ANP and BNP Exert Anti-Inflammatory Action via NPR-1/cGMP Axis by Interfering with Canonical, Non-Canonical, and Alternative Routes of Inflammasome Activation in Human THP1 Cells




Abstract
1. Introduction
2. Results and Discussion
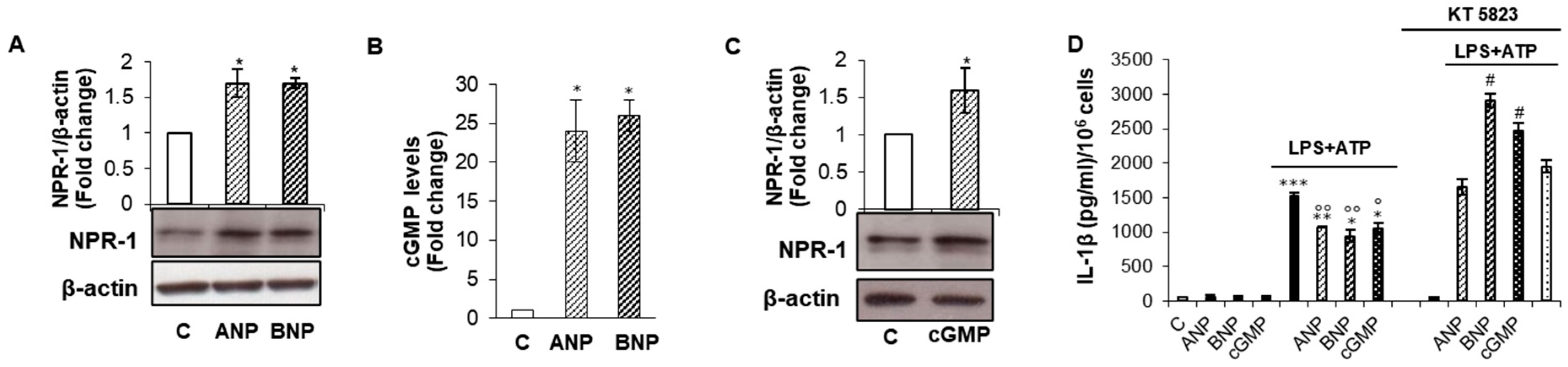
2.1. cGMP/PKG-I Axis Drives ANP/BNP Inhibitory Effect on IL-1β Secretion
2.2. cGMP/PKG-I Axis is Involved in the Priming Mechanism Controlling IL-1β Production
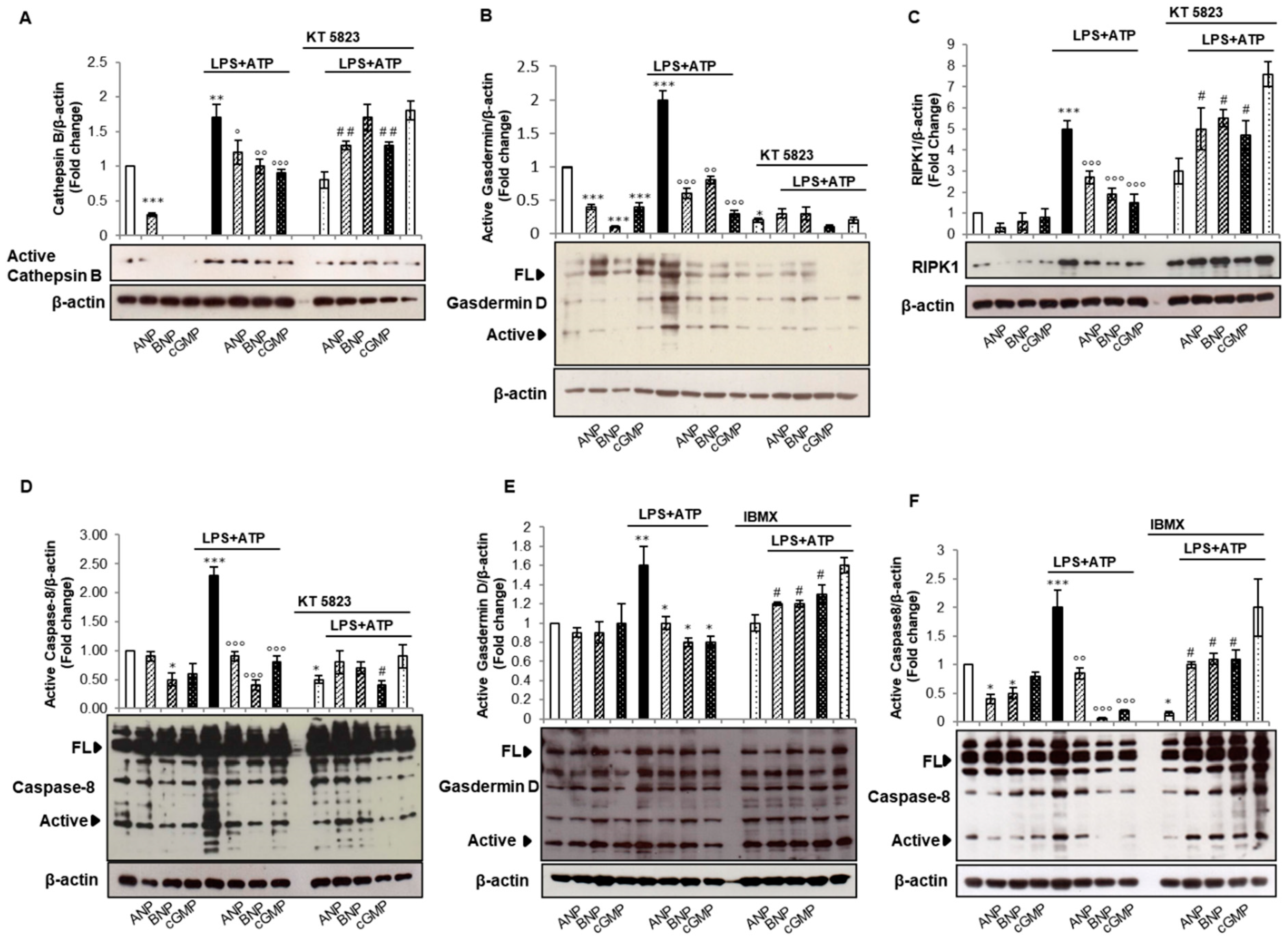
2.3. cGMP/PKG-I Axis Is Involved in the Activation Step of IL-1β Maturation
2.4. cGMP but Not PKG-I Is Involved in Non-Canonical and Alternative Inflammasome Activation
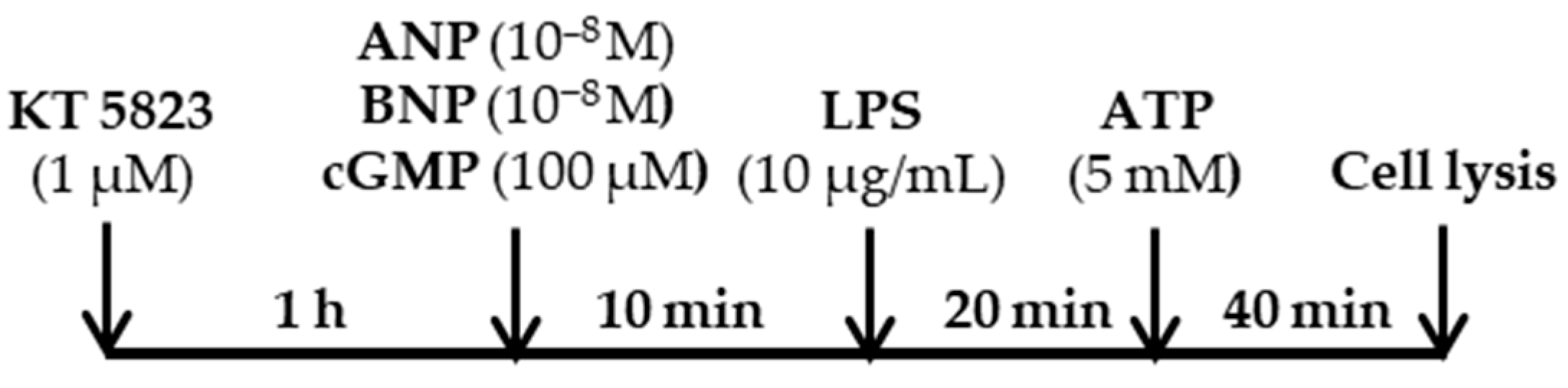
3. Materials and Methods
3.1. Reagents
3.2. Cell Culture and Drug Treatments
3.3. Measurements of Secreted IL-1β
3.4. cGMP Measurements
3.5. Western Blot Analysis
3.6. Chemical Crosslinking of ASC Oligomers
3.7. Phosphorylation Site Prediction
3.8. Statistical analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lowe, D.G.; Chang, M.S.; Hellmiss, R.; Chen, E.; Singh, S.; Garbers, D.L.; Goeddel, D.V. Human atrial natriuretic peptide receptor defines a new paradigm for second messenger signal transduction. EMBO J. 1989, 8, 1377–13784. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic peptides: Their structures, receptors, physiologic functions and therapeutic applications. Handb. Exp. Pharmacol. 2009, 191, 341–366. [Google Scholar]
- Vinnakota, S.; Chen, H.H. The Importance of Natriuretic Peptides in Cardiometabolic Diseases. J. Endocr. Soc. 2020, 4, bvaa052. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, T.; Kuwahara, K.; Nakao, K. Current biochemistry, molecular biology, and clinical relevance of natriuretic peptides. J. Cardiol. 2011, 57, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Gruden, G.; Landi, A.; Bruno, G. Natriuretic peptides, heart, and adipose tissue: New findings and future developments for diabetes research. Diabetes Care 2014, 37, 2899–2908. [Google Scholar] [CrossRef]
- Kuhn, M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef]
- Fish-Trotter, H.; Ferguson, J.F.; Patel, N.; Arora, P.; Allen, N.B.; Bachmann, K.N.; Daniels, L.B.; Reilly, M.P.; Lima, J.A.C.; Wang, T.J.; et al. Inflammation and Circulating Natriuretic Peptide Levels. Circ. Heart Fail. 2020, 13, e006570. [Google Scholar] [CrossRef]
- Zhang, J.; Li, M.; Yang, Y.; Yan, Y.; Li, J.; Qu, J.; Wang, J. NPR-A: A Therapeutic Target in Inflammation and Cancer. Crit. Rev. Eukaryot. Gene. Expr. 2015, 25, 41–46. [Google Scholar] [CrossRef]
- de Bold, A.J. Cardiac natriuretic peptides gene expression and secretion in inflammation. J. Investig. Med. 2009, 57, 736. [Google Scholar]
- Ogawa, T.; de Bold, A.J. Brain natriuretic Peptide production and secretion in inflammation. J. Transplant. 2012, 2012, 962347. [Google Scholar] [CrossRef]
- Mezzasoma, L.; Cagini, L.; Antognelli, C.; Puma, F.; Pacifico, E.; Talesa, V.N. TNF-α regulates natriuretic peptides and aquaporins in human bronchial epithelial cells BEAS-2B. Mediat. Inflamm. 2013, 2013, 159349. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosyula, S.; Wang, Z.; Murad, H.; Vallabhajosyula, S.; Sundaragiri, P.R.; Kashani, K.; Miller, W.L.; Jaffe, A.L.; Vallabhajosyula, S. Natriuretic Peptides to Predict Short-Term Mortality in Patients With Sepsis: A Systematic Review and Meta-analysis. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 4, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, X.; Xu, W.; Behera, S.; Hellermann, G.; Kumar, A.; Lockey, R.F.; Mohapatra, S.; Mohapatra, S.S. Natriuretic peptide receptor a as a novel anticancer target. Cancer Res. 2008, 68, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, W.; Mohapatra, S.; Kong, X.; Li, X.; Lockey, R.F.; Mohapatra, S.S. Prevention of airway inflammation with topical cream containing imiquimod and small interfering RNA for natriuretic peptide receptor. Genet. Vaccines Ther. 2008, 6, 7. [Google Scholar] [CrossRef]
- Das, S.; Periyasamy, R.; Pandey, K.N. Activation of IKK/NF-κB provokes renal inflammatory responses in guanylyl cyclase/natriuretic peptide receptor-A gene-knockout mice. Physiol. Genom. 2012, 44, 430–442. [Google Scholar] [CrossRef][Green Version]
- Vellaichamy, E.; Khurana, M.L.; Fink, J.; Pandey, K.N. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J. Biol. Chem. 2005, 280, 19230–19242. [Google Scholar] [CrossRef]
- Vellaichamy, E.; Kaur, K.; Pandey, K.N. Enhanced activation of pro-inflammatory cytokines in mice lacking natriuretic peptide receptor-A. Peptides 2007, 28, 893–899. [Google Scholar] [CrossRef][Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Mezzasoma, L.; Antognelli, C.; Talesa, V.N. Atrial natriuretic peptide down-regulates LPS/ATP-mediated IL-1β release by inhibiting NF-kB, NLRP3 inflammasome and Caspase-1 activation in THP-1 cells. Immunol. Res. 2016, 64, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Mezzasoma, L.; Antognelli, C.; Talesa, V.N. A Novel Role for Brain Natriuretic Peptide: Inhibition of IL-1β Secretion via Downregulation of NF-kB/Erk 1/2 and NALP3/ASC/Caspase-1 Activation in Human THP-1 Monocyte. Mediat. Inflamm. 2017, 2017, 5858315. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Jin, S.; Xiang, P.; Liu, J.; Yang, Y.; Hu, S.; Sheng, J.; He, Q.; Yu, W.; Han, W.; Jin, J.; et al. Activation of cGMP/PKG/p65 signaling associated with PDE5-Is downregulates CCL5 secretion by CD8 + T cells in benign prostatic hyperplasia. Prostate 2019, 79, 909–919. [Google Scholar] [CrossRef]
- Lee, M.L.; Sulistyowati, E.; Hsu, J.H.; Huang, B.Y.; Dai, Z.K.; Wu, B.N.; Chao, Y.Y.; Yeh, J.L. KMUP-1 Ameliorates Ischemia-Induced Cardiomyocyte Apoptosis through the NO⁻cGMP⁻MAPK Signaling Pathways. Molecules 2019, 24, 1376. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory Caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasome. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 2017, 171, 1057–1071.e11. [Google Scholar] [CrossRef]
- Ghonime, M.G.; Shamaa, O.R.; Das, S.; Eldomany, R.A.; Fernandes-Alnemri, T.; Alnemri, E.S.; Gavrilin, M.A.; Wewers, M.D. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J. Immunol. 2014, 192, 3881–3888. [Google Scholar] [CrossRef]
- Nakao, K.; Morii, N.; Itoh, H.; Nakao, K.; Morii, N.; Itoh, H.; Yamada, T.; Shiono, S.; Sugawara, A.; Saito, Y.; et al. Atrial natriuretic polypeptide in the brain: Implication of central cardiovascular control. J. Hypertens. Suppl. 1986, 4, S492–S496. [Google Scholar] [PubMed]
- de Lemos, J.A.; McGuire, D.K.; Drazner, M.H. B-type natriuretic peptide in cardiovascular disease. Lancet 2003, 362, 316–322. [Google Scholar] [CrossRef]
- D’Elia, E.; Iacovoni, A.; Vaduganathan, M.; Lorini, F.L.; Perlini, S.; Senni, M. Neprilysin inhibition in heart failure: Mechanisms and substrates beyond modulating natriuretic peptides. Eur. J. Heart Fail. 2017, 19, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. GPS 2.0, a Tool to Predict Kinase-specific Phosphorylation Sites in Hierarchy. Mol. Cell. Proteom. 2008, 7, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Meszaros, G.; He, W.T.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gámez, M.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef]
- Mezzasoma, L.; Costanzi, E.; Scarpelli, P.; Talesa, V.N.; Bellezza, I. Extracellular Vesicles from Human Advanced-Stage Prostate Cancer Cells Modify the Inflammatory Response of Microenvironment-Residing Cells. Cancers 2019, 11, 1276. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends. Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Eby, M.T.; Jasmin, A.; Kumar, A.; Liu, L.; Hood, L. Activation of the NF-kappaB pathway by Caspase 8 and its homologs. Oncogene 2000, 19, 4451–4460. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Kanneganti, T.D. Novel roles for Caspase-8 in IL-1β and inflammasome regulation. Am. J. Pathol. 2015, 185, 17–25. [Google Scholar] [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and Caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846b. [Google Scholar] [CrossRef]
- Banerjee, C.; Singh, A.; Das, T.K.; Raman, R.; Shrivastava, A.; Mazumder, S. Ameliorating ER-stress attenuates Aeromonas hydrophila-induced mitochondrial dysfunctioning and Caspase mediated HKM apoptosis in Clarias batrachus. Sci. Rep. 2014, 4, 5820. [Google Scholar] [CrossRef] [PubMed]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zeng, L.; Zhu, S.; Liu, J.; Zeh, H.J.; Kroemer, G.; Wang, H.; Billiar, T.R.; Jiang, J.; Tang, D.; et al. cAMP metabolism controls Caspase-11 inflammasome activation and pyroptosis in sepsis. Sci. Adv. 2019, 5, eaav5562. [Google Scholar] [CrossRef]
- Frantz, S.; Falcao-Pires, I.; Balligand, J.L.; Bauersachs, J.; Brutsaert, D.; Ciccarelli, M.; Dawson, D.; de Windt, L.J.; Giacca, M.; Hamdani, N.; et al. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur. J. Heart Fail. 2018, 20, 445–459. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. PARADIGM-HF Investigators and Committees. Angiotensin–neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Giczewska, A.; Sliwa, K.; Edelmann, F.; Refsgaard, J.; Bocchi, E.; Ezekowitz, J.A.; Hernandez, A.F.; O’Connor, C.M.; Roessig, L.; et al. Clinical Outcomes and Response to Vericiguat According to Index Heart Failure Event: Insights from the VICTORIA Trial. JAMA Cardiol. 2020, e206455. [Google Scholar] [CrossRef]
- Mancini, A.; Tantucci, M.; Mazzocchetti, P.; de Iure, A.; Durante, V.; Macchioni, L.; Giampà, C.; Alvino, A.; Gaetani, L.; Costa, C.; et al. Microglial activation and the nitric oxide/cGMP/PKG pathway underlie enhanced neuronal vulnerability to mitochondrial dysfunction in experimental multiple sclerosis. Neurobiol. Dis. 2018, 113, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S. Functional crosstalk between non-canonical caspase-11 and canonical NLRP3 inflammasomes during infection-mediated inflammation. Immunology 2020, 159, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Feng, Y.; Lyons, J.D.; Berger, S.C.; Otani, S.; DeLaney, A.; Tharp, G.K.; Maner-Smith, K.; Burd, E.M.; Schaeffer, M.; et al. Caspase-8 Collaborates with Caspase-11 to Drive Tissue Damage and Execution of Endotoxic Shock. Immunity 2018, 49, 42–55.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, J.; Zeng, Y.; Chen, K.; Wang, C.; Yang, S.; Sun, N.; Chen, H.; Duan, K.; Zeng, G. Pyroptosis: A pro-inflammatory type of cell death in cardiovascular disease. Clin. Chim. Acta 2020, 510, 62–72. [Google Scholar] [CrossRef]
- Ridke, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Shah, A. Novel Coronavirus-Induced NLRP3 Inflammasome Activation: A Potential Drug Target in the Treatment of COVID-19. Front. Immunol. 2020, 11, 1021. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Code | Kinase | Peptide | Score | Cutoff |
|---|---|---|---|---|---|
| 169 | T | AGC/PKG/PRKG1 | VSLNKRYTRLRLIKE | 10,511 | 10,046 |
| 295 | S | AGC/PKG/PRKG1 | HKIVRKPSRILFLMD | 11,906 | 10,046 |
| 955 | S | AGC/PKG/PRKG1 | SQSLRKLSLGNNDLG | 12,266 | 10,046 |
| 102 | S | AGC/PKG/PRKG2 | GSDNARVSNPTVICQ | 64.2 | 55,934 |
| 295 | S | AGC/PKG/PRKG2 | HKIVRKPSRILFLMD | 59,621 | 55,934 |
| 436 | S | AGC/PKG/PRKG2 | GKSLAQTSKTTTAVY | 60,347 | 55,934 |
| 728 | S | AGC/PKG/PRKG2 | VNSHLTSSFCRGLFS | 56,211 | 55,934 |
| 955 | S | AGC/PKG/PRKG2 | SQSLRKLSLGNNDLG | 57,236 | 55,934 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezzasoma, L.; Talesa, V.N.; Romani, R.; Bellezza, I. ANP and BNP Exert Anti-Inflammatory Action via NPR-1/cGMP Axis by Interfering with Canonical, Non-Canonical, and Alternative Routes of Inflammasome Activation in Human THP1 Cells. Int. J. Mol. Sci. 2021, 22, 24. https://doi.org/10.3390/ijms22010024
Mezzasoma L, Talesa VN, Romani R, Bellezza I. ANP and BNP Exert Anti-Inflammatory Action via NPR-1/cGMP Axis by Interfering with Canonical, Non-Canonical, and Alternative Routes of Inflammasome Activation in Human THP1 Cells. International Journal of Molecular Sciences. 2021; 22(1):24. https://doi.org/10.3390/ijms22010024
Chicago/Turabian StyleMezzasoma, Letizia, Vincenzo Nicola Talesa, Rita Romani, and Ilaria Bellezza. 2021. "ANP and BNP Exert Anti-Inflammatory Action via NPR-1/cGMP Axis by Interfering with Canonical, Non-Canonical, and Alternative Routes of Inflammasome Activation in Human THP1 Cells" International Journal of Molecular Sciences 22, no. 1: 24. https://doi.org/10.3390/ijms22010024
APA StyleMezzasoma, L., Talesa, V. N., Romani, R., & Bellezza, I. (2021). ANP and BNP Exert Anti-Inflammatory Action via NPR-1/cGMP Axis by Interfering with Canonical, Non-Canonical, and Alternative Routes of Inflammasome Activation in Human THP1 Cells. International Journal of Molecular Sciences, 22(1), 24. https://doi.org/10.3390/ijms22010024