How the AHR Became Important in Cancer: The Role of Chronically Active AHR in Cancer Aggression

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
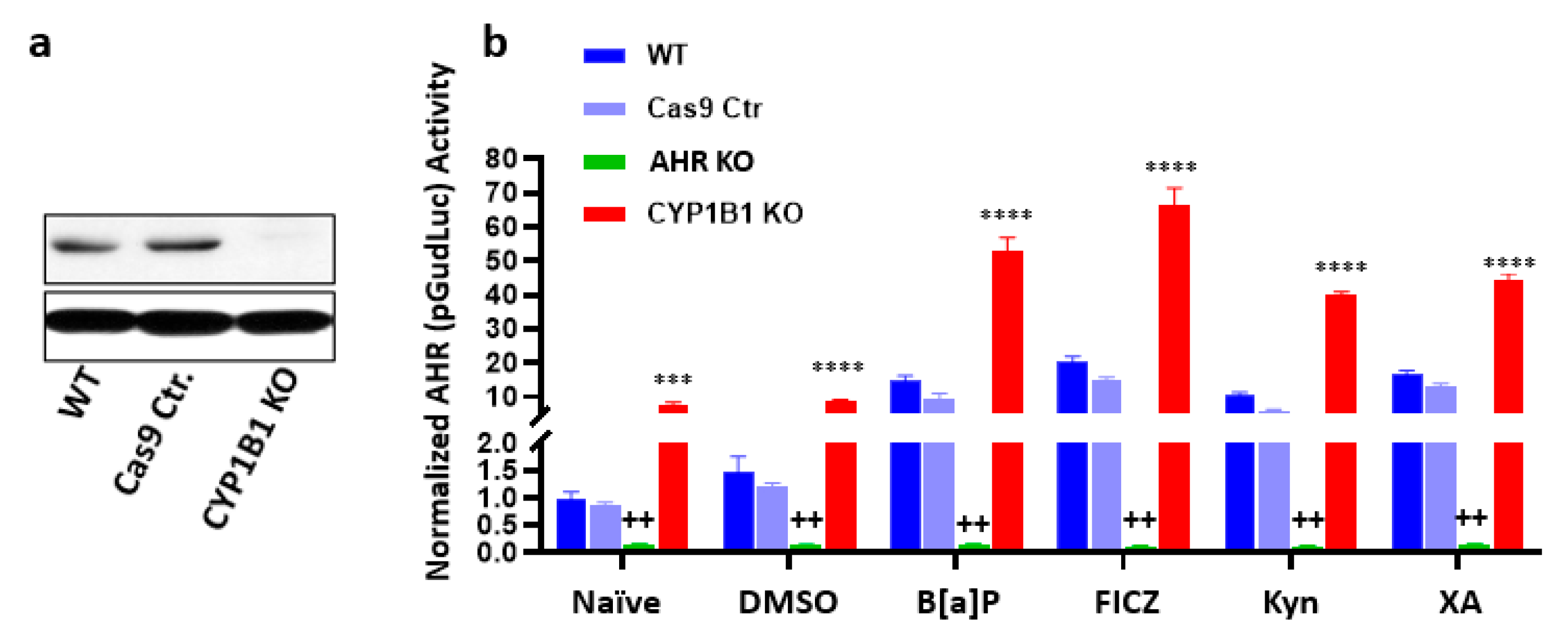{kind=link}
Abstract
1. The First Hints of a Role for the AHR in Cancer: Carcinogenic Environmental AHR Ligands
2. AHR Transcriptional Signaling
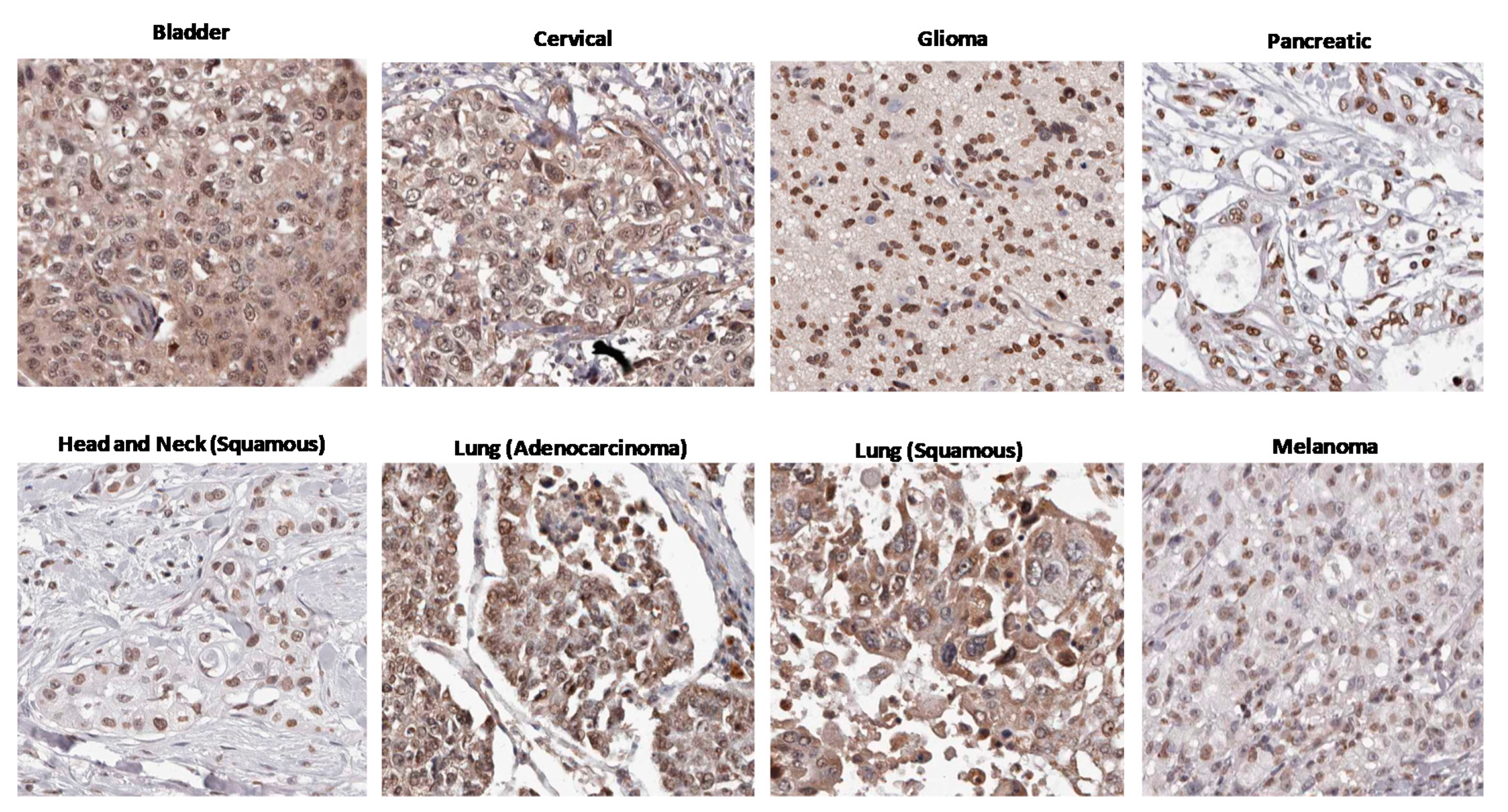
3. Circumstantial Evidence: High AHR Expression in Many Cancers
4. Evidence Builds: An Association between Chronic “Constitutive” AHR Activity and Cancer Patient Outcomes
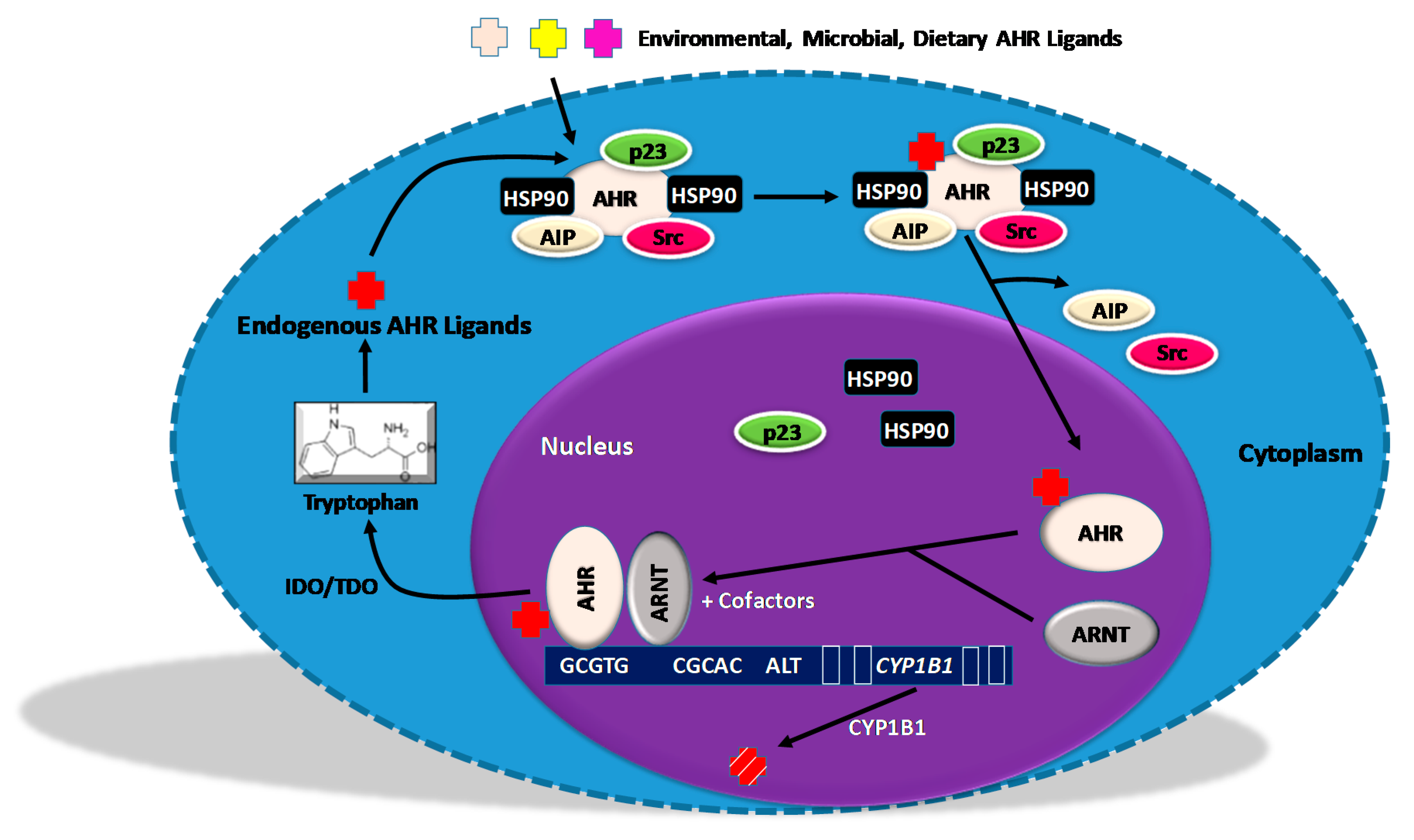
5. Regulators of AHR Activity
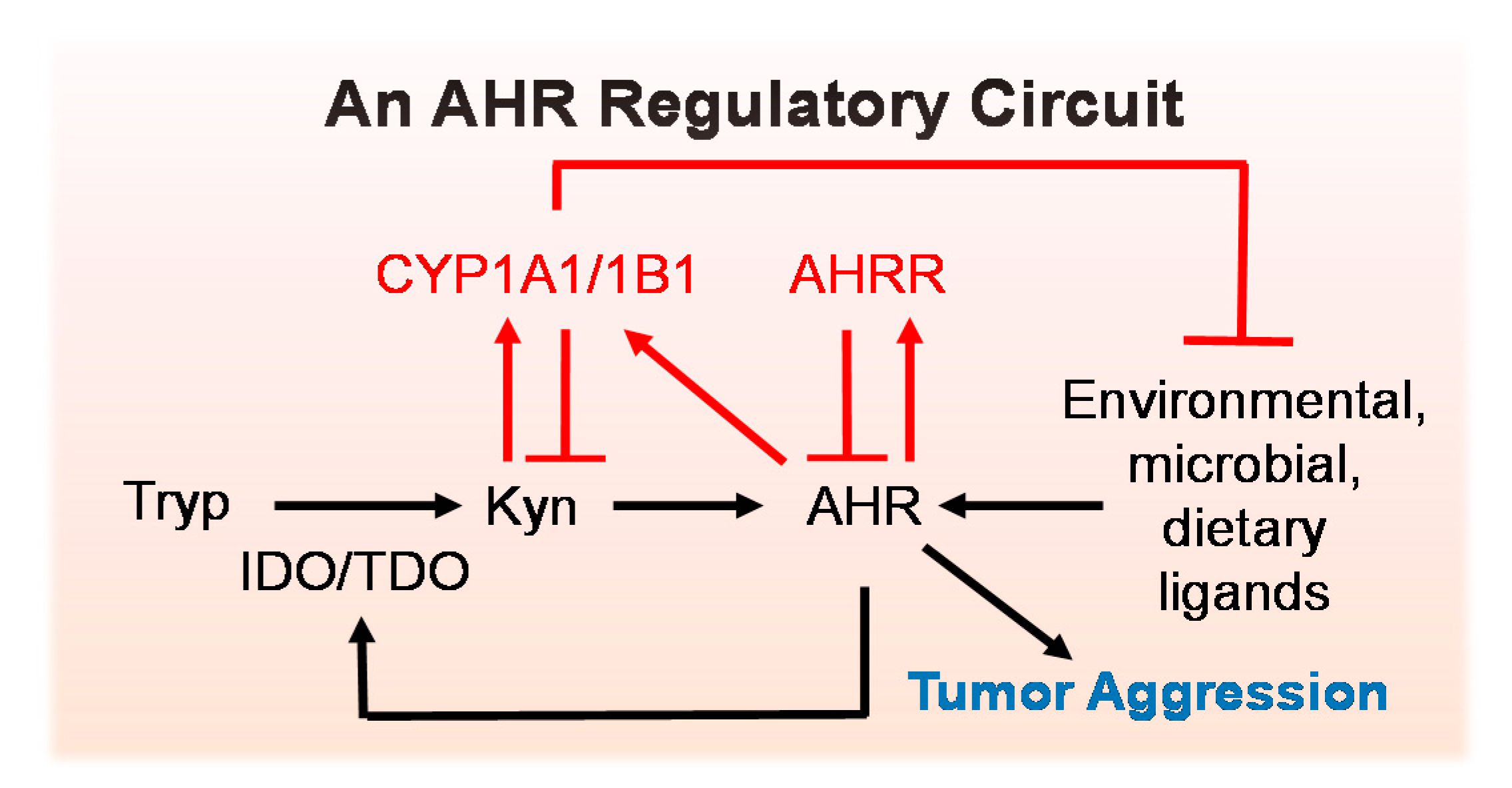
5.1. An AHR Amplification Loop: A Possible Driver of “Constitutively Active” AHR
5.2. Negative Regulators of the AHR Amplification Loop
6. Consequences of Chronic AHR Activity in Cancer
6.1. AHR-Mediated Cell Migration and Invasion
6.2. AHR-Mediated Epithelial-to-Mesenchymal Transition (EMT) and Metastasis
6.3. AHR Role in Cancer Stem Cell (CSC) Development
6.4. The AHR’s Role in Malignant Cell Apoptosis
7. Caveats: Interspecies Differences
8. Conclusions
9. Materials and Methods
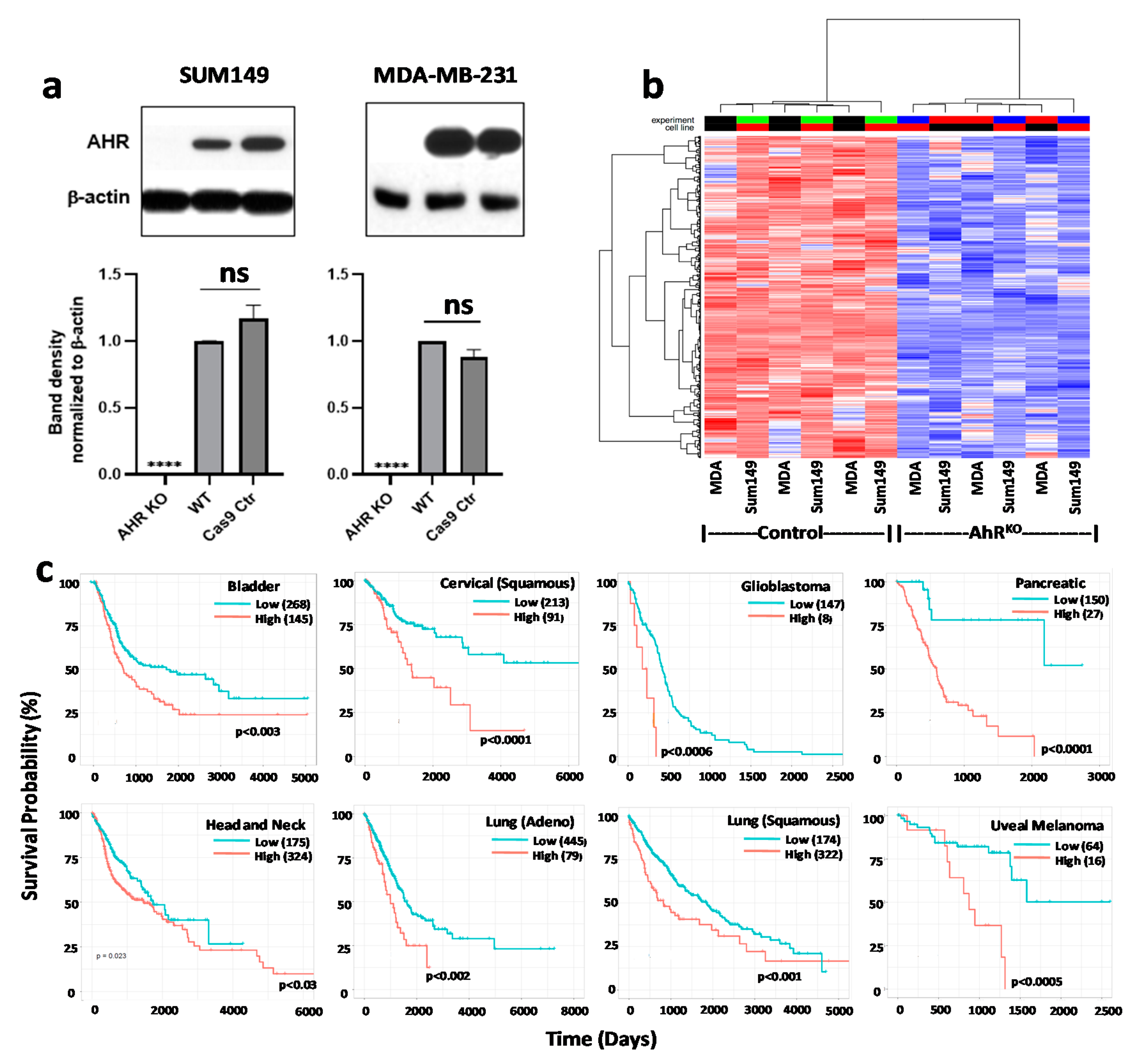
9.1. Generation of AHR or CYP1B1 Knockout MDA-MB-231 and SUM149 Cell Lines with CRISPR-Cas9 Gene Editing
9.2. Western Blotting
9.3. AHR-Driven Reporter Assay
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AHR | Aryl hydrocarbon receptor |
| AHRR | AHR repressor |
| AIP/XAP2 | Immunophilin-like AHR-interacting protein/Hepatitis B virus X-Associated Protein 2 |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| bHLH-PAS | Basic helix-loop-helix-Per Arnt Sim |
| FICZ | 6-formylindolo[3,2-b]carbazole |
| Kyn | Kynurenine |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
References
- Poland, A.; Glover, E. Chlorinated dibenzo-p-dioxins: Potent inducers of delta-aminolevulinic acid synthetase and aryl hydrocarbon hydroxylase. II. A study of the structure-activity relationship. Mol. Pharmacol. 1973, 9, 736–747. [Google Scholar] [PubMed]
- Poland, A.; Glover, E. Studies on the mechanism of toxicity of the chlorinated dibenzo-p-dioxins. Environ. Health Perspect. 1973, 5, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.; Glover, E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: A potent inducer of -aminolevulinic acid synthetase. Science 1973, 179, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Gielen, J.E. Genetic regulation of aryl hydrocarbon hydroxylase induction in the mouse. Fed. Proc. USA 1972, 31, 1315–1325. [Google Scholar]
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar] [PubMed]
- Poland, A.; Glover, E.; Robinson, J.; Nebert, D. Genetic expression of aryl hydrocarbon hydroxylase activity: Induction of monooxygenase activites and cytochrome P1-450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons. J. Biol. Chem. 1974, 249, 5599–5606. [Google Scholar]
- Ayres, S.M.; Webb, K.B.; Evans, R.G.; Mikes, J. Is 2,3,7,8-TCDD (dioxin) a carcinogen for humans? Environ. Health Perspect. 1985, 62, 329–335. [Google Scholar] [CrossRef][Green Version]
- Catalani, S. IARC revision on dioxin and some dioxin-like compounds. G. Ital. Med. Lav. Ergon. 2010, 32, 79–81. [Google Scholar]
- Perdew, G.H.; Poland, A. Purification of the Ah receptor from C57BL/6J mouse liver. J. Biol. Chem. 1988, 263, 9848–9852. [Google Scholar]
- Bradfield, C.A.; Glover, E.; Poland, A. Purification and N-terminal amino acid sequence of the Ah receptor from the C57BL/6J mouse. Mol. Pharmacol. 1991, 39, 13–19. [Google Scholar]
- Burbach, K.M.; Poland, A.; Bradfield, C.A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. USA 1992, 89, 8185–8189. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Sogawa, K.; Watanabe, N.; Chujoh, Y.; Matsushita, N.; Gotoh, O.; Funae, Y.; Fujii-Kuriyama, Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem. Biophys. Res. Commun. 1992, 184, 246–253. [Google Scholar] [CrossRef]
- Shimizu, Y.; Nakatsuru, Y.; Ichinose, M.; Takahashi, Y.; Kume, H.; Mimura, J.; Fujii-Kuriyama, Y.; Ishikawa, T. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, T.; Fujimura, T.; Aiba, S. Aryl Hydrocarbon Receptor Modulates Carcinogenesis and Maintenance of Skin Cancers. Front. Med. (Lausanne) 2019, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Ide, F.; Kishi, R.; Akutagawa, T.; Sakai, S.; Nakamura, M.; Ishikawa, T.; Fujii-Kuriyama, Y.; Nakatsuru, Y. Aryl hydrocarbon receptor plays a significant role in mediating airborne particulate-induced carcinogenesis in mice. Environ. Sci. Technol. 2007, 41, 3775–3780. [Google Scholar] [CrossRef] [PubMed]
- Juricek, L.; Bui, L.C.; Busi, F.; Pierre, S.; Guyot, E.; Lamouri, A.; Dupret, J.M.; Barouki, R.; Coumoul, X.; Rodrigues-Lima, F. Activation of the aryl hydrocarbon receptor by carcinogenic aromatic amines and modulatory effects of their N-acetylated metabolites. Arch. Toxicol. 2015, 89, 2403–2412. [Google Scholar] [CrossRef]
- Glazer, L.; Hahn, M.E.; Aluru, N. Delayed effects of developmental exposure to low levels of the aryl hydrocarbon receptor agonist 3,3′,4,4′,5-pentachlorobiphenyl (PCB126) on adult zebrafish behavior. Neurotoxicology 2015, 52, 134–143. [Google Scholar] [CrossRef]
- Ovesen, J.L.; Schnekenburger, M.; Puga, A. Aryl hydrocarbon receptor ligands of widely different toxic equivalency factors induce similar histone marks in target gene chromatin. Toxicol. Sci. 2011, 121, 123–131. [Google Scholar] [CrossRef]
- Zhang, S.; Rowlands, C.; Safe, S. Ligand-dependent interactions of the Ah receptor with coactivators in a mammalian two-hybrid assay. Toxicol. Appl. Pharmacol. 2007, 227, 196–206. [Google Scholar] [CrossRef]
- Shimada, T.; Hayes, C.L.; Yamazaki, H.; Amin, S.; Hecht, S.S.; Guengerich, F.P.; Sutter, T.R. Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res. 1996, 56, 2979–2984. [Google Scholar]
- Tang, Y.M.; Wo, Y.Y.; Stewart, J.; Hawkins, A.L.; Griffin, C.A.; Sutter, T.R.; Greenlee, W.F. Isolation and characterization of the human cytochrome P450 CYP1B1 gene. J. Biol. Chem. 1996, 271, 28324–28330. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.; Lax, S.; Wyde, M.; Crofts, F.; Lucier, C.; Sutter, T. CYP1B1 and CYP1A1 exhibit differential inducibility by TCDD in a Sprague-Dawley rat tumor promotion model. Fund. Appl. Toxicol. 1997, 36, 215. [Google Scholar]
- Sutter, T.R.; Tang, Y.M.; Hayes, C.L.; Wo, Y.Y.; Jabs, E.W.; Li, X.; Yin, H.; Cody, C.W.; Greenlee, W.F. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J. Biol. Chem. 1994, 269, 13092–13099. [Google Scholar] [PubMed]
- Crespi, C.L.; Penman, B.W.; Steimel, D.T.; Smith, T.; Yang, C.S.; Sutter, T.R. Development of a human lymphoblastoid cell line constitutively expressing human CYP1B1 cDNA: Substrate specificity with model substrates and promutagens. Mutagenesis 1997, 12, 83–89. [Google Scholar] [CrossRef][Green Version]
- Shimada, T.; Oda, Y.; Gillam, E.M.; Guengerich, F.P.; Inoue, K. Metabolic activation of polycyclic aromatic hydrocarbons and other procarcinogens by cytochromes P450 1A1 and P450 1B1 allelic variants and other human cytochromes P450 in Salmonella typhimurium NM2009. Drug. Metab. Dispos. 2001, 29, 1176–1182. [Google Scholar]
- Thier, R.; Bruning, T.; Roos, P.H.; Bolt, H.M. Cytochrome P450 1B1, a new keystone in gene-environment interactions related to human head and neck cancer? Arch. Toxicol. 2002, 76, 249–256. [Google Scholar] [CrossRef]
- Buters, J.; Quintanilla-Martinez, L.; Schober, W.; Soballa, V.J.; Hintermair, J.; Wolff, T.; Gonzalez, F.J.; Greim, H. CYP1B1 determines susceptibility to low doses of 7,12-dimethylbenz[a]anthracene-induced ovarian cancers in mice: Correlation of CYP1B1-mediated DNA adducts with carcinogenicity. Carcinogenesis 2003, 24, 327–334. [Google Scholar] [CrossRef]
- Buters, J.T.; Mahadevan, B.; Quintanilla-Martinez, L.; Gonzalez, F.J.; Greim, H.; Baird, W.M.; Luch, A. Cytochrome P450 1B1 determines susceptibility to dibenzo[a,l]pyrene-induced tumor formation. Chem. Res. Toxicol. 2002, 15, 1127–1135. [Google Scholar] [CrossRef]
- Heidel, S.M.; Holston, K.; Buters, J.T.; Gonzalez, F.J.; Jefcoate, C.R.; Czupyrynski, C.J. Bone marrow stromal cell cytochrome P4501B1 is required for pre-B cell apoptosis induced by 7,12-dimethylbenz[a]anthracene. Mol. Pharmacol. 1999, 56, 1317–1323. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Sullivan, A.P.; Sebastian, A.; Perry, G.H.; Jablonski, N.G.; Perdew, G.H. Divergent Ah Receptor Ligand Selectivity during Hominin Evolution. Mol. Biol. Evol. 2016, 33, 2648–2658. [Google Scholar] [CrossRef]
- Embrechts, J.; Lemiere, F.; Van Dongen, W.; Esmans, E.L.; Buytaert, P.; Van Marck, E.; Kockx, M.; Makar, A. Detection of estrogen DNA-adducts in human breast tumor tissue and healthy tissue by combined nano LC-nano ES tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2003, 14, 482–491. [Google Scholar] [CrossRef]
- Markushin, Y.; Zhong, W.; Cavalieri, E.L.; Rogan, E.G.; Small, G.J.; Yeung, E.S.; Jankowiak, R. Spectral characterization of catechol estrogen quinone (CEQ)-derived DNA adducts and their identification in human breast tissue extract. Chem. Res. Toxicol. 2003, 16, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Yepez, S.; Tirado-Rodriguez, A.; Montecillo-Aguado, M.R.; Yang, J.; Hammock, B.D.; Hankinson, O. Aryl Hydrocarbon Receptor-Dependent inductions of omega-3 and omega-6 polyunsaturated fatty acid metabolism act inversely on tumor progression. Sci. Rep. 2020, 10, 7843. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.E.; Karchner, S.I.; Shapiro, M.A.; Perera, S.A. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc. Natl. Acad. Sci. USA 1997, 94, 13743–13748. [Google Scholar] [CrossRef]
- Hahn, M.E.; Woodin, B.R.; Stegeman, J.J.; Tillitt, D.E. Aryl hydrocarbon receptor function in early vertebrates: Inducibility of cytochrome P450 1A in agnathan and elasmobranch fish. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1998, 120, 67–75. [Google Scholar]
- Hahn, M.E.; Karchner, S.I.; Evans, B.R.; Franks, D.G.; Merson, R.R.; Lapseritis, J.M. Unexpected diversity of aryl hydrocarbon receptors in non-mammalian vertebrates: Insights from comparative genomics. J. Exp. Zoolog. A Comp. Exp. Biol. 2006, 305, 693–706. [Google Scholar] [CrossRef]
- Hahn, M.E. The aryl hydrocarbon receptor: A comparative perspective. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1998, 121, 23–53. [Google Scholar] [CrossRef]
- Hahn, M.E. Aryl hydrocarbon receptors: Diversity and evolution. Chem. Biol. Interact. 2002, 141, 131–160. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Kumar, M.B.; Perdew, G.H. Nuclear receptor coactivator SRC-1 interacts with the Q-rich subdomain of the AhR and modulates its transactivation potential. Gene Expr. 1999, 8, 273–286. [Google Scholar]
- Ma, Q.; Whitlock, J.P., Jr. A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Chem. 1997, 272, 8878–8884. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.; Cuthill, S.; Wikstroem, A.-C.; Poellinger, L.; Gustafsson, J.-A. Association of the Dioxin Receptor with Mr 90,000 Heat Shock protein. Biochem. Biophys. Res. Commun. 1988, 155, 801–807. [Google Scholar] [CrossRef]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J. Biol. Chem. 1999, 274, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Pappas, B.; Yang, Y.; Wang, Y.; Kim, K.; Chung, H.J.; Cheung, M.; Ngo, K.; Shinn, A.; Chan, W.K. p23 protects the human aryl hydrocarbon receptor from degradation via a heat shock protein 90-independent mechanism. Biochem. Pharmacol. 2018, 152, 34–44. [Google Scholar] [CrossRef]
- Pongratz, I.; Grant, G.; Mason, F.; Poellinger, L. Dual roles of the 90-kDa heat shock protein hsp90 in modulating fuctional activites of the dioxin receptor. J. Biol. Chem. 1992, 267, 13728–13733. [Google Scholar]
- Reyes, H.; Reisz-Porszasz, S.; Hankinson, O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 1992, 256, 1193–1195. [Google Scholar] [CrossRef]
- Reisz-Porszasz, S.; Probst, M.R.; Fukunaga, B.N.; Hankinson, O. Identification of functional domains of the aryl hydrocarbon receptor nuclear translocator protein (ARNT). Mol. Cell. Biol. 1994, 14, 6075–6086. [Google Scholar] [CrossRef]
- Harper, P.A.; Giannone, J.V.; Okey, A.B.; Denison, M.S. In vitro transformation of the human Ah receptor and its binding to a dioxin response element. Mol. Pharmacol. 1992, 42, 603–612. [Google Scholar]
- Beischlag, T.V.; Wang, S.; Rose, D.W.; Torchia, J.; Reisz-Porszasz, S.; Muhammad, K.; Nelson, W.E.; Probst, M.R.; Rosenfeld, M.G.; Hankinson, O. Recruitment of the NCoA/SRC-1/p160 family of transcriptional coactivators by the aryl hydrocarbon receptor/aryl hydrocarbon receptor nuclear translocator complex. Mol. Cell Biol. 2002, 22, 4319–4333. [Google Scholar] [CrossRef]
- Denison, M.S.; Fisher, J.M.; Whitlock, J.P., Jr. Protein-DNA interactions at recognition sites for the dioxin-Ah receptor complex. J. Biol. Chem 1989, 264, 16478–16482. [Google Scholar]
- Carlson, D.B.; Perdew, G.H. A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J. Biochem. Mol. Toxicol. 2002, 16, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Romieu-Mourez, R.; Sherr, D.H.; Sonenshein, G.E. The RelA NF-kB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000, 19, 5498–5506. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Li, W.; Wu, D.; Miller, J.K.; Sweeney, C.; Lazennec, G.; Fujisawa, Y.; Matsumura, F. Interaction of aryl hydrocarbon receptor and NF-kappaB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch. Biochem. Biophys. 2011, 512, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J. Pharmacol. Exp. Ther. 2013, 345, 419–429. [Google Scholar] [CrossRef]
- Wang, F.; Hoivik, D.; Pollenz, R.; Safe, S. Functional and physical interactions between the estrogen receptor Sp1 and nuclear aryl hydrocarbon receptor complexes. Nucleic Acids Res. 1998, 26, 3044–3052. [Google Scholar] [CrossRef]
- Baker, J.R.; Sakoff, J.A.; McCluskey, A. The aryl hydrocarbon receptor (AhR) as a breast cancer drug target. Med. Res. Rev. 2020, 40, 972–1001. [Google Scholar] [CrossRef]
- Marlowe, J.L.; Fan, Y.; Chang, X.; Peng, L.; Knudsen, E.S.; Xia, Y.; Puga, A. The aryl hydrocarbon receptor binds to E2F1 and inhibits E2F1-induced apoptosis. Mol. Biol. Cell 2008, 19, 3263–3271. [Google Scholar] [CrossRef]
- Puga, A.; Xia, Y.; Elferink, C. Role of the aryl hydrocarbon receptor in cell cycle regulation. Chem. Biol. Interact. 2002, 141, 117–130. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef]
- Shashar, M.; Belghasem, M.E.; Matsuura, S.; Walker, J.; Richards, S.; Alousi, F.; Rijal, K.; Kolachalama, V.B.; Balcells, M.; Odagi, M.; et al. Targeting STUB1-tissue factor axis normalizes hyperthrombotic uremic phenotype without increasing bleeding risk. Sci. Transl. Med. 2017, 9, eaam8475. [Google Scholar] [CrossRef]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Raufman, J.P.; Xie, G. Src-mediated cross-talk between farnesoid X and epidermal growth factor receptors inhibits human intestinal cell proliferation and tumorigenesis. PLoS ONE 2012, 7, e48461. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Peng, Z.; Raufman, J.P. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1006–G1015. [Google Scholar] [CrossRef] [PubMed]
- Endler, A.; Chen, L.; Shibasaki, F. Coactivator recruitment of AhR/ARNT1. Int. J. Mol. Sci. 2014, 15, 11100–11110. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef]
- Dou, H.; Duan, Y.; Zhang, X.; Yu, Q.; Di, Q.; Song, Y.; Li, P.; Gong, Y. Aryl hydrocarbon receptor (AhR) regulates adipocyte differentiation by assembling CRL4B ubiquitin ligase to target PPARgamma for proteasomal degradation. J. Biol. Chem. 2019, 294, 18504–18515. [Google Scholar] [CrossRef]
- Hestermann, E.V.; Brown, M. Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Mol. Cell Biol. 2003, 23, 7920–7925. [Google Scholar] [CrossRef]
- Quintana, F.J.; Sherr, D.H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013, 65, 1148–1161. [Google Scholar] [CrossRef]
- Matikainen, T.; Perez, G.I.; Jurisicova, A.; Pru, J.K.; Schlezinger, J.J.; Ryu, H.Y.; Laine, J.; Sakai, T.; Korsmeyer, S.J.; Casper, R.F.; et al. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat. Genet. 2001, 28, 355–360. [Google Scholar] [CrossRef]
- Matikainen, T.M.; Moriyama, T.; Morita, Y.; Perez, G.I.; Korsmeyer, S.J.; Sherr, D.H.; Tilly, J.L. Ligand activation of the aromatic hydrocarbon receptor transcription factor drives Bax-dependent apoptosis in developing fetal ovarian germ cells. Endocrinology 2002, 143, 615–620. [Google Scholar] [CrossRef]
- Chevallier, A.; Mialot, A.; Petit, J.M.; Fernandez-Salguero, P.; Barouki, R.; Coumoul, X.; Beraneck, M. Oculomotor deficits in aryl hydrocarbon receptor null mouse. PLoS ONE 2013, 8, e53520. [Google Scholar] [CrossRef] [PubMed]
- Lahvis, G.P.; Lindell, S.L.; Thomas, R.S.; McCuskey, R.S.; Murphy, C.; Glover, E.; Bentz, M.; Southard, J.; Bradfield, C.A. Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 10442–10447. [Google Scholar] [CrossRef] [PubMed]
- Lahvis, G.P.; Pyzalski, R.W.; Glover, E.; Pitot, H.C.; McElwee, M.K.; Bradfield, C.A. The aryl hydrocarbon receptor is required for developmental closure of the ductus venosus in the neonatal mouse. Mol. Pharmacol. 2005, 67, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Salguero, P.M.; Ward, J.M.; Sundberg, J.P.; Gonzalez, F.J. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 1997, 34, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Kurita, H.; Carreira, V.S.; Fan, Y.; Jiang, M.; Naticchioni, M.; Koch, S.; Rubinstein, J.; Puga, A. Ah receptor expression in cardiomyocytes protects adult female mice from heart dysfunction induced by TCDD exposure. Toxicology 2016, 355–356, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, J.; Ko, C.I.; Fan, Y.; Carreira, V.; Chen, Y.; Xia, Y.; Medvedovic, M.; Puga, A. Disruption of aryl hydrocarbon receptor homeostatic levels during embryonic stem cell differentiation alters expression of homeobox transcription factors that control cardiomyogenesis. Environ. Health Perspect. 2013, 121, 1334–1343. [Google Scholar] [CrossRef]
- Leung, A.; Zulick, E.; Skvir, N.; Vanuytsel, K.; Morrison, T.A.; Naing, Z.H.; Wang, Z.; Dai, Y.; Chui, D.H.K.; Steinberg, M.H.; et al. Notch and Aryl Hydrocarbon Receptor Signaling Impact Definitive Hematopoiesis from Human Pluripotent Stem Cells. Stem Cells 2018. [Google Scholar] [CrossRef]
- Smith, B.W.; Rozelle, S.S.; Leung, A.; Ubellacker, J.; Parks, A.; Nah, S.K.; French, D.; Gadue, P.; Monti, S.; Chui, D.H.; et al. The aryl hydrocarbon receptor directs hematopoietic progenitor cell expansion and differentiation. Blood 2013, 122, 376–385. [Google Scholar] [CrossRef]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Jux, B.; Kadow, S.; Luecke, S.; Rannug, A.; Krutmann, J.; Esser, C. The aryl hydrocarbon receptor mediates UVB radiation-induced skin tanning. J. Investig. Dermatol. 2011, 131, 203–210. [Google Scholar] [CrossRef]
- Trikha, P.; Lee, D.A. The role of AhR in transcriptional regulation of immune cell development and function. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188335. [Google Scholar] [CrossRef] [PubMed]
- Sherr, D.H.; Monti, S. The role of the aryl hydrocarbon receptor in normal and malignant B cell development. Semin. Immunopathol. 2013, 35, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Hayashibara, T.; Yamada, Y.; Mori, N.; Harasawa, H.; Sugahara, K.; Miyanishi, T.; Kamihira, S.; Tomonaga, M. Possible involvement of aryl hydrocarbon receptor (AhR) in adult T-cell leukemia (ATL) leukemogenesis: Constitutive activation of AhR in ATL. Biochem. Biophys. Res. Commun. 2003, 300, 128–134. [Google Scholar] [CrossRef]
- Yang, X.; Solomon, S.; Fraser, L.R.; Trombino, A.F.; Liu, D.; Sonenshein, G.E.; Hestermann, E.V.; Sherr, D.H. Constitutive regulation of CYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J. Cell. Biochem. 2008, 104, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Trombino, A.F.; Near, R.I.; Matulka, R.A.; Yang, S.; Hafer, L.J.; Toselli, P.A.; Kim, D.W.; Rogers, A.E.; Sonenshein, G.E.; Sherr, D.H. Expression of the aryl hydrocarbon receptor/transcription factor (AhR) and AhR-regulated CYP1 gene transcripts in a rat model of mammary tumorigenesis. Breast Cancer Res. Treat. 2000, 63, 117–131. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Smith, K.; John, K.; Krishnegowda, G.; Amin, S.G.; Perdew, G.H. Ah receptor antagonism represses head and neck tumor cell aggressive phenotype. Mol. Cancer Res. 2012, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Schroeder, J.C.; Perdew, G.H. Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol. Carcinog. 2011, 50, 173–183. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2020, 22, 729–740. [Google Scholar] [CrossRef]
- Ishida, M.; Mikami, S.; Kikuchi, E.; Kosaka, T.; Miyajima, A.; Nakagawa, K.; Mukai, M.; Okada, Y.; Oya, M. Activation of the aryl hydrocarbon receptor pathway enhances cancer cell invasion by upregulating the MMP expression and is associated with poor prognosis in upper urinary tract urothelial cancer. Carcinogenesis 2010, 31, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Chang, H.; Tsai, W.T.; Wu, M.H.; Liao, Y.S.; Chen, J.T.; Su, J.M. Overexpression of aryl hydrocarbon receptor in human lung carcinomas. Toxicol. Pathol. 2003, 31, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Chang, H.; Ho, W.L.; Wu, M.H.; Su, J.M. Association of aryl hydrocarbon receptor and cytochrome P4501B1 expressions in human non-small cell lung cancers. Lung Cancer 2003, 42, 255–261. [Google Scholar] [CrossRef]
- Koliopanos, A.; Kleeff, J.; Xiao, Y.; Safe, S.; Zimmermann, A.; Buchler, M.W.; Friess, H. Increased arylhydrocarbon receptor expression offers a potential therapeutic target for pancreatic cancer. Oncogene 2002, 21, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rocken, C.; Klein-Hitpass, L.; Gotze, T.; Leodolter, A.; Malfertheiner, P.; Ebert, M.P. Microarray analysis of gene expression in metastatic gastric cancer cells after incubation with the methylation inhibitor 5-aza-2′-deoxycytidine. Clin. Exp. Metastasis 2004, 21, 389–397. [Google Scholar] [CrossRef]
- Peng, T.L.; Chen, J.; Mao, W.; Song, X.; Chen, M.H. Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol. 2009, 10, 27. [Google Scholar] [CrossRef]
- Bogoevska, V.; Wolters-Eisfeld, G.; Hofmann, B.T.; El Gammal, A.T.; Mercanoglu, B.; Gebauer, F.; Vashist, Y.K.; Bogoevski, D.; Perez, D.; Gagliani, N.; et al. HRG/HER2/HER3 signaling promotes AhR-mediated Memo-1 expression and migration in colorectal cancer. Oncogene 2017, 36, 2394–2404. [Google Scholar] [CrossRef]
- Wang, Z.; Monti, S.; Sherr, D.H. The diverse and important contributions of the AHR to cancer and cancer immunity. Curr. Opin. Toxicol. 2017, 2, 102–107. [Google Scholar] [CrossRef]
- Su, J.M.; Lin, P.; Chang, H. Prognostic value of nuclear translocation of aryl hydrocarbon receptor for non-small cell lung cancer. Anticancer Res. 2013, 33, 3953–3961. [Google Scholar]
- Vacher, S.; Castagnet, P.; Chemlali, W.; Lallemand, F.; Meseure, D.; Pocard, M.; Bieche, I.; Perrot-Applanat, M. High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PLoS ONE 2018, 13, e0190619. [Google Scholar] [CrossRef]
- Andersson, P.; McGuire, J.; Rubio, C.; Gradin, K.; Whitelaw, M.L.; Pettersson, S.; Hanberg, A.; Poellinger, L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 9990–9995. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.V.; Andersson, P.; Gradin, K.; Stein, P.; Dieckmann, A.; Pettersson, S.; Hanberg, A.; Poellinger, L. The dioxin/aryl hydrocarbon receptor mediates downregulation of osteopontin gene expression in a mouse model of gastric tumourigenesis. Oncogene 2005, 24, 3216–3222. [Google Scholar] [CrossRef] [PubMed]
- Moennikes, O.; Loeppen, S.; Buchmann, A.; Andersson, P.; Ittrich, C.; Poellinger, L.; Schwarz, M. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004, 64, 4707–4710. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Cai, X.; Guo, Y.; Xu, M.; Tian, J.; Locker, J.; Xie, W. Constitutive Activation of the Human Aryl Hydrocarbon Receptor in Mice Promotes Hepatocarcinogenesis Independent of Its Coactivator Gadd45b. Toxicol. Sci. 2019, 167, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Richmond, O.; Ghotbaddini, M.; Allen, C.; Walker, A.; Zahir, S.; Powell, J.B. The aryl hydrocarbon receptor is constitutively active in advanced prostate cancer cells. PLoS ONE 2014, 9, e95058. [Google Scholar] [CrossRef] [PubMed]
- Stanford, E.A.; Ramirez-Cardenas, A.; Wang, Z.; Novikov, O.; Alamoud, K.; Koutrakis, P.; Mizgerd, J.P.; Genco, C.A.; Kukuruzinska, M.; Monti, S.; et al. Role for the aryl hydrocarbon receptor and diverse ligands in oral squamous cell carcinoma migration and tumorigenesis. Mol. Cancer Res. 2016, 14, 696–706. [Google Scholar] [CrossRef]
- Narasimhan, S.; Stanford Zulick, E.; Novikov, O.; Parks, A.J.; Schlezinger, J.J.; Wang, Z.; Laroche, F.; Feng, H.; Mulas, F.; Monti, S.; et al. Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor. Int. J. Mol. Sci. 2018, 19, 1388. [Google Scholar] [CrossRef]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2- human breast cancer cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef]
- Parks, A.J.; Pollastri, M.P.; Hahn, M.E.; Stanford, E.A.; Novikov, O.; Franks, D.G.; Haigh, S.E.; Narasimhan, S.; Ashton, T.D.; Hopper, T.G.; et al. In silico identification of an aryl hydrocarbon receptor antagonist with biological activity in vitro and in vivo. Mol. Pharmacol. 2014, 86, 593–608. [Google Scholar] [CrossRef]
- Corre, S.; Tardif, N.; Mouchet, N.; Leclair, H.M.; Boussemart, L.; Gautron, A.; Bachelot, L.; Perrot, A.; Soshilov, A.; Rogiers, A.; et al. Sustained activation of the Aryl hydrocarbon Receptor transcription factor promotes resistance to BRAF-inhibitors in melanoma. Nat. Commun. 2018, 9, 4775. [Google Scholar] [CrossRef]
- Leclair, H.M.; Tardif, N.; Paris, A.; Galibert, M.D.; Corre, S. Role of Flavonoids in the Prevention of AhR-Dependent Resistance During Treatment with BRAF Inhibitors. Int. J. Mol. Sci 2020, 21, 5025. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. HPA029722. Available online: https://www.proteinatlas.org/ENSG00000106546-AHR/pathology/urothelial+cancer#img (accessed on 25 October 2020).
- The Human Protein Atlas. HPA029722. Available online: https://www.proteinatlas.org/ENSG00000106546-AHR/pathology/cervical+cancer#img (accessed on 25 October 2020).
- The Human Protein Atlas. HPA029723. Available online: https://www.proteinatlas.org/ENSG00000106546-AHR/pathology/glioma#img (accessed on 25 October 2020).
- The Human Protein Atlas. HPA029723. Available online: https://www.proteinatlas.org/ENSG00000106546-AHR/pathology/pancreatic+cancer#img (accessed on 25 October 2020).
- The Human Protein Atlas. HPA029723. Available online: https://www.proteinatlas.org/ENSG00000106546-AHR/pathology/head+and+neck+cancer#img (accessed on 25 October 2020).
- The Human Protein Atlas. HPA029722. Available online: https://www.proteinatlas.org/ENSG00000106546-AHR/pathology/lung+cancer#img (accessed on 25 October 2020).
- The Human Protein Atlas. HPA029723. Available online: https://www.proteinatlas.org/ENSG00000106546-AHR/pathology/melanoma#img (accessed on 25 October 2020).
- Mohamed, H.T.; Gadalla, R.; El-Husseiny, N.; Hassan, H.; Wang, Z.; Ibrahim, S.A.; El-Shinawi, M.; Sherr, D.H.; Mohamed, M.M. Inflammatory breast cancer: Activation of the aryl hydrocarbon receptor and its target CYP1B1 correlates closely with Wnt5a/b-beta-catenin signalling, the stem cell phenotype and disease progression. J. Adv. Res. 2019, 16, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, U.; Zhang, X.; Kuhn, C.; Jalaguier, S.; Colinge, J.; Pfender, K.; Mayr, D.; Ditsch, N.; Harbeck, N.; Mahner, S.; et al. The Prognostic Impact of the Aryl Hydrocarbon Receptor (AhR) in Primary Breast Cancer Depends on the Lymph Node Status. Int. J. Mol. Sci. 2019, 20, 1016. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.A.; Murray, G.I.; Ah-See, A.K.; Greenlee, W.F.; Marcus, C.B.; Burke, M.D.; Melvin, W.T. Differential expression of CYP1A1 and CYP1B1 in human breast cancer. Biochem. Soc. Trans. 1996, 24, 327S. [Google Scholar] [CrossRef] [PubMed]
- Murray, G.I.; Taylor, M.C.; McFadyen, M.C.; McKay, J.A.; Greenlee, W.F.; Burke, M.D.; Melvin, W.T. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997, 57, 3026–3031. [Google Scholar] [PubMed]
- Ma, J.X.; Zhang, K.L.; Liu, X.; Ma, Y.L.; Pei, L.N.; Zhu, Y.F.; Zhou, L.; Chen, X.Y.; Kong, Q.Y.; Li, H.; et al. Concurrent expression of aryl hydrocarbon receptor and CYP1A1 but not CYP1A1 MspI polymorphism is correlated with gastric cancers raised in Dalian, China. Cancer Lett. 2006, 240, 253–260. [Google Scholar] [CrossRef]
- Spink, D.C.; Spink, B.C.; Cao, J.Q.; DePasquale, J.A.; Pentecost, B.T.; Fasco, M.J.; Li, Y.; Sutter, T.R. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis 1998, 19, 291–298. [Google Scholar] [CrossRef]
- McKay, J.A.; Melvin, W.T.; Ah-See, A.K.; Ewen, S.W.; Greenlee, W.F.; Marcus, C.B.; Burke, M.D.; Murray, G.I. Expression of cytochrome P450 CYP1B1 in breast cancer. FEBS Lett. 1995, 374, 270–272. [Google Scholar] [CrossRef]
- Chang, H.; Su, J.M.; Huang, C.C.; Liu, L.C.; Tsai, C.H.; Chou, M.C.; Lin, P. Using a combination of cytochrome P450 1B1 and beta-catenin for early diagnosis and prevention of colorectal cancer. Cancer Detect. Prev. 2005, 29, 562–569. [Google Scholar] [CrossRef]
- Nukaya, M.; Lin, B.C.; Glover, E.; Moran, S.M.; Kennedy, G.D.; Bradfield, C.A. The aryl hydrocarbon receptor-interacting protein (AIP) is required for dioxin-induced hepatotoxicity but not for the induction of the Cyp1a1 and Cyp1a2 genes. J. Biol. Chem. 2010, 285, 35599–35605. [Google Scholar] [CrossRef]
- Flies, A.; Ahmadi, T.; Parks, A.J.; Prokaeva, T.; Weng, L.; Rolfe, S.S.; Seldin, D.C.; Sherr, D.H. Immunoglobulin light chain, Blimp-1 and cytochrome P4501B1 peptides as potential vaccines for AL amyloidosis. Immunol. Cell Biol. 2012, 90, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Gribben, J.G.; Ryan, D.P.; Boyajian, R.; Urban, R.G.; Hedley, M.L.; Beach, K.; Nealon, P.; Matulonis, U.; Campos, S.; Gilligan, T.D.; et al. Unexpected association between induction of immunity to the universal tumor antigen CYP1B1 and response to next therapy. Clin. Cancer Res. 2005, 11, 4430–4436. [Google Scholar] [CrossRef] [PubMed]
- Luby, T.M. Targeting cytochrome P450 CYP1B1 with a therapeutic cancer vaccine. Expert Rev. Vaccines 2008, 7, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, M.C.; Murray, G.I. Cytochrome P450 1B1: A novel anticancer therapeutic target. Future Oncol. 2005, 1, 259–263. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Abnet, C.C.; Fagundes, R.B.; Strickland, P.T.; Kamangar, F.; Roth, M.J.; Taylor, P.R.; Dawsey, S.M. The influence of genetic polymorphisms in Ahr, CYP1A1, CYP1A2, CYP1B1, GST M1, GST T1 and UGT1A1 on urine 1-hydroxypyrene glucuronide concentrations in healthy subjects from Rio Grande do Sul, Brazil. Carcinogenesis 2007, 28, 112–117. [Google Scholar] [CrossRef]
- Anttila, S.; Lei, X.D.; Elovaara, E.; Karjalainen, A.; Sun, W.; Vainio, H.; Hankinson, O. An uncommon phenotype of poor inducibility of CYP1A1 in human lung is not ascribable to polymorphisms in the AHR, ARNT, or CYP1A1 genes. Pharmacogenetics 2000, 10, 741–751. [Google Scholar] [CrossRef]
- Cauchi, S.; Stucker, I.; Solas, C.; Laurent-Puig, P.; Cenee, S.; Hemon, D.; Jacquet, M.; Kremers, P.; Beaune, P.; Massaad-Massade, L. Polymorphisms of human aryl hydrocarbon receptor (AhR) gene in a French population: Relationship with CYP1A1 inducibility and lung cancer. Carcinogenesis 2001, 22, 1819–1824. [Google Scholar] [CrossRef][Green Version]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci 2010, 115, 89–97. [Google Scholar] [CrossRef]
- Lowe, M.M.; Mold, J.E.; Kanwar, B.; Huang, Y.; Louie, A.; Pollastri, M.P.; Wang, C.; Patel, G.; Franks, D.G.; Schlezinger, J.; et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS ONE 2014, 9, e87877. [Google Scholar] [CrossRef]
- Seok, S.H.; Ma, Z.X.; Feltenberger, J.B.; Chen, H.; Chen, H.; Scarlett, C.; Lin, Z.; Satyshur, K.A.; Cortopassi, M.; Jefcoate, C.R.; et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J. Biol. Chem. 2018, 293, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Van Baren, N.; Van den Eynde, B.J. Tryptophan-degrading enzymes in tumoral immune resistance. Front. Immunol. 2015, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Simones, T.; Shepherd, D.M. Consequences of AhR activation in steady-state dendritic cells. Toxicol. Sci. 2011, 119, 293–307. [Google Scholar] [CrossRef]
- Li, Q.; Harden, J.L.; Anderson, C.D.; Egilmez, N.K. Tolerogenic Phenotype of IFN-gamma-Induced IDO+ Dendritic Cells Is Maintained via an Autocrine IDO-Kynurenine/AhR-IDO Loop. J. Immunol. 2016, 197, 962–970. [Google Scholar] [CrossRef]
- Hwang, S.J.; Hwang, Y.J.; Yun, M.O.; Kim, J.H.; Oh, G.S.; Park, J.H. Indoxyl 3-sulfate stimulates Th17 differentiation enhancing phosphorylation of c-Src and STAT3 to worsen experimental autoimmune encephalomyelitis. Toxicol. Lett. 2013, 220, 109–117. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef]
- Benson, J.M.; Shepherd, D.M. Dietary ligands of the aryl hydrocarbon receptor induce anti-inflammatory and immunoregulatory effects on murine dendritic cells. Toxicol. Sci. 2011, 124, 327–338. [Google Scholar] [CrossRef]
- Wincent, E.; Amini, N.; Luecke, S.; Glatt, H.; Bergman, J.; Crescenzi, C.; Rannug, A.; Rannug, U. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem. 2009, 284, 2690–2696. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Mori, Y.; Matsui, S.; Takigami, H.; Fujino, J.; Kitagawa, H.; Miller, C.A., 3rd; Kato, T.; Saeki, K.; Matsuda, T. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J. Biol. Chem. 2001, 276, 31475–31478. [Google Scholar] [CrossRef] [PubMed]
- Schlezinger, J.J.; Bernard, P.L.; Haas, A.; Grandjean, P.; Weihe, P.; Sherr, D.H. Direct assessment of cumulative aryl hydrocarbon receptor agonist activity in sera from experimentally exposed mice and environmentally exposed humans. Environ. Health Perspect. 2010, 118, 693–698. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Angeli, D.; Ferrell, J.E., Jr.; Sontag, E.D. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback systems. Proc. Natl. Acad. Sci. USA 2004, 101, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Avendano, M.S.; Leidy, C.; Pedraza, J.M. Tuning the range and stability of multiple phenotypic states with coupled positive-negative feedback loops. Nat. Commun. 2013, 4, 2605. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kato, A.; Mitrophanov, A.Y.; Groisman, E.A. A connector of two-component regulatory systems promotes signal amplification and persistence of expression. Proc. Natl. Acad. Sci. USA 2007, 104, 12063–12068. [Google Scholar] [CrossRef] [PubMed]
- Mitrophanov, A.Y.; Groisman, E.A. Positive feedback in cellular control systems. Bioessays 2008, 30, 542–555. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y.; Hadley, T.J.; Groisman, E.A. Positive autoregulation shapes response timing and intensity in two-component signal transduction systems. J. Mol. Biol. 2010, 401, 671–680. [Google Scholar] [CrossRef]
- Baba, T.; Mimura, J.; Gradin, K.; Kuroiwa, A.; Watanabe, T.; Matsuda, Y.; Inazawa, J.; Sogawa, K.; Fujii-Kuriyama, Y. Structure and expression of the Ah receptor repressor gene. J. Biol. Chem. 2001, 276, 33101–33110. [Google Scholar] [CrossRef]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef]
- Evans, B.R.; Karchner, S.I.; Allan, L.L.; Pollenz, R.S.; Tanguay, R.L.; Jenny, M.J.; Sherr, D.H.; Hahn, M.E. Repression of aryl hydrocarbon receptor (AHR) signaling by AHR repressor: Role of DNA binding and competition for AHR nuclear translocator. Mol. Pharmacol. 2008, 73, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.E.; Allan, L.L.; Sherr, D.H. Regulation of constitutive and inducible AHR signaling: Complex interactions involving the AHR repressor. Biochem. Pharmacol. 2009, 77, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Cuesta, N.; Murty, V.; Woodson, K.; Adams, L.; Gonzalez, N.; Martinez, A.; Narayan, G.; Kirsch, I.; Franklin, W.; et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J. Clin. Investig. 2008, 118, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Levine-Fridman, A.; Chen, L.; Elferink, C.J. Cytochrome P4501A1 promotes G1 phase cell cycle progression by controlling aryl hydrocarbon receptor activity. Mol. Pharmacol. 2004, 65, 461–469. [Google Scholar] [CrossRef]
- Stanford, E.A.; Wang, Z.; Novikov, O.; Mulas, F.; Landesman-Bollag, E.; Monti, S.; Smith, B.W.; Seldin, D.C.; Murphy, G.J.; Sherr, D.H. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 2016, 14, 20. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Mulero-Navarro, S.; Pozo-Guisado, E.; Perez-Mancera, P.A.; Alvarez-Barrientos, A.; Catalina-Fernandez, I.; Hernandez-Nieto, E.; Saenz-Santamaria, J.; Martinez, N.; Rojas, J.M.; Sanchez-Garcia, I.; et al. Immortalized mouse mammary fibroblasts lacking dioxin receptor have impaired tumorigenicity in a subcutaneous mouse xenograph model. J. Biol. Chem. 2005, 280, 28731–28741. [Google Scholar] [CrossRef]
- Diry, M.; Tomkiewicz, C.; Koehle, C.; Coumoul, X.; Bock, K.W.; Barouki, R.; Transy, C. Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene 2006, 25, 5570–5574. [Google Scholar] [CrossRef]
- Goode, G.D.; Ballard, B.R.; Manning, H.C.; Freeman, M.L.; Kang, Y.; Eltom, S.E. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int. J. Cancer 2013, 133, 2769–2780. [Google Scholar] [CrossRef]
- Haque, M.; Francis, J.; Sehgal, I. Aryl hydrocarbon exposure induces expression of MMP-9 in human prostate cancer cell lines. Cancer Lett. 2005, 225, 159–166. [Google Scholar] [CrossRef]
- Ide, H.; Lu, Y.; Yu, J.; Noguchi, T.; Kanayama, M.; Muto, S.; Yamaguchi, R.; Kawato, S.; Horie, S. Aryl hydrocarbon receptor signaling involved in the invasiveness of LNCaP cells. Hum. Cell 2017, 30, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.; Godwin, T.D.; Yap, A.S.; Guilford, P.J. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Salomon, A.; Thiery, J.P. Host microenvironment in breast cancer development: Epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res. 2003, 5, 101–106. [Google Scholar] [CrossRef]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.T.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell-Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef]
- Brooks, J.; Eltom, S.E. Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr. Cancer Drug Targets 2011, 11, 654–669. [Google Scholar] [CrossRef]
- Zhu, P.; Yu, H.; Zhou, K.; Bai, Y.; Qi, R.; Zhang, S. 3,3′-Diindolylmethane modulates aryl hydrocarbon receptor of esophageal squamous cell carcinoma to reverse epithelial-mesenchymal transition through repressing RhoA/ROCK1-mediated COX2/PGE2 pathway. J. Exp. Clin. Cancer Res. 2020, 39, 113. [Google Scholar] [CrossRef]
- Zhu, P.; Zhou, K.; Lu, S.; Bai, Y.; Qi, R.; Zhang, S. Modulation of aryl hydrocarbon receptor inhibits esophageal squamous cell carcinoma progression by repressing COX2/PGE2/STAT3 axis. J. Cell Commun. Signal. 2020, 14, 175–192. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Xiang, L.; Lee, S.J.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, E384–E393. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, L.; Xiao, J.; Qi, X.K.; Zhang, J.; Li, X.; Wang, Z.; Lian, Y.F.; Xiang, T.; Zhang, Y.; et al. SHROOM2 inhibits tumor metastasis through RhoA-ROCK pathway-dependent and -independent mechanisms in nasopharyngeal carcinoma. Cell Death Dis. 2019, 10, 58. [Google Scholar] [CrossRef]
- Liu, J.; Seibold, S.A.; Rieke, C.J.; Song, I.; Cukier, R.I.; Smith, W.L. Prostaglandin endoperoxide H synthases: Peroxidase hydroperoxide specificity and cyclooxygenase activation. J. Biol. Chem. 2007, 282, 18233–18244. [Google Scholar] [CrossRef] [PubMed]
- Che, D.; Zhang, S.; Jing, Z.; Shang, L.; Jin, S.; Liu, F.; Shen, J.; Li, Y.; Hu, J.; Meng, Q.; et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/beta-catenin signalling pathway. Mol. Immunol. 2017, 90, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, X.; Jiang, M.; Bi, Y.; Xu, J.; Han, M. Lipopolysaccharide induces inflammation and facilitates lung metastasis in a breast cancer model via the prostaglandin E2-EP2 pathway. Mol. Med. Rep. 2015, 11, 4454–4462. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Yang, Y.; Che, Q.; Jiang, F.; Wang, H.; Chen, Z.; Zhu, M.; Tong, H.; Zhang, H.; Yan, X.; et al. Prostaglandin E2 (PGE2) promotes proliferation and invasion by enhancing SUMO-1 activity via EP4 receptor in endometrial cancer. Tumour Biol. 2016, 37, 12203–12211. [Google Scholar] [CrossRef]
- Reyes-Reyes, E.M.; Ramos, I.N.; Tavera-Garcia, M.A.; Ramos, K.S. The aryl hydrocarbon receptor agonist benzo(a)pyrene reactivates LINE-1 in HepG2 cells through canonical TGF-beta1 signaling: Implications in hepatocellular carcinogenesis. Am. J. Cancer Res. 2016, 6, 1066–1077. [Google Scholar]
- Warusavitarne, J.; McDougall, F.; de Silva, K.; Barnetson, R.; Messina, M.; Robinson, B.G.; Schnitzler, M. Restoring TGFbeta function in microsatellite unstable (MSI-H) colorectal cancer reduces tumourigenicity but increases metastasis formation. Int. J. Colorectal Dis. 2009, 24, 139–144. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef]
- Kwon, Y.J.; Baek, H.S.; Ye, D.J.; Shin, S.; Kim, D.; Chun, Y.J. CYP1B1 Enhances Cell Proliferation and Metastasis through Induction of EMT and Activation of Wnt/beta-Catenin Signaling via Sp1 Upregulation. PLoS ONE 2016, 11, e0151598. [Google Scholar] [CrossRef]
- Mengoni, M.; Braun, A.D.; Gaffal, E.; Tuting, T. The aryl hydrocarbon receptor promotes inflammation-induced dedifferentiation and systemic metastatic spread of melanoma cells. Int. J. Cancer 2020, 147, 2902–2913. [Google Scholar] [CrossRef]
- Murante, F.G.; Gasiewicz, T.A. Hemopoietic progenitor cells are sensitive targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J mice. Toxicol. Sci. 2000, 54, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.P.; Casado, F.L.; Opanashuk, L.A.; Gasiewicz, T.A. The aryl hydrocarbon receptor has a normal function in the regulation of hematopoietic and other stem/progenitor cell populations. Biochem. Pharmacol. 2009, 77, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.P.; Garrett, R.W.; Casado, F.L.; Gasiewicz, T.A. Aryl hydrocarbon receptor-null allele mice have hematopoietic stem/progenitor cells with abnormal characteristics and functions. Stem Cells Dev. 2011, 20, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, T.S.; Silverstone, A.E.; Baggs, R.B.; Quimby, F.W.; Staples, J.E.; Gasiewicz, T.A. A chimeric aryl hydrocarbon receptor knockout mouse model indicates that aryl hydrocarbon receptor activation in hematopoietic cells contributes to the hepatic lesions induced by 2,3,7, 8- tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 1999, 158, 33–40. [Google Scholar] [CrossRef]
- Singh, K.P.; Bennett, J.A.; Casado, F.L.; Walrath, J.L.; Welle, S.L.; Gasiewicz, T.A. Loss of aryl hydrocarbon receptor promotes gene changes associated with premature hematopoietic stem cell exhaustion and development of a myeloproliferative disorder in aging mice. Stem Cells Dev. 2014, 23, 95–106. [Google Scholar] [CrossRef]
- Ko, C.I.; Wang, Q.; Fan, Y.; Xia, Y.; Puga, A. Pluripotency factors and Polycomb Group proteins repress aryl hydrocarbon receptor expression in murine embryonic stem cells. Stem Cell Res. 2014, 12, 296–308. [Google Scholar] [CrossRef]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010, 329, 1345–1348. [Google Scholar] [CrossRef]
- Wagner, J.E., Jr.; Brunstein, C.G.; Boitano, A.E.; DeFor, T.E.; McKenna, D.; Sumstad, D.; Blazar, B.R.; Tolar, J.; Le, C.; Jones, J.; et al. Phase I/II Trial of StemRegenin-1 Expanded Umbilical Cord Blood Hematopoietic Stem Cells Supports Testing as a Stand-Alone Graft. Cell Stem Cell 2016, 18, 144–155. [Google Scholar] [CrossRef]
- Tan, K.P.; Wang, B.; Yang, M.; Boutros, P.C.; Macaulay, J.; Xu, H.; Chuang, A.I.; Kosuge, K.; Yamamoto, M.; Takahashi, S.; et al. Aryl hydrocarbon receptor is a transcriptional activator of the human breast cancer resistance protein (BCRP/ABCG2). Mol. Pharmacol. 2010, 78, 175–185. [Google Scholar] [CrossRef]
- Wu, C.; Yu, S.; Tan, Q.; Guo, P.; Liu, H. Role of AhR in regulating cancer stem cell-like characteristics in choriocarcinoma. Cell Cycle 2018, 17, 2309–2320. [Google Scholar] [CrossRef]
- Yan, B.; Liu, S.; Shi, Y.; Liu, N.; Chen, L.; Wang, X.; Xiao, D.; Liu, X.; Mao, C.; Jiang, Y.; et al. Activation of AhR with nuclear IKKalpha regulates cancer stem-like properties in the occurrence of radioresistance. Cell Death Dis. 2018, 9, 490. [Google Scholar] [CrossRef] [PubMed]
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and beta-Catenin and Akt activation. Mol. Cancer 2017, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: Potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2010, 16, 45–55. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Oberg, M.; Bergander, L.; Hakansson, H.; Rannug, U.; Rannug, A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol. Sci. 2005, 85, 935–943. [Google Scholar] [CrossRef]
- Wincent, E.; Bengtsson, J.; Mohammadi Bardbori, A.; Alsberg, T.; Luecke, S.; Rannug, U.; Rannug, A. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 4479–4484. [Google Scholar] [CrossRef]
- Sasaki-Kudoh, E.; Kudo, I.; Kakizaki, Y.; Hosaka, M.; Ikeda, S.; Uemura, S.; Grave, E.; Togashi, S.; Sugawara, T.; Shimizu, H.; et al. Cisplatin Inhibits AhR Activation. Am. J. Mol. Biol. 2018, 8, 69–82. [Google Scholar] [CrossRef]
- Gao, W.Q.; Ma, J.; Sun, L.L.; Li, Q.; Zhu, R.Y.; Jin, J. Paclitaxel-mediated human aryl hydrocarbon receptor mRNA translation by an internal ribosomal entry site-dependent mechanism. Oncol. Rep. 2017, 38, 3211–3219. [Google Scholar] [CrossRef][Green Version]
- Neuzil, J.; Stantic, M.; Zobalova, R.; Chladova, J.; Wang, X.; Prochazka, L.; Dong, L.; Andera, L.; Ralph, S.J. Tumour-initiating cells vs. cancer ’stem’ cells and CD133: What’s in the name? Biochem. Biophys. Res. Commun. 2007, 355, 855–859. [Google Scholar] [CrossRef]
- Ly, M.; Rentas, S.; Vujovic, A.; Wong, N.; Moreira, S.; Xu, J.; Holzapfel, N.; Bhatia, S.; Tran, D.; Minden, M.D.; et al. Diminished AHR Signaling Drives Human Acute Myeloid Leukemia Stem Cell Maintenance. Cancer Res. 2019, 79, 5799–5811. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Kolluri, S.K.; Jin, U.H.; Safe, S. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch. Toxicol. 2017, 91, 2497–2513. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Morales, J.L.; Flaveny, C.A.; Dinatale, B.C.; Chiaro, C.; Gowdahalli, K.; Amin, S.; Perdew, G.H. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol. Pharmacol. 2010, 77, 247–254. [Google Scholar] [CrossRef]
- Zhao, B.; Degroot, D.E.; Hayashi, A.; He, G.; Denison, M.S. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol. Sci. 2010, 117, 393–403. [Google Scholar] [CrossRef]
- Safe, S. Molecular biology of the Ah receptor and its role in carcinogenesis. Toxicol. Lett. 2001, 120, 1–7. [Google Scholar] [CrossRef]
- Safe, S.; Cheng, Y.; Jin, U.H. The Aryl Hydrocarbon Receptor (AhR) as a Drug Target for Cancer Chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef]
- Safe, S.; Qin, C.; McDougal, A. Development of selective aryl hydrocarbon receptor modulators for treatment of breast cancer. Expert Opin. Investig. Drugs 1999, 8, 1385–1396. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Jang, H.S.; Pearce, M.; Kerkvliet, N.I.; Kolluri, S.K. The aryl hydrocarbon receptor is required for induction of p21cip1/waf1 expression and growth inhibition by SU5416 in hepatoma cells. Oncotarget 2017, 8, 25211–25225. [Google Scholar] [CrossRef]
- Jin, U.H.; Lee, S.O.; Pfent, C.; Safe, S. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer 2014, 14, 498. [Google Scholar] [CrossRef]
- Schlezinger, J.J.; Liu, D.; Farago, M.; Seldin, D.C.; Belguise, K.; Sonenshein, G.E.; Sherr, D.H. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol. Chem. 2006, 387, 1175–1187. [Google Scholar] [CrossRef]
- Rhile, M.J.; Nagarkatti, M.; Nagarkatti, P.S. Role of Fas apoptosis and MHC genes in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunotoxicity of T cells. Toxicology 1996, 110, 153–167. [Google Scholar] [CrossRef]
- Park, K.T.; Mitchell, K.A.; Huang, G.; Elferink, C.J. The aryl hydrocarbon receptor predisposes hepatocytes to Fas-mediated apoptosis. Mol. Pharmacol. 2005, 67, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Robles, R.; Morita, Y.; Mann, K.K.; Perez, G.I.; Yang, S.; Matikainen, T.; Sherr, D.H.; Tilly, J.L. The aryl hydrocarbon receptor, a basic helix-loop-helix transcription factor of the PAS gene family, is required for normal ovarian germ cell dynamics in the mouse. Endocrinology 2000, 141, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Li, W.; Sciullo, E.; Newman, J.; Hammock, B.; Reader, J.R.; Tuscano, J.; Matsumura, F. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am. J. Pathol. 2007, 171, 1538–1548. [Google Scholar] [CrossRef]
- Bekki, K.; Vogel, H.; Li, W.; Ito, T.; Sweeney, C.; Haarmann-Stemmann, T.; Matsumura, F.; Vogel, C.F. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic. Biochem. Physiol. 2015, 120, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, Y.; Li, W.; Wu, D.; Wong, P.; Vogel, C.; Dong, B.; Kung, H.J.; Matsumura, F. Ligand-independent activation of the arylhydrocarbon receptor by ETK (Bmx) tyrosine kinase helps MCF10AT1 breast cancer cells to survive in an apoptosis-inducing environment. Biol. Chem. 2011, 392, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Vogeley, C.; Esser, C.; Tuting, T.; Krutmann, J.; Haarmann-Stemmann, T. Role of the Aryl Hydrocarbon Receptor in Environmentally Induced Skin Aging and Skin Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 6005. [Google Scholar] [CrossRef] [PubMed]
- Frauenstein, K.; Sydlik, U.; Tigges, J.; Majora, M.; Wiek, C.; Hanenberg, H.; Abel, J.; Esser, C.; Fritsche, E.; Krutmann, J.; et al. Evidence for a novel anti-apoptotic pathway in human keratinocytes involving the aryl hydrocarbon receptor, E2F1, and checkpoint kinase 1. Cell Death Differ. 2013, 20, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Pollet, M.; Shaik, S.; Mescher, M.; Frauenstein, K.; Tigges, J.; Braun, S.A.; Sondenheimer, K.; Kaveh, M.; Bruhs, A.; Meller, S.; et al. The AHR represses nucleotide excision repair and apoptosis and contributes to UV-induced skin carcinogenesis. Cell Death Differ. 2018, 25, 1823–1836. [Google Scholar] [CrossRef]
- Leja-Szpak, A.; Goralska, M.; Link-Lenczowski, P.; Czech, U.; Nawrot-Porabka, K.; Bonior, J.; Jaworek, J. The Opposite Effect of L-kynurenine and Ahr Inhibitor Ch223191 on Apoptotic Protein Expression in Pancreatic Carcinoma Cells (Panc-1). Anticancer Agents Med. Chem. 2019, 19, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Perdew, G.H. Transgenic Humanized AHR Mouse Reveals Differences between Human and Mouse AHR Ligand Selectivity. Mol. Cell Pharmacol. 2009, 1, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Motohashi, H.; Hosoya, T.; Nakajima, O.; Takahashi, S.; Ohsako, S.; Aoki, Y.; Nishimura, N.; Tohyama, C.; Fujii-Kuriyama, Y.; et al. Distinct response to dioxin in an arylhydrocarbon receptor (AHR)-humanized mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 5652–5657. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.; Reen, R.K.; Kusnadi, A.; Perdew, G.H. The mouse and human Ah receptor differ in recognition of LXXLL motifs. Arch. Biochem. Biophys. 2008, 471, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Murray, I.A.; Perdew, G.H. Differential Gene Regulation by the Human and Mouse Aryl Hydrocarbon Receptor. Toxicol. Sci. 2009, 114, 217–225. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Snyder, M.; Kenison, J.E.; Yang, K.; Lara, B.; Lydell, E.; Bennani, K.; Novikov, O.; Federico, A.; Monti, S.; et al. How the AHR Became Important in Cancer: The Role of Chronically Active AHR in Cancer Aggression. Int. J. Mol. Sci. 2021, 22, 387. https://doi.org/10.3390/ijms22010387
Wang Z, Snyder M, Kenison JE, Yang K, Lara B, Lydell E, Bennani K, Novikov O, Federico A, Monti S, et al. How the AHR Became Important in Cancer: The Role of Chronically Active AHR in Cancer Aggression. International Journal of Molecular Sciences. 2021; 22(1):387. https://doi.org/10.3390/ijms22010387
Chicago/Turabian StyleWang, Zhongyan, Megan Snyder, Jessica E. Kenison, Kangkang Yang, Brian Lara, Emily Lydell, Kawtar Bennani, Olga Novikov, Anthony Federico, Stefano Monti, and et al. 2021. "How the AHR Became Important in Cancer: The Role of Chronically Active AHR in Cancer Aggression" International Journal of Molecular Sciences 22, no. 1: 387. https://doi.org/10.3390/ijms22010387
APA StyleWang, Z., Snyder, M., Kenison, J. E., Yang, K., Lara, B., Lydell, E., Bennani, K., Novikov, O., Federico, A., Monti, S., & Sherr, D. H. (2021). How the AHR Became Important in Cancer: The Role of Chronically Active AHR in Cancer Aggression. International Journal of Molecular Sciences, 22(1), 387. https://doi.org/10.3390/ijms22010387