.; Nguyen, T.H.; Boyer, S.H.; Grote, M.P.; Sun, Z.; Chen, M.; Craigo, W.A.; van Poelje, P.D.; MacKenna, D.A.; et al. A potent and selective AMPK activator that inhibits de novo lipogenesis. ACS Med. Chem. Lett. 2010, 1, 478–482. [Google Scholar] [CrossRef] [Green Version]
- Langendorf, C.G.; Ngoei, K.R.; Scott, J.W.; Ling, N.X.; Issa, S.M.; Gorman, M.A.; Parker, M.W.; Sakamoto, K.; Oakhill, J.S.; Kemp, B.E. Structural basis of allosteric and synergistic activation of AMPK by furan-2-phosphonic derivative C2 binding. Nat. Commun. 2016, 7, 10912. [Google Scholar] [CrossRef]
- Hunter, R.W.; Foretz, M.; Bultot, L.; Fullerton, M.D.; Deak, M.; Ross, F.A.; Hawley, S.A.; Shpiro, N.; Viollet, B.; Barron, D.; et al. Mechanism of action of compound-13: An alpha1-selective small molecule activator of AMPK. Chem. Biol. 2014, 21, 866–879. [Google Scholar] [CrossRef] [Green Version]
- Tuli, H.S.; Sharma, A.K.; Sandhu, S.S.; Kashyap, D. Cordycepin: A bioactive metabolite with therapeutic potential. Life Sci. 2013, 93, 863–869. [Google Scholar] [CrossRef]
- Wong, Y.Y.; Moon, A.; Duffin, R.; Barthet-Barateig, A.; Meijer, H.A.; Clemens, M.J.; de Moor, C.H. Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. J. Biol. Chem. 2010, 285, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Kai, Q.; Gao, J.; Lian, Z.Q.; Wu, C.M.; Wu, C.A.; Zhu, H.B. Cordycepin prevents hyperlipidemia in hamsters fed a high-fat diet via activation of AMP-activated protein kinase. J. Pharmacol. Sci. 2010, 113, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Tsou, P.; Zheng, B.; Hsu, C.H.; Sasaki, A.T.; Cantley, L.C. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011, 13, 476–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Shitan, N. Secondary metabolites in plants: Transport and self-tolerance mechanisms. Biosci. Biotechnol. Biochem. 2016, 80, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, M.H.; Fogarty, S.; Stevenson, C.; Gallagher, A.M.; Palit, P.; Hawley, S.A.; Hardie, D.G.; Coxon, G.D.; Waigh, R.D.; Tate, R.J.; et al. Mechanisms underlying the metabolic actions of galegine that contribute to weight loss in mice. Br. J. Pharmacol. 2008, 153, 1669–1677. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, J. Galega, Goat’s Rue. In Theatricum Botanicum; Thomas Cotes: London, UK, 1640; pp. 417–418. [Google Scholar]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin activates AMPK through the lysosomal pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Li, J.Y.; Gosby, A.; To, S.W.; Cheng, Z.; Miyoshi, H.; Taketo, M.M.; Cooney, G.J.; Kraegen, E.W.; James, D.E.; et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: A mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 2008, 57, 1414–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.L.; Yu, R.T.; Gong, J.; Feng, Y.; Dai, Y.L.; Hu, F.; Hu, Y.H.; Tao, Y.D.; Leng, Y. Arctigenin, a natural compound, activates AMP-activated protein kinase via inhibition of mitochondria complex I and ameliorates metabolic disorders in ob/ob mice. Diabetologia 2012, 55, 1469–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Anand, S.K.; Singh, N.; Dwivedi, U.N.; Kakkar, P. Berbamine induced AMPK activation regulates mTOR/SREBP-1c axis and Nrf2/ARE pathway to allay lipid accumulation and oxidative stress in steatotic HepG2 cells. Eur. J. Pharmacol. 2020, 882, 173244. [Google Scholar] [CrossRef]
- Li, J.; Liu, M.; Yu, H.; Wang, W.; Han, L.; Chen, Q.; Ruan, J.; Wen, S.; Zhang, Y.; Wang, T. Mangiferin improves hepatic lipid metabolism mainly through its metabolite-norathyriol by modulating SIRT-1/AMPK/SREBP-1c signaling. Front. Pharmacol. 2018, 9, 201. [Google Scholar] [CrossRef]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Goransson, O.; McBride, A.; Hawley, S.A.; Ross, F.A.; Shpiro, N.; Foretz, M.; Viollet, B.; Hardie, D.G.; Sakamoto, K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 32549–32560. [Google Scholar] [CrossRef] [Green Version]
- Sanders, M.J.; Ali, Z.S.; Hegarty, B.D.; Heath, R.; Snowden, M.A.; Carling, D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J. Biol. Chem. 2007, 282, 32539–32548. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.W.; van Denderen, B.J.; Jorgensen, S.B.; Honeyman, J.E.; Steinberg, G.R.; Oakhill, J.S.; Iseli, T.J.; Koay, A.; Gooley, P.R.; Stapleton, D.; et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem. Biol. 2008, 15, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef]
- Esquejo, R.M.; Salatto, C.T.; Delmore, J.; Albuquerque, B.; Reyes, A.; Shi, Y.; Moccia, R.; Cokorinos, E.; Peloquin, M.; Monetti, M.; et al. Activation of liver AMPK with PF-06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine 2018, 31, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salatto, C.T.; Miller, R.A.; Cameron, K.O.; Cokorinos, E.; Reyes, A.; Ward, J.; Calabrese, M.; Kurumbail, R.; Rajamohan, F.; Kalgutkar, A.S.; et al. Selective activation of AMPK b1-containing isoforms improves kidney function in a rat model of diabetic nephropathy. J. Pharmacol. Exp. Ther. 2017, 361, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjobsted, R.; Jorgensen, N.O.; Tran, J.L.; Jatkar, A.; Cialdea, K.; Esquejo, R.M.; et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 2017, 25, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.F.; Rajamohan, F.; Harris, M.S.; Caspers, N.L.; Magyar, R.; Withka, J.M.; Wang, H.; Borzilleri, K.A.; Sahasrabudhe, P.V.; Hoth, L.R.; et al. Structural basis for AMPK activation: Natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure 2014, 22, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Langendorf, C.G.; Kemp, B.E. Choreography of AMPK activation. Cell Res. 2015, 25, 5–6. [Google Scholar] [CrossRef]
- Reymond, P.; Farmer, E.E. Jasmonate and salicylate as global signals for defense gene expression. Curr. Opin. Plant Biol. 1998, 1, 404–411. [Google Scholar] [CrossRef]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Pinkosky, S.L.; Scott, J.W.; Desjardins, E.M.; Smith, B.K.; Day, E.A.; Ford, R.J.; Langendorf, C.G.; Ling, N.X.Y.; Nero, T.L.; Loh, K.; et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK beta1 isoforms. Nat. Metab. 2020, 2, 873–881. [Google Scholar] [CrossRef]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Clark, H.; Carling, D.; Saggerson, D. Covalent activation of heart AMP-activated protein kinase in response to physiological concentrations of long-chain fatty acids. Eur. J. Biochem. 2004, 271, 2215–2224. [Google Scholar] [CrossRef]
- Suchankova, G.; Tekle, M.; Saha, A.K.; Ruderman, N.B.; Clarke, S.D.; Gettys, T.W. Dietary polyunsaturated fatty acids enhance hepatic AMP-activated protein kinase activity in rats. Biochem. Biophys. Res. Commun. 2005, 326, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Steinberg, G.R.; Chen, Z.P.; Kemp, B.E.; Febbraio, M.A. Fatty acids stimulate AMP-activated protein kinase and enhance fatty acid oxidation in L6 myotubes. J. Physiol. 2006, 574 Pt 1, 139–147. [Google Scholar] [CrossRef]
- Za’tara, G.; Bar-Tana, J.; Kalderon, B.; Suter, M.; Morad, E.; Samovski, D.; Neumann, D.; Hertz, R. AMPK activation by long chain fatty acyl analogs. Biochem. Pharmacol. 2008, 76, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase: Fuel gauge of the mammalian cell? Eur. J. Biochem. 1997, 246, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MRC Kinase Profiling Inhibitor Database. Available online: http://www.kinase-screen.mrc.ac.uk/kinase-inhibitors (accessed on 30 September 2020).
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Hoglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Dandapani, M.; Hardie, D.G. AMP-activated protein kinase: Friend or foe in cancer? Annu. Rev. Cancer Biol. 2020, 4, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 protein kinases of the AMPK subfamily, including the MARK/PAR-1 kinases. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaleel, M.; McBride, A.; Lizcano, J.M.; Deak, M.; Toth, R.; Morrice, N.A.; Alessi, D.R. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 2005, 579, 1417–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, J.M.; Svensson, R.U.; Lou, H.J.; Winslow, M.M.; Turk, B.E.; Shaw, R.J. An AMPK-independent signaling pathway downstream of the LKB1 tumor suppressor controls Snail1 and metastatic potential. Mol. Cell 2014, 55, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK Is a negative regulator of the Warburg effect and suppresses tumor growth In vivo. Cell Metab. 2012, 17, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houde, V.P.; Donzelli, S.; Sacconi, A.; Galic, S.; Hammill, J.A.; Bramson, J.L.; Foster, R.A.; Tsakiridis, T.; Kemp, B.E.; Grasso, G.; et al. AMPK beta1 reduces tumor progression and improves survival in p53 null mice. Mol. Oncol. 2017, 11, 1143–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penfold, L.; Woods, A.; Muckett, P.; Nikitin, A.Y.; Kent, T.R.; Zhang, S.; Graham, R.; Pollard, A.; Carling, D. CAMKK2 promotes prostate cancer independently of AMPK via increased lipogenesis. Cancer Res. 2018, 78, 6747–6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vara-Ciruelos, D.; Dandapani, M.; Russell, F.M.; Grzes, K.M.; Atrih, A.; Foretz, M.; Viollet, B.; Lamont, D.J.; Cantrell, D.A.; Hardie, D.G. Phenformin, but not metformin, delays development of T cell acute lymphoblastic leukemia/lymphoma via cell-autonomous AMPK activation. Cell Rep. 2019, 27, 690–698. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Decensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev. Res. 2010, 3, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; McBurnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem. J. 2008, 412, 211–221. [Google Scholar] [CrossRef]
- Crofford, O.B. Metformin. N. Engl. J. Med. 1995, 333, 588–589. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.H.; White, M.A.; Potts, P.R. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, I.K.; Yao, Y.; Kim, G.; Xia, W.; Kim, H.; Kim, S.J.; Park, M.K.; Hwang, J.P.; Gonzalez-Billalabeitia, E.; Hung, M.C.; et al. A UBE2O-AMPKalpha2 axis that promotes tumor initiation and progression offers opportunities for therapy. Cancer Cell 2017, 31, 208–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic analysis reveals AMPK Is required to support tumor growth in murine Kras-dependent lung cancer models. Cell Metab. 2019, 29, 285–302. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Monteverde, T.; Muthalagu, N.; Port, J.; Murphy, D.J. Evidence of cancer promoting roles for AMPK and related kinases. FEBS J. 2015, 282, 4658–4671. [Google Scholar] [CrossRef] [Green Version]
- Phoenix, K.N.; Devarakonda, C.V.; Fox, M.M.; Stevens, L.E.; Claffey, K.P. AMPKalpha2 suppresses murine embryonic fibroblast transformation and tumorigenesis. Genes Cancer 2012, 3, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Network, T.C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Speidel, D. The role of DNA damage responses in p53 biology. Arch. Toxicol. 2015, 89, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.W.; Park, S.B.; Lee, S.J.; Seo, M.S.; Trosko, J.E.; Kang, K.S. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS ONE 2011, 6, e28068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Hu, C.; Zhang, W.; Shen, Y.; Wang, J.; Hu, F.; Yu, P. Metformin inhibits the proliferation, metastasis, and cancer stem-like sphere formation in osteosarcoma MG63 cells in vitro. Tumour Biol. 2015, 36, 9873–9883. [Google Scholar] [CrossRef]
- Yuan, X.; Wei, W.; Bao, Q.; Chen, H.; Jin, P.; Jiang, W. Metformin inhibits glioma cells stemness and epithelial-mesenchymal transition via regulating YAP activity. Biomed. Pharmacother. 2018, 102, 263–270. [Google Scholar] [CrossRef]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Maehara, O.; Ohnishi, S.; Asano, A.; Suda, G.; Natsuizaka, M.; Nakagawa, K.; Kobayashi, M.; Sakamoto, N.; Takeda, H. Metformin regulates the expression of CD133 through the AMPK-CEBPbeta pathway in hepatocellular carcinoma cell lines. Neoplasia 2019, 21, 545–556. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71, 3196–3201. [Google Scholar] [CrossRef] [Green Version]
- Fasih, A.; Elbaz, H.A.; Huttemann, M.; Konski, A.A.; Zielske, S.P. Radiosensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat. Res. 2014, 182, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Martin, A.; Vellon, L.; Quiros, P.M.; Cufi, S.; Ruiz de Galarreta, E.; Oliveras-Ferraros, C.; Martin, A.G.; Martin-Castillo, B.; Lopez-Otin, C.; Menendez, J.A. Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells. Cell Cycle 2012, 11, 974–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Bort, A.; Sanchez, B.G.; Mateos-Gomez, P.A.; Vara-Ciruelos, D.; Rodriguez-Henche, N.; Diaz-Laviada, I. Targeting AMP-activated kinase impacts hepatocellular cancer stem cells induced by long-term treatment with sorafenib. Mol. Oncol. 2019, 13, 1311–1331. [Google Scholar] [CrossRef]
- Sengupta, S.; Nagalingam, A.; Muniraj, N.; Bonner, M.Y.; Mistriotis, P.; Afthinos, A.; Kuppusamy, P.; Lanoue, D.; Cho, S.; Korangath, P.; et al. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem-like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene 2017, 36, 5709–5721. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jin, J.; Wan, F.; Zhao, L.; Chu, H.; Chen, C.; Liao, G.; Liu, J.; Yu, Y.; Teng, H.; et al. AMPK Promotes SPOP-Mediated NANOG Degradation to Regulate Prostate Cancer Cell Stemness. Dev. Cell 2019, 48, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russell, F.M.; Hardie, D.G. AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer? Int. J. Mol. Sci. 2021, 22, 186. https://doi.org/10.3390/ijms22010186
Russell FM, Hardie DG. AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer? International Journal of Molecular Sciences. 2021; 22(1):186. https://doi.org/10.3390/ijms22010186
Chicago/Turabian StyleRussell, Fiona M., and David Grahame Hardie. 2021. "AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer?" International Journal of Molecular Sciences 22, no. 1: 186. https://doi.org/10.3390/ijms22010186
APA StyleRussell, F. M., & Hardie, D. G. (2021). AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer? International Journal of Molecular Sciences, 22(1), 186. https://doi.org/10.3390/ijms22010186