Functional Comparison between VP64-dCas9-VP64 and dCas9-VP192 CRISPR Activators in Human Embryonic Kidney Cells




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
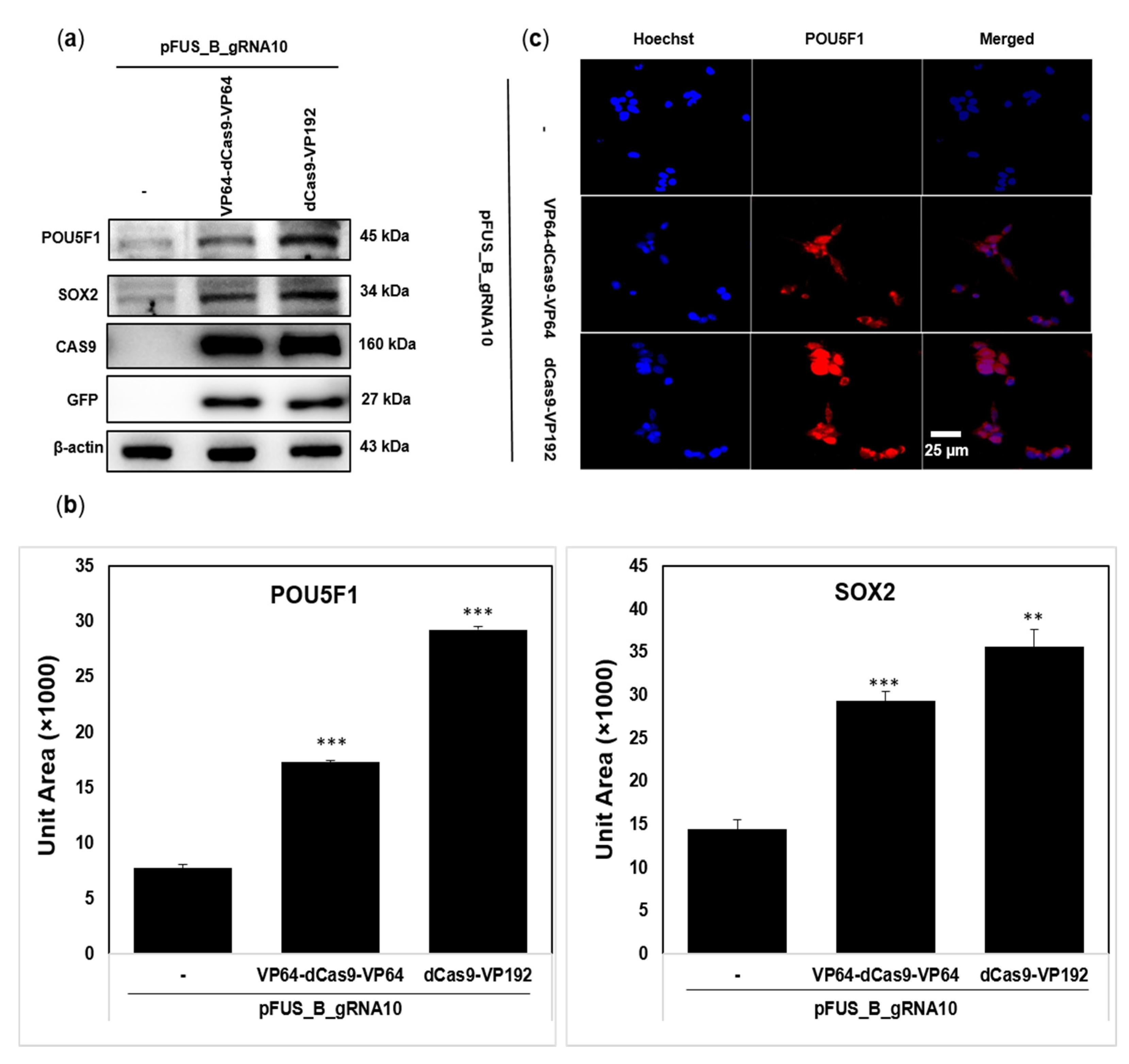
2.1. dCas9-VP192 Activator Is More Efficient Than VP64-dCas9-VP64 to Induce mRNA Expression
2.2. dCas9-VP192 Activator Is Also Efficient at Protein Level Than VP64-dCas9-VP64
3. Discussion
4. Materials and Methods
4.1. Cell Line and Culturing Condition
4.2. Designing gRNAs
4.3. Construction and Confirmation of Array and Multiplexed CRISPR Plasmids
4.4. Transfection
4.5. Real Time RT-PCR (qRT-PCR)
4.6. Western Blotting
4.7. Immunocytochemical Staining
4.8. Off-Target Prediction
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hebbes, T.R.; Thorne, A.W.; Crane-Robinson, C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988, 7, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.-I.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javaid, N.; Choi, S. Acetylation-and methylation-related epigenetic proteins in the context of their targets. Genes 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schotta, G.; Ebert, A.; Krauss, V.; Fischer, A.; Hoffmann, J.; Rea, S.; Jenuwein, T.; Dorn, R.; Reuter, G. Central role of Drosophila SU (VAR) 3–9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 2002, 21, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, S.I.; Moazed, D. Heterochromatin and epigenetic control of gene expression. Science 2003, 301, 798–802. [Google Scholar] [CrossRef] [Green Version]
- Méchali, M.; Yoshida, K.; Coulombe, P.; Pasero, P. Genetic and epigenetic determinants of DNA replication origins, position and activation. Curr. Opin. Genet. Dev. 2013, 23, 124–131. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. An epigenetic code for DNA damage repair pathways? Biochem. Cell Biol. 2005, 83, 270–285. [Google Scholar] [CrossRef]
- Balboa, D.; Weltner, J.; Eurola, S.; Trokovic, R.; Wartiovaara, K.; Otonkoski, T. Conditionally stabilized dCas9 activator for controlling gene expression in human cell reprogramming and differentiation. Stem Cell Rep. 2015, 5, 448–459. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Chen, M.; Liu, Y.; Qi, L.S.; Ding, S. CRISPR-based chromatin remodeling of the endogenous Oct4 or Sox2 locus enables reprogramming to pluripotency. Cell Stem Cell 2018, 22, 252–261.e4. [Google Scholar] [CrossRef] [Green Version]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Blancafort, P.; Segal, D.J.; Barbas, C.F. Designing transcription factor architectures for drug discovery. Mol. Pharmacol. 2004, 66, 1361–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sera, T. Zinc-finger-based artificial transcription factors and their applications. Adv. Drug Deliv. Rev. 2009, 61, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Segal, D.J.; Dreier, B.; Barbas, C.F. Toward controlling gene expression at will: Specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl. Acad. Sci. USA 1998, 95, 14628–14633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerli, R.R.; Dreier, B.; Barbas, C.F. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. USA 2000, 97, 1495–1500. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.M.; Arlotta, P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011, 29, 149. [Google Scholar] [CrossRef] [Green Version]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Maeder, M.L.; Linder, S.J.; Reyon, D.; Angstman, J.F.; Fu, Y.; Sander, J.D.; Joung, J.K. Robust, synergistic regulation of human gene expression using TALE activators. Nat. Methods 2013, 10, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Perez-Pinera, P.; Ousterout, D.G.; Brunger, J.M.; Farin, A.M.; Glass, K.A.; Guilak, F.; Crawford, G.E.; Hartemink, A.J.; Gersbach, C.A. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat. Methods 2013, 10, 239. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Char, S.N.; Neelakandan, A.K.; Nahampun, H.; Frame, B.; Main, M.; Spalding, M.H.; Becraft, P.W.; Meyers, B.C.; Walbot, V.; Wang, K. An Agrobacterium-delivered CRISPR/Cas9 system for high-frequency targeted mutagenesis in maize. Plant Biotechnol. J. 2017, 15, 257–268. [Google Scholar] [CrossRef] [PubMed]
- García-Tuñón, I.; Hernández-Sánchez, M.; Ordoñez, J.L.; Alonso-Pérez, V.; Álamo-Quijada, M.; Benito, R.; Guerrero, C.; Hernández-Rivas, J.M.; Sánchez-Martín, M. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget 2017, 8, 26027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef] [PubMed]
- Maddalo, D.; Manchado, E.; Concepcion, C.P.; Bonetti, C.; Vidigal, J.A.; Han, Y.-C.; Ogrodowski, P.; Crippa, A.; Rekhtman, N.; de Stanchina, E. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 2014, 516, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhang, L.; Stevens, J.; Gibson, G. CRISPR/Cas9 mediated generation of stable chondrocyte cell lines with targeted gene knockouts; analysis of an aggrecan knockout cell line. Bone 2014, 69, 118–125. [Google Scholar] [CrossRef]
- Tang, S.; Chen, T.; Yu, Z.; Zhu, X.; Yang, M.; Xie, B.; Li, N.; Cao, X.; Wang, J. RasGRP3 limits Toll-like receptor-triggered inflammatory response in macrophages by activating Rap1 small GTPase. Nat. Commun. 2014, 5, 4657. [Google Scholar] [CrossRef]
- Jing, W.; Zhang, X.; Sun, W.; Hou, X.; Yao, Z.; Zhu, Y. CRISPR/CAS9-mediated genome editing of miRNA-155 inhibits proinflammatory cytokine production by RAW264. 7 cells. BioMed Res. Int. 2015, 2015, 326042. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-W.; Lai, Y.-S.; Westin, E.; Khodadadi-Jamayran, A.; Pawlik, K.M.; Lamb Jr, L.S.; Goldman, F.D.; Townes, T.M. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-enhanced gene targeting. Cell Rep. 2015, 12, 1668–1677. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [Green Version]
- Seeger, C.; Sohn, J.A. Targeting hepatitis B virus with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.-J. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, S.; Hua, L.; Takahashi, Y.; Narita, S.; Liu, Y.-H.; Li, Y. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by CRISPR/Cas9. Biochem. Biophys. Res. Commun. 2014, 450, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.-S.; Wang, Z.-M.; Wong, N.-H.M.; Zhang, Z.-Q.; Cheng, T.-F.; Lui, W.-Y.; Chan, C.-P.; Jin, D.-Y. Suppression of Epstein-Barr virus DNA load in latently infected nasopharyngeal carcinoma cells by CRISPR/Cas9. Virus Res. 2018, 244, 296–303. [Google Scholar] [CrossRef]
- Kistler, K.E.; Vosshall, L.B.; Matthews, B.J. Genome engineering with CRISPR-Cas9 in the mosquito Aedes aegypti. Cell Rep. 2015, 11, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.C.; Platt, R.J.; Goldfless, S.J.; Zhang, F.; Niles, J.C. Efficient CRISPR-Cas9–mediated genome editing in Plasmodium falciparum. Nat. Methods 2014, 11, 915–918. [Google Scholar] [CrossRef]
- Ghorbal, M.; Gorman, M.; Macpherson, C.R.; Martins, R.M.; Scherf, A.; Lopez-Rubio, J.-J. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat. Biotechnol. 2014, 32, 819. [Google Scholar] [CrossRef]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Pankowicz, F.P.; Barzi, M.; Legras, X.; Hubert, L.; Mi, T.; Tomolonis, J.A.; Ravishankar, M.; Sun, Q.; Yang, D.; Borowiak, M. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat. Commun. 2016, 7, 12642. [Google Scholar] [CrossRef]
- Halim, D.; Wilson, M.P.; Oliver, D.; Brosens, E.; Verheij, J.B.; Han, Y.; Nanda, V.; Lyu, Q.; Doukas, M.; Stoop, H. Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc. Natl. Acad. Sci. USA 2017, 114, E2739–E2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Macovei, A.; Sevilla, N.R.; Cantos, C.; Jonson, G.B.; Slamet-Loedin, I.; Čermák, T.; Voytas, D.F.; Choi, I.R.; Chadha-Mohanty, P. Novel alleles of rice eIF4G generated by CRISPR/Cas9-targeted mutagenesis confer resistance to Rice tungro spherical virus. Plant Biotechnol. J. 2018, 16, 1918–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, A.; Arndell, T.; Borisjuk, N.; Sharma, N.; Watson-Haigh, N.S.; Tucker, E.J.; Baumann, U.; Langridge, P.; Whitford, R. CRISPR/Cas9-mediated knockout of Ms1 enables the rapid generation of male-sterile hexaploid wheat lines for use in hybrid seed production. Plant Biotechnol. J. 2019, 17, 1905–1913. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Zhang, Y.; Kleinstiver, B.P.; Guo, J.A.; Aryee, M.J.; Miller, J.; Malzahn, A.; Zarecor, S.; Lawrence-Dill, C.J.; Joung, J.K. Activities and specificities of CRISPR/Cas9 and Cas12a nucleases for targeted mutagenesis in maize. Plant Biotechnol. J. 2019, 17, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Manghwar, H.; Sun, L.; Wang, P.; Wang, G.; Sheng, H.; Zhang, J.; Liu, H.; Qin, L.; Rui, H. Whole genome sequencing reveals rare off-target mutations and considerable inherent genetic or/and somaclonal variations in CRISPR/Cas9-edited cotton plants. Plant Biotechnol. J. 2019, 17, 858–868. [Google Scholar] [CrossRef]
- Baltes, N.J.; Gil-Humanes, J.; Cermak, T.; Atkins, P.A.; Voytas, D.F. DNA replicons for plant genome engineering. Plant Cell 2014, 26, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Liang, D.; Wang, Y.; Bai, M.; Tang, W.; Bao, S.; Yan, Z.; Li, D.; Li, J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013, 13, 659–662. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, K.; Kaneko, T.; Voigt, B.; Mashimo, T. Allele-specific genome editing and correction of disease-associated phenotypes in rats using the CRISPR–Cas platform. Nat. Commun. 2014, 5, 4240. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Tu, L.-C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016, 34, 528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.K.; Hwang, J.S.; Chung, T.Y.; Shin, Y.J. SOX2 activation using CRISPR/dCas9 promotes wound healing in corneal endothelial cells. Stem Cells 2018, 36, 1851–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, e2579–e2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 1225829. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W. RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nat. Methods 2013, 10, 973. [Google Scholar] [CrossRef] [Green Version]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180. [Google Scholar] [CrossRef] [Green Version]
- Westra, E.R.; Semenova, E.; Datsenko, K.A.; Jackson, R.N.; Wiedenheft, B.; Severinov, K.; Brouns, S.J. Type IE CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 2013, 9, e1003742. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Björklund, S.; Li, Y.; Sayre, M.H.; Kornberg, R.D. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell 1994, 77, 599–608. [Google Scholar] [CrossRef]
- Chavez, A.; Tuttle, M.; Pruitt, B.W.; Ewen-Campen, B.; Chari, R.; Ter-Ovanesyan, D.; Haque, S.J.; Cecchi, R.J.; Kowal, E.J.; Buchthal, J. Comparison of Cas9 activators in multiple species. Nat. Methods 2016, 13, 563. [Google Scholar] [CrossRef] [Green Version]
- Piatek, A.; Ali, Z.; Baazim, H.; Li, L.; Abulfaraj, A.; Al-Shareef, S.; Aouida, M.; Mahfouz, M.M. RNA-guided transcriptional regulation in planta via synthetic dC as9-based transcription factors. Plant Biotechnol. J. 2015, 13, 578–589. [Google Scholar] [CrossRef]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Zou, Q.; Lai, S.; Zhang, Q.; Li, L.; Yan, Q.; Zhou, X.; Zhong, H.; Lai, L. Conversion of embryonic stem cells into extraembryonic lineages by CRISPR-mediated activators. Sci. Rep. 2016, 6, 19648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.B.; Adler, A.F.; Wang, H.-G.; D’Ippolito, A.M.; Hutchinson, H.A.; Reddy, T.E.; Pitt, G.S.; Leong, K.W.; Gersbach, C.A. Targeted epigenetic remodeling of endogenous loci by CRISPR/Cas9-based transcriptional activators directly converts fibroblasts to neuronal cells. Cell Stem Cell 2016, 19, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Ji, H.; Kabadi, A.M.; Gersbach, C.A.; Christoforou, N.; Leong, K.W. A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Rep. 2014, 3, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326. [Google Scholar] [CrossRef] [Green Version]
- Haenfler, J.M.; Skariah, G.; Rodriguez, C.M.; Monteiro da Rocha, A.; Parent, J.M.; Smith, G.D.; Todd, P.K. Targeted reactivation of fmr1 transcription in fragile x syndrome embryonic stem cells. Front. Mol. Neurosci. 2018, 11, 282. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Lei, Y.; Wong, W.-K.; Liu, S.; Lee, K.-C.; He, X.; You, W.; Zhou, R.; Guo, J.-T.; Chen, X. Direct activation of human and mouse Oct4 genes using engineered TALE and Cas9 transcription factors. Nucleic Acids Res. 2014, 42, 4375–4390. [Google Scholar] [CrossRef]
- Vad-Nielsen, J.; Lin, L.; Bolund, L.; Nielsen, A.L.; Luo, Y. Golden Gate Assembly of CRISPR gRNA expression array for simultaneously targeting multiple genes. Cell. Mol. Life Sci. 2016, 73, 4315–4325. [Google Scholar] [CrossRef]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [Green Version]
- Kabadi, A.M.; Ousterout, D.G.; Hilton, I.B.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014, 42, e147. [Google Scholar] [CrossRef] [Green Version]
- Xiong, K.; Zhou, Y.; Blichfeld, K.A.; Hyttel, P.; Bolund, L.; Freude, K.K.; Luo, Y. RNA-guided activation of pluripotency genes in human fibroblasts. Cell. Reprogram. 2017, 19, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution—trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.; Elledge, S.J.; Hannon, G.J. Lessons from Nature: MicroRNA-based shRNA libraries. Nat. Methods 2006, 3, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef]
- Sigoillot, F.D.; Lyman, S.; Huckins, J.F.; Adamson, B.; Chung, E.; Quattrochi, B.; King, R.W. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat. Methods 2012, 9, 363. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas III, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347. [Google Scholar] [CrossRef]
- Koike-Yusa, H.; Li, Y.; Tan, E.-P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inui, M.; Miyado, M.; Igarashi, M.; Tamano, M.; Kubo, A.; Yamashita, S.; Asahara, H.; Fukami, M.; Takada, S. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci. Rep. 2014, 4, 5396. [Google Scholar] [CrossRef]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Rebar, E.J.; Huang, Y.; Hickey, R.; Nath, A.K.; Meoli, D.; Nath, S.; Chen, B.; Xu, L.; Liang, Y.; Jamieson, A.C. Induction of angiogenesis in a mouse model using engineered transcription factors. Nat. Med. 2002, 8, 1427–1432. [Google Scholar] [CrossRef]
- Beltran, A.; Parikh, S.; Liu, Y.; Cuevas, B.; Johnson, G.; Futscher, B.W.; Blancafort, P. Re-activation of a dormant tumor suppressor gene maspin by designed transcription factors. Oncogene 2007, 26, 2791–2798. [Google Scholar] [CrossRef] [Green Version]
- Bartsevich, V.V.; Miller, J.C.; Case, C.C.; Pabo, C.O. Engineered zinc finger proteins for controlling stem cell fate. Stem Cells 2003, 21, 632–637. [Google Scholar] [CrossRef]
- Bultmann, S.; Morbitzer, R.; Schmidt, C.S.; Thanisch, K.; Spada, F.; Elsaesser, J.; Lahaye, T.; Leonhardt, H. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res. 2012, 40, 5368–5377. [Google Scholar] [CrossRef] [Green Version]
- Blancafort, P.; Magnenat, L.; Barbas, C.F. Scanning the human genome with combinatorial transcription factor libraries. Nat. Biotechnol. 2003, 21, 269–274. [Google Scholar] [CrossRef]
- Park, K.-S.; Lee, D.-k.; Lee, H.; Lee, Y.; Jang, Y.-S.; Kim, Y.H.; Yang, H.-Y.; Lee, S.-I.; Seol, W.; Kim, J.-S. Phenotypic alteration of eukaryotic cells using randomized libraries of artificial transcription factors. Nat. Biotechnol. 2003, 21, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA–guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [Green Version]
- Hunt, M.A.; Currie, M.J.; Robinson, B.A.; Dachs, G.U. Optimizing transfection of primary human umbilical vein endothelial cells using commercially available chemical transfection reagents. J. Biomol. Tech. 2010, 21, 66. [Google Scholar] [PubMed]
- Schenborn, E.T.; Goiffon, V. DEAE-dextran transfection of mammalian cultured cells. In Transcription Factor Protocols; Springer: Berlin/Heidelberg, Germany, 2000; pp. 147–153. [Google Scholar]
- Holmen, S.L.; Vanbrocklin, M.W.; Eversole, R.R.; Stapleton, S.R.; Ginsberg, L.C. Efficient lipid-mediated transfection of DNA into primary rat hepatocytes. Cell. Dev. Biol. Anim. 1995, 31, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Washbourne, P.; McAllister, A.K. Techniques for gene transfer into neurons. Curr. Opin. Neurobiol. 2002, 12, 566–573. [Google Scholar] [CrossRef]
- Kim, T.K.; Eberwine, J.H. Mammalian cell transfection: The present and the future. Anal. Bioanal. Chem. 2010, 397, 3173–3178. [Google Scholar] [CrossRef] [Green Version]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, D.; Xiong, X.; Yan, B.; Xie, W.; Sheen, J.; Li, J.-F. A potent Cas9-derived gene activator for plant and mammalian cells. Nat. Plants 2017, 3, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, S.H.; LaFrance, B.; Kaplan, M.; Doudna, J.A. Conformational control of DNA target cleavage by CRISPR–Cas9. Nature 2015, 527, 110–113. [Google Scholar] [CrossRef]
- Huai, C.; Li, G.; Yao, R.; Zhang, Y.; Cao, M.; Kong, L.; Jia, C.; Yuan, H.; Chen, H.; Lu, D. Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat. Commun. 2017, 8, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagdas, Y.S.; Chen, J.S.; Sternberg, S.H.; Doudna, J.A.; Yildiz, A. A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci. Adv. 2017, 3, eaao0027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, I.B.; D’ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Gurdon, J.B.; Elsdale, T.R.; Fischberg, M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature 1958, 182, 64–65. [Google Scholar] [CrossRef]
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef]
- Wakayama, T.; Perry, A.C.; Zuccotti, M.; Johnson, K.R.; Yanagimachi, R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 1998, 394, 369–374. [Google Scholar] [CrossRef]
- Byrne, J.; Pedersen, D.; Clepper, L.; Nelson, M.; Sanger, W.; Gokhale, S.; Wolf, D.; Mitalipov, S. Producing primate embryonic stem cells by somatic cell nuclear transfer. Nature 2007, 450, 497–502. [Google Scholar] [CrossRef]
- Tada, M.; Takahama, Y.; Abe, K.; Nakatsuji, N.; Tada, T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr. Biol. 2001, 11, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Cowan, C.A.; Atienza, J.; Melton, D.A.; Eggan, K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science 2005, 309, 1369–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchieu, J.; Jaenisch, R. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007, 448, 318–324. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Park, I.-H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Lowry, W.; Richter, L.; Yachechko, R.; Pyle, A.; Tchieu, J.; Sridharan, R.; Clark, A.; Plath, K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. USA 2008, 105, 2883–2888. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2008, 2, 10–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, W.; Zhou, H.; Wei, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, Q.; Yin, X.; Yang, W.; Du, Y.; Hou, P.; Ge, J.; Liu, C.; Zhang, W.; Zhang, X. Generation of iPSCs from mouse fibroblasts with a single gene, Oct4, and small molecules. Cell Res. 2011, 21, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtfeld, M.; Hochedlinger, K. Induced pluripotency: History, mechanisms, and applications. Genes Dev. 2010, 24, 2239–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vad-Nielsen, J.; Lin, L.; Jensen, K.T.; Nielsen, A.L.; Luo, Y. A golden gate-based protocol for assembly of multiplexed gRNA expression arrays for CRISPR/Cas9. Bio Protoc. 2016, 6, e2059. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javaid, N.; Pham, T.L.H.; Choi, S. Functional Comparison between VP64-dCas9-VP64 and dCas9-VP192 CRISPR Activators in Human Embryonic Kidney Cells. Int. J. Mol. Sci. 2021, 22, 397. https://doi.org/10.3390/ijms22010397
Javaid N, Pham TLH, Choi S. Functional Comparison between VP64-dCas9-VP64 and dCas9-VP192 CRISPR Activators in Human Embryonic Kidney Cells. International Journal of Molecular Sciences. 2021; 22(1):397. https://doi.org/10.3390/ijms22010397
Chicago/Turabian StyleJavaid, Nasir, Thuong L. H. Pham, and Sangdun Choi. 2021. "Functional Comparison between VP64-dCas9-VP64 and dCas9-VP192 CRISPR Activators in Human Embryonic Kidney Cells" International Journal of Molecular Sciences 22, no. 1: 397. https://doi.org/10.3390/ijms22010397
APA StyleJavaid, N., Pham, T. L. H., & Choi, S. (2021). Functional Comparison between VP64-dCas9-VP64 and dCas9-VP192 CRISPR Activators in Human Embryonic Kidney Cells. International Journal of Molecular Sciences, 22(1), 397. https://doi.org/10.3390/ijms22010397