Intermittent Hypoxic Conditioning Rescues Cognition and Mitochondrial Bioenergetic Profile in the Triple Transgenic Mouse Model of Alzheimer’s Disease

 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results
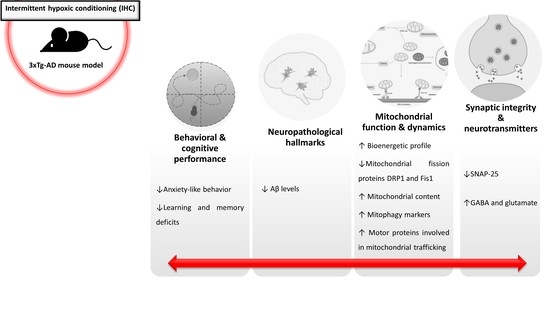
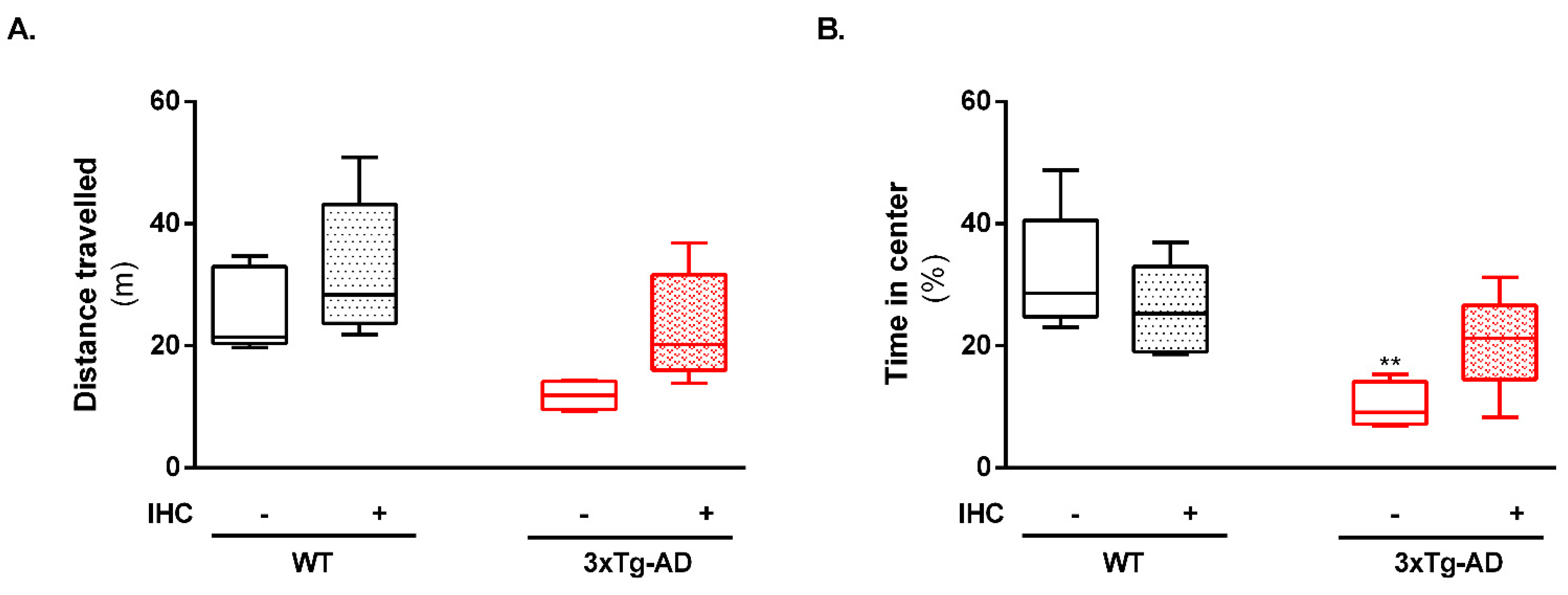
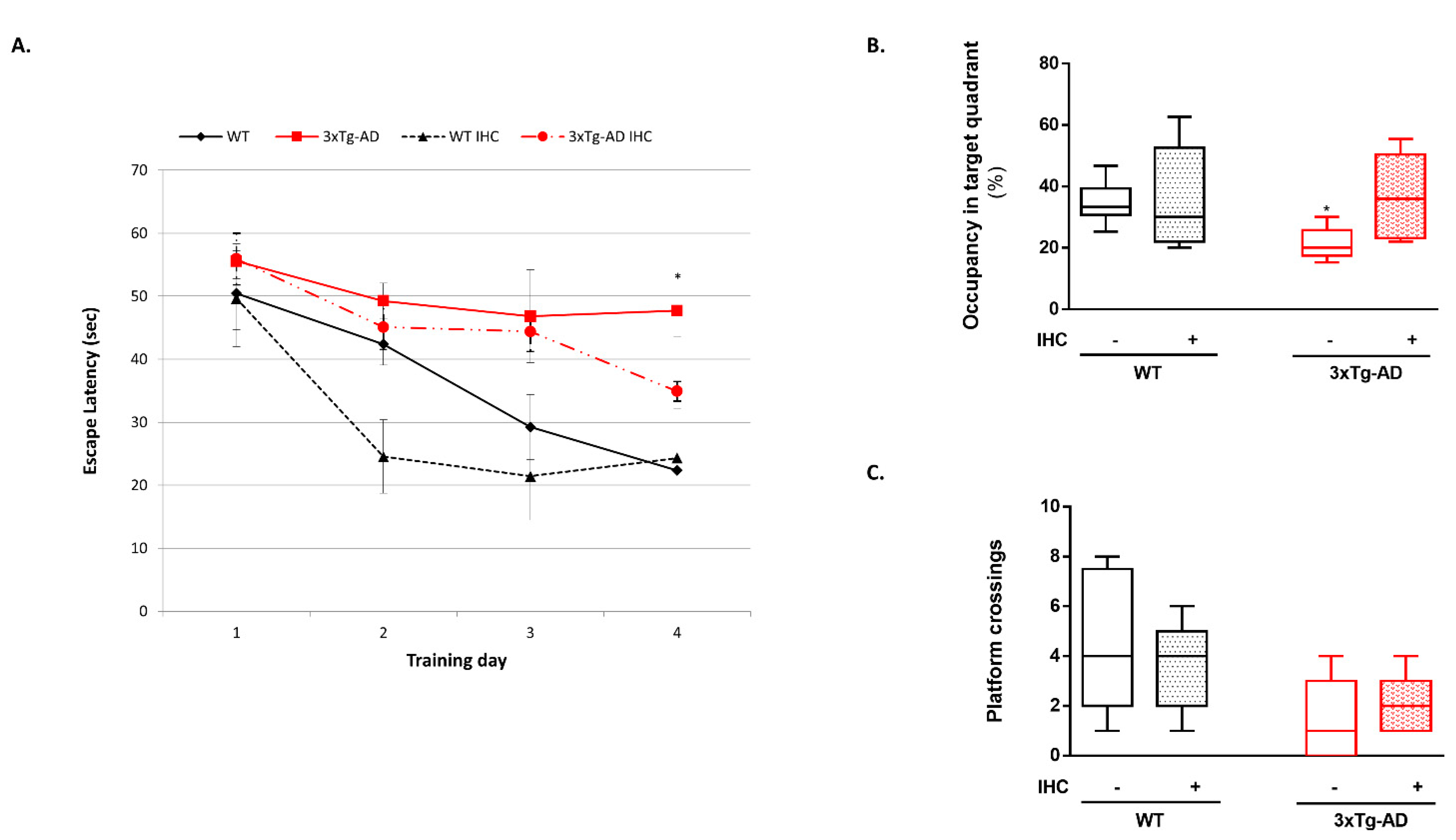
2.1. IHC Partially Prevents Behavioral and Cognitive Alterations in 3×Tg-AD Mice
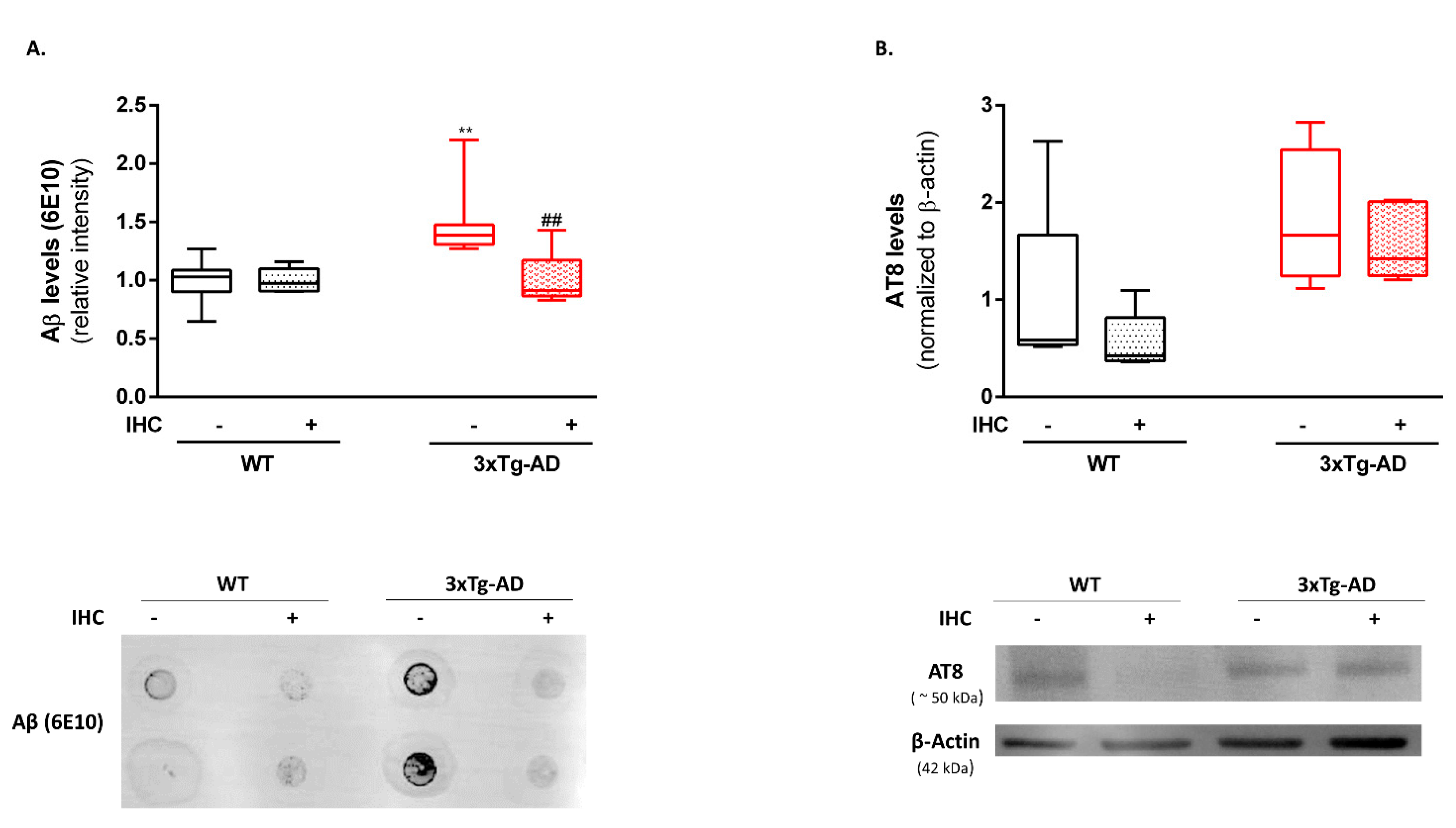
2.2. HP Abrogates Aβ Pathology in the Brain Cortex of 3×Tg-AD Mice
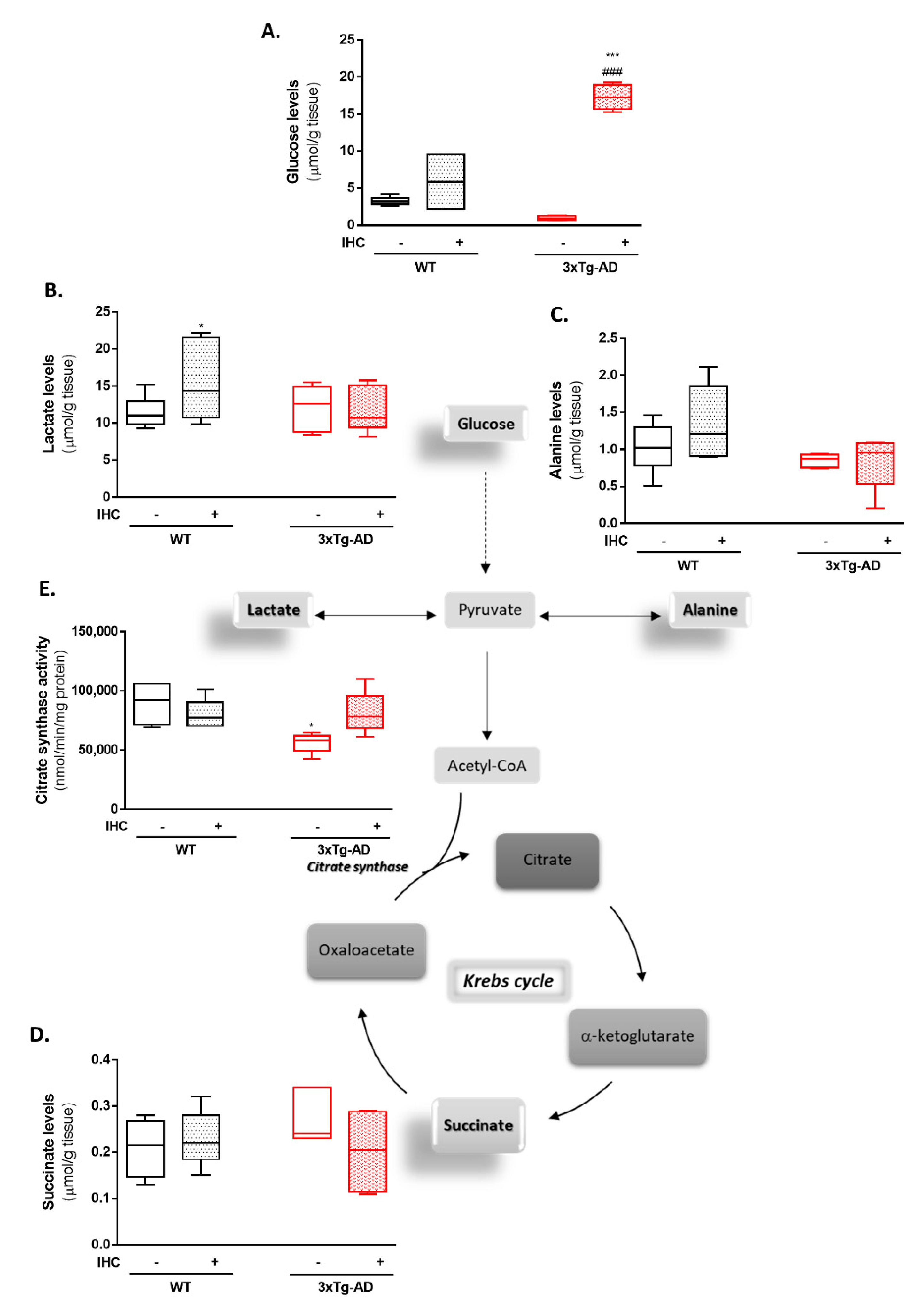
2.3. IHC Ameliorates Mitochondrial Bioenergetic Function in the Brain Cortex of 3×Tg-AD Mice
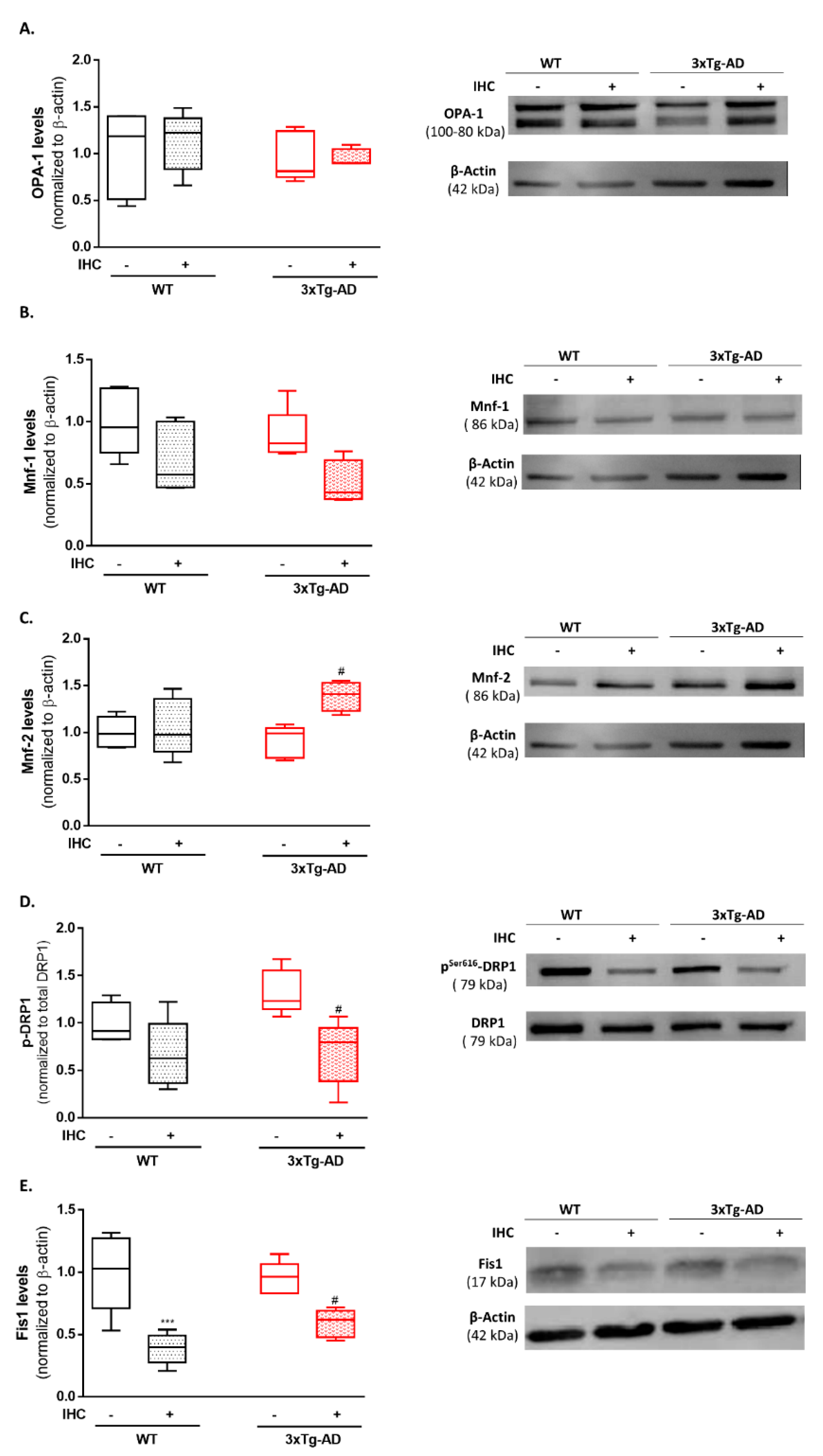
2.4. IHC Reduces the Levels of Mitochondrial Fission-Related Proteins in the Brain Cortex of 3×Tg-AD Mice
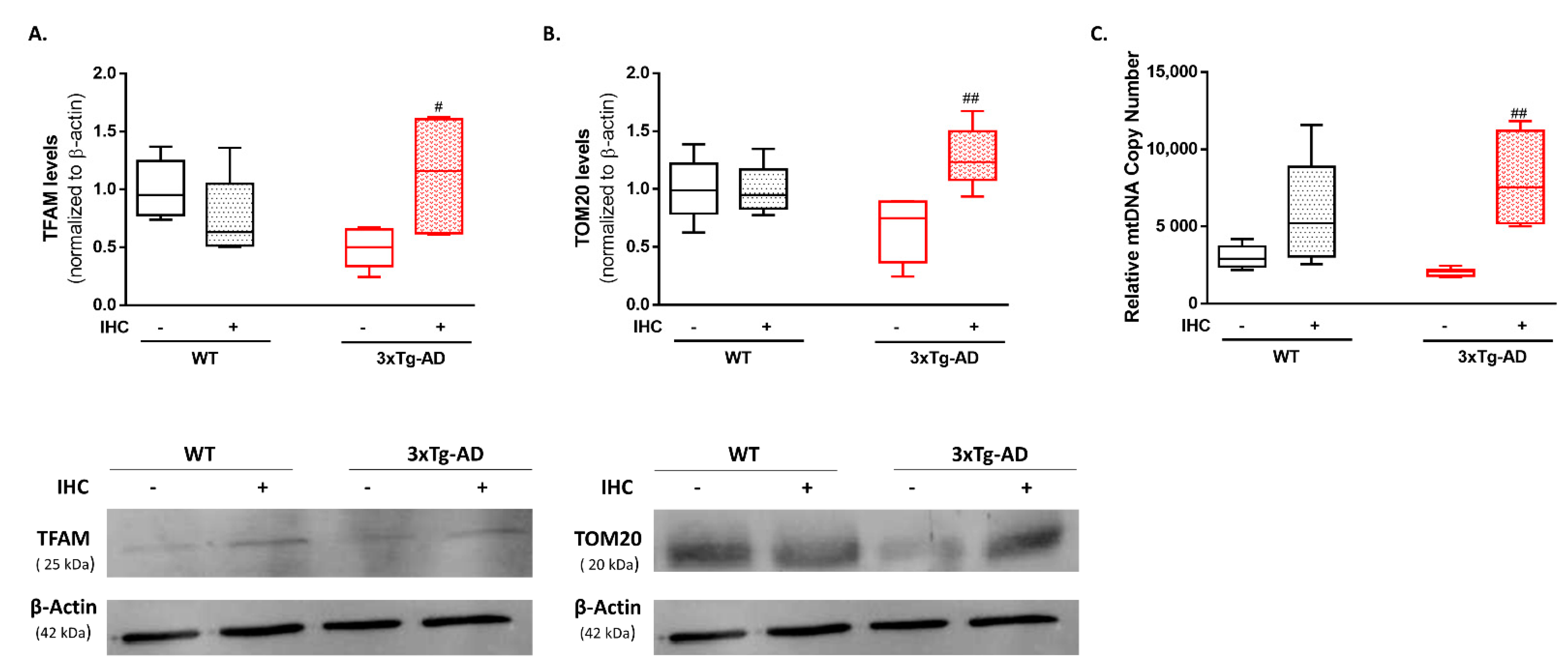
2.5. IHC Promotes Mitochondrial Biogenesis Increasing Mitochondrial Content in the Brain Cortex of 3×Tg-AD Mice
2.6. IHC Enhances the Molecular Machinery Behind Mitophagy in the Brain Cortex of 3×Tg-AD Mice
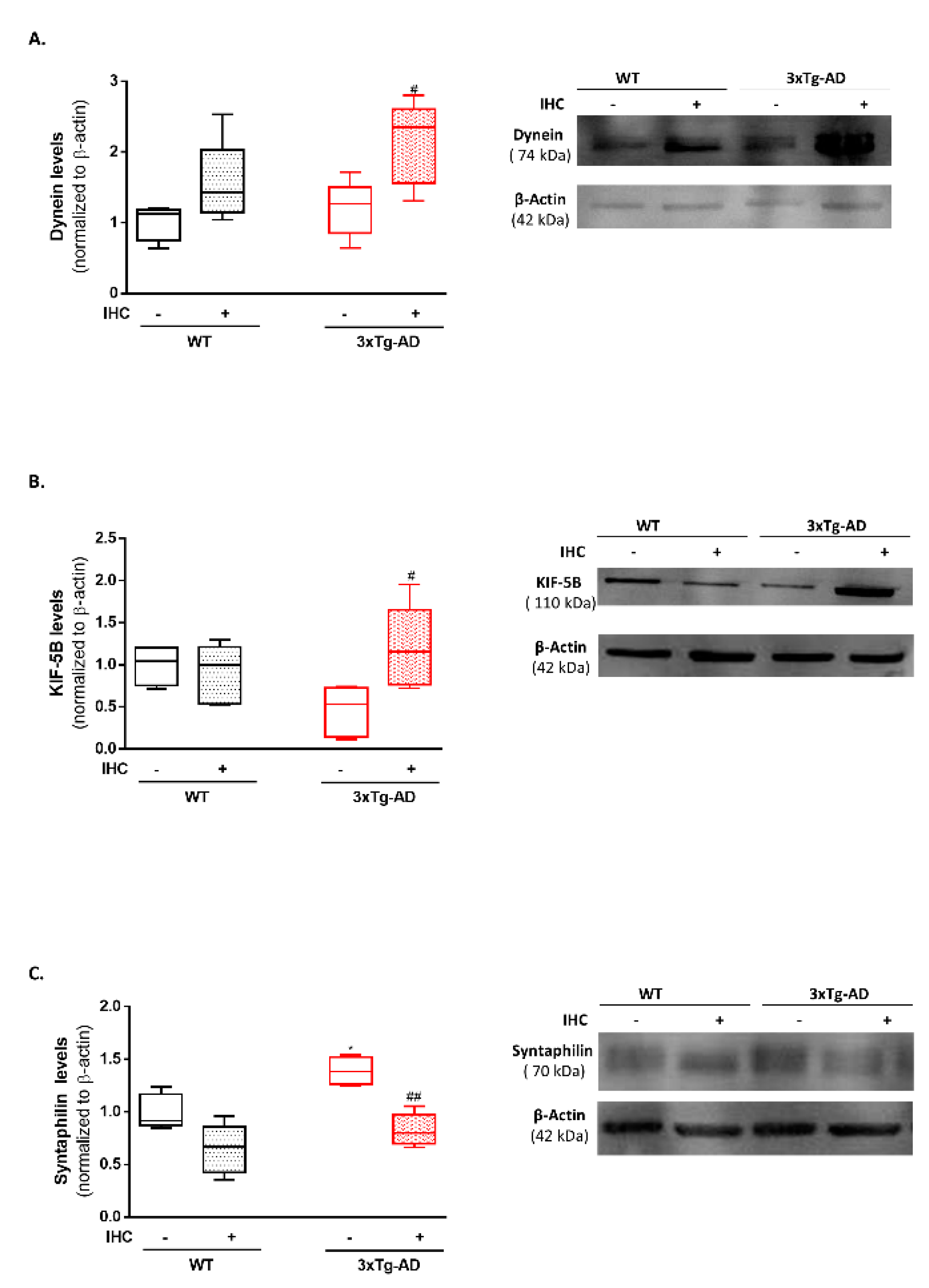
2.7. IHC Increases Mitochondrial Motor Proteins in the Brain Cortex of 3×Tg-AD Mice
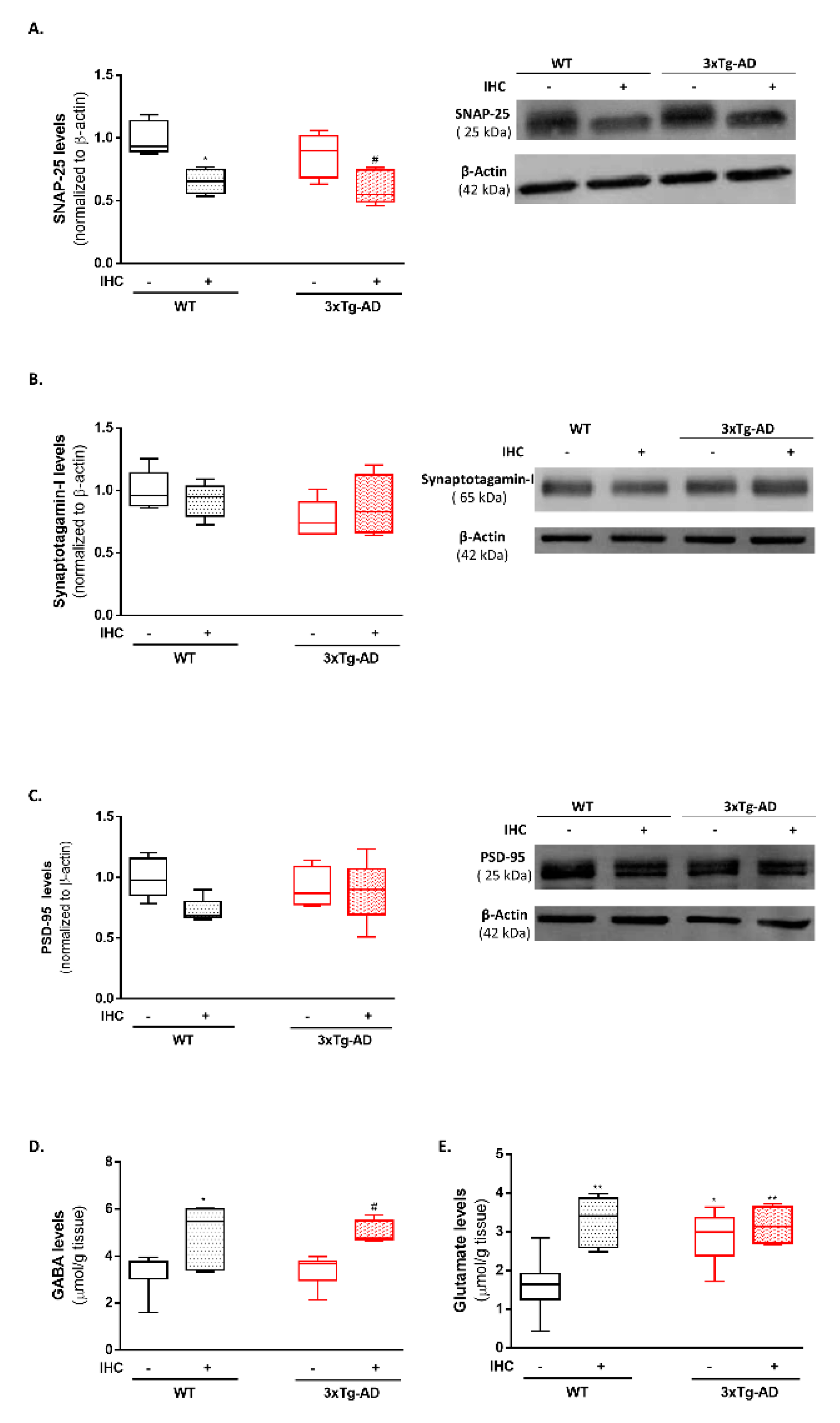
2.8. IHC Downregulates SNAP-25 and Increases Glutamate and GABA Levels and in the Brain Cortex of 3×Tg-AD Mice
3. Discussion
4. Materials and Methods
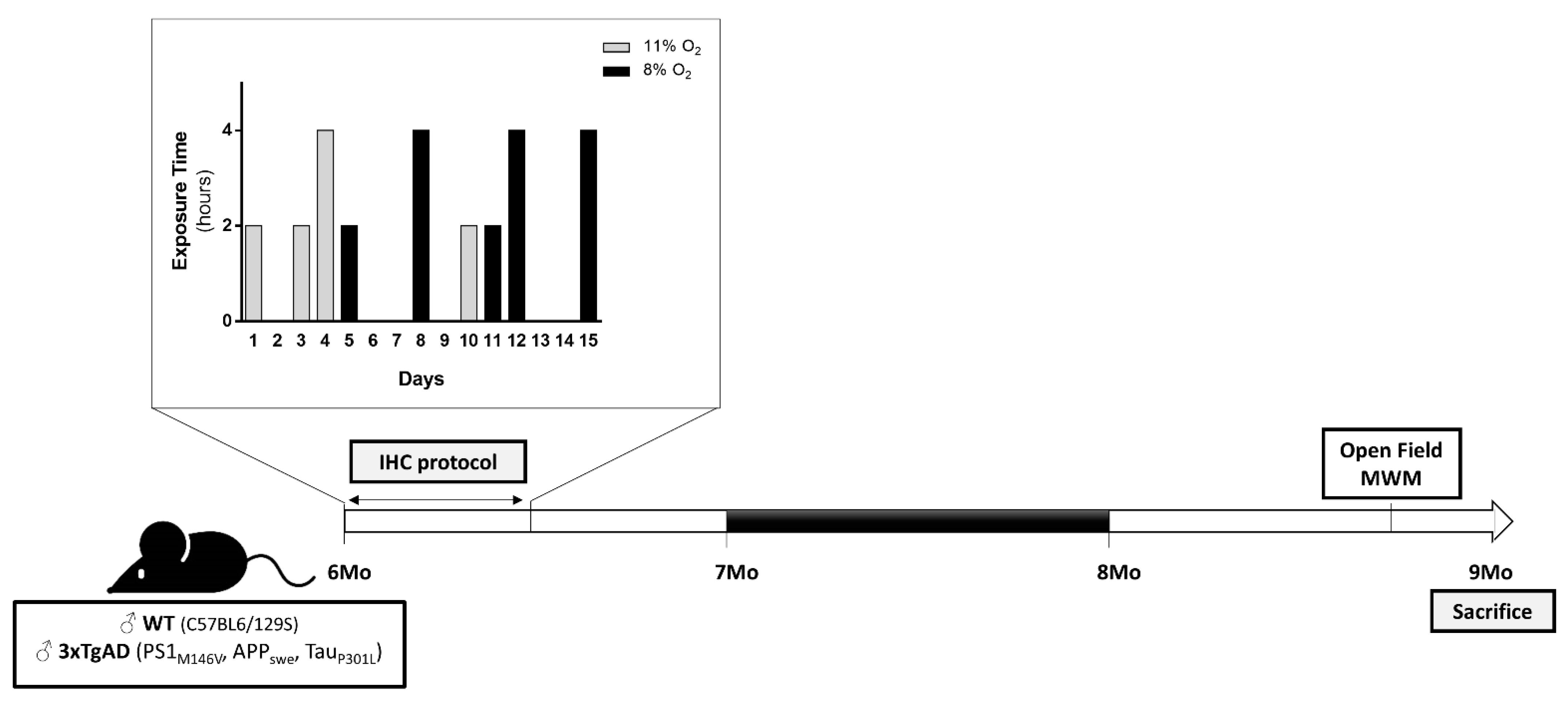
4.1. Animals and Ethics Statements
4.2. Behavioral Analyses
4.2.1. Open Field Test
4.2.2. MWM Test
4.3. NMR Spectroscopy
4.4. Preparation of Mitochondrial Fraction
4.5. Measurements of ΔΨm
4.6. Mitochondrial Respiration Measurements
4.7. Citrate Synthase Activity
4.8. Determination of Mitochondrial DNA Copy Number
4.9. Western Blot Analysis
4.10. Immuno-Dot-Blot Assay
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hickman, R.A.; Faustin, A.; Wisniewski, T. Alzheimer Disease and Its Growing Epidemic: Risk Factors, Biomarkers, and the Urgent Need for Therapeutics. Neurol. Clin. 2016, 34, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.D. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical epidemiology of Alzheimer’s disease: Assessing sex and gender differences. Clin. Epidemiol. 2014, 6, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Jack, C.R., Jr.; Morris, J.C.; Sperling, R.; Frolich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimers Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Cardoso, S.; Carvalho, C.; Correia, S.C.; Seica, R.M.; Moreira, P.I. Alzheimer’s Disease: From Mitochondrial Perturbations to Mitochondrial Medicine. Brain Pathol. 2016, 26, 632–647. [Google Scholar] [CrossRef]
- Dezfulian, C.; Garrett, M.; Gonzalez, N.R. Clinical application of preconditioning and postconditioning to achieve neuroprotection. Transl. Stroke Res. 2013, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Leak, R.K. Conditioning Against the Pathology of Parkinson’s disease. Cond. Med. 2018, 1, 143–162. [Google Scholar] [PubMed]
- Wang, Y.; Reis, C.; Applegate, R., 2nd; Stier, G.; Martin, R.; Zhang, J.H. Ischemic conditioning-induced endogenous brain protection: Applications pre-, per- or post-stroke. Exp. Neurol. 2015, 272, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Agathokleous, E. Building Biological Shields via Hormesis. Trends Pharm. Sci. 2019, 40, 8–10. [Google Scholar] [CrossRef]
- Vinciguerra, A.; Cuomo, O.; Cepparulo, P.; Anzilotti, S.; Brancaccio, P.; Sirabella, R.; Guida, N.; Annunziato, L.; Pignataro, G. Models and methods for conditioning the ischemic brain. J. Neurosci. Methods 2018, 310, 63–74. [Google Scholar] [CrossRef]
- Manukhina, E.B.; Goryacheva, A.V.; Barskov, I.V.; Viktorov, I.V.; Guseva, A.A.; Pshennikova, M.G.; Khomenko, I.P.; Mashina, S.Y.; Pokidyshev, D.A.; Malyshev, I.Y. Prevention of neurodegenerative damage to the brain in rats in experimental Alzheimer’s disease by adaptation to hypoxia. Neurosci. Behav. Physiol. 2010, 40, 737–743. [Google Scholar] [CrossRef]
- Boengler, K.; Heusch, G.; Schulz, R. Mitochondria in postconditioning. Antioxid. Redox Signal. 2011, 14, 863–880. [Google Scholar] [CrossRef]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” target of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef]
- Correia, S.C.; Cardoso, S.; Santos, R.X.; Carvalho, C.; Santos, M.S.; Perry, G.; Smith, M.A.; Moreira, P.I. New insights into the mechanisms of mitochondrial preconditioning-triggered neuroprotection. Curr. Pharm. Des. 2011, 17, 3381–3389. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012, 1441, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 943–972. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 2020, 4, NS20200008. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y. Emerging Concepts and Functions of Autophagy as a Regulator of Synaptic Components and Plasticity. Cells 2019, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in Neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Perry, G.; Moreira, P.I. Mitochondrial traffic jams in Alzheimer’s disease—pinpointing the roadblocks. Biochim. Biophys. Acta 2016, 1862, 1909–1917. [Google Scholar] [CrossRef]
- Zheng, Y.R.; Zhang, X.N.; Chen, Z. Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 2019, 25, 876–886. [Google Scholar] [CrossRef]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef]
- Meng, S.X.; Wang, B.; Li, W.T. Intermittent hypoxia improves cognition and reduces anxiety-related behavior in APP/PS1 mice. Brain Behav. 2020, 10, e01513. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, X.; Schenck, H.; Hall, J.R.; Ross, S.E.; Kline, G.P.; Chen, S.; Mallet, R.T.; Chen, P. Intermittent Hypoxia Training for Treating Mild Cognitive Impairment: A Pilot Study. Am. J. Alzheimers Dis. Dement. 2020, 35, 1533317519896725. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Mathotaarachchi, S.; Mohades, S.; Benedet, A.L.; Chung, C.O.; Shin, M.; Wang, S.; Beaudry, T.; Kang, M.S.; Soucy, J.P.; et al. Amyloid-beta and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer’s disease. Mol. Psychiatry 2017, 22, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Mathotaarachchi, S.; Shin, M.; Benedet, A.L.; Mohades, S.; Wang, S.; Beaudry, T.; Kang, M.S.; Soucy, J.P.; Labbe, A.; et al. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimers Dement. J. Alzheimers Assoc. 2017, 13, 644–653. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Shiota, S.; Takekawa, H.; Matsumoto, S.E.; Takeda, K.; Nurwidya, F.; Yoshioka, Y.; Takahashi, F.; Hattori, N.; Tabira, T.; Mochizuki, H.; et al. Chronic intermittent hypoxia/reoxygenation facilitate amyloid-beta generation in mice. J. Alzheimers Dis. JAD 2013, 37, 325–333. [Google Scholar] [CrossRef]
- Ashok, B.S.; Ajith, T.A.; Sivanesan, S. Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin. Exp. Pharmacol. Physiol. 2017, 44, 327–334. [Google Scholar] [CrossRef]
- Zhang, F.; Niu, L.; Li, S.; Le, W. Pathological Impacts of Chronic Hypoxia on Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 902–909. [Google Scholar] [CrossRef]
- Azari, N.P.; Pettigrew, K.D.; Schapiro, M.B.; Haxby, J.V.; Grady, C.L.; Pietrini, P.; Salerno, J.A.; Heston, L.L.; Rapoport, S.I.; Horwitz, B. Early detection of Alzheimer’s disease: A statistical approach using positron emission tomographic data. J. Cereb. Blood Flow Metab. 1993, 13, 438–447. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Mastrogiacomo, F.; Bergeron, C.; Kish, S.J. Brain alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. J. Neurochem. 1993, 61, 2007–2014. [Google Scholar] [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Proenca, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Valla, J.; Schneider, L.; Niedzielko, T.; Coon, K.D.; Caselli, R.; Sabbagh, M.N.; Ahern, G.L.; Baxter, L.; Alexander, G.; Walker, D.G.; et al. Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion 2006, 6, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Schmitt, K.; Gotz, J. Mitochondrial dysfunction—the beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-beta toxicity. Alzheimers Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef]
- Chen, J.; Liao, W.; Gao, W.; Huang, J.; Gao, Y. Intermittent hypoxia protects cerebral mitochondrial function from calcium overload. Acta Neurol. Belg. 2013, 113, 507–513. [Google Scholar] [CrossRef]
- Ju, X.; Mallet, R.T.; Downey, H.F.; Metzger, D.B.; Jung, M.E. Intermittent hypoxia conditioning protects mitochondrial cytochrome c oxidase of rat cerebellum from ethanol withdrawal stress. J. Appl. Physiol. 2012, 112, 1706–1714. [Google Scholar] [CrossRef]
- Baloyannis, S.J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimers Dis. JAD 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. TEM 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.B.; Pineda, F.J.; Hardwick, J.M. Mitochondrial fission and fusion dynamics: The long and short of it. Cell Death Differ. 2008, 15, 1147–1152. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Huang, S.; Wu, L.; Wang, Y.; Hu, G.; Li, G.; Zhang, H.; Yu, H.; Swerdlow, R.H.; Chen, J.X.; et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Et Biophys. Acta 2014, 1842, 220–231. [Google Scholar] [CrossRef]
- Espino de la Fuente-Munoz, C.; Rosas-Lemus, M.; Moreno-Castilla, P.; Bermudez-Rattoni, F.; Uribe-Carvajal, S.; Arias, C. Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 8727. [Google Scholar] [CrossRef]
- Das, R.; Chakrabarti, O. Mitochondrial hyperfusion: A friend or a foe. Biochem. Soc. Trans. 2020, 48, 631–644. [Google Scholar] [CrossRef]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Gutsaeva, D.R.; Carraway, M.S.; Suliman, H.B.; Demchenko, I.T.; Shitara, H.; Yonekawa, H.; Piantadosi, C.A. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J. Neurosci. 2008, 28, 2015–2024. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. CB 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef] [PubMed]
- Martin-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; Garcia-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Martin-Maestro, P.; Gargini, R.; Sproul, A.A.; Garcia, E.; Anton, L.C.; Noggle, S.; Arancio, O.; Avila, J.; Garcia-Escudero, V. Mitophagy Failure in Fibroblasts and iPSC-Derived Neurons of Alzheimer’s Disease-Associated Presenilin 1 Mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef]
- Zhou, M.; Xia, Z.Y.; Lei, S.Q.; Leng, Y.; Xue, R. Role of mitophagy regulated by Parkin/DJ-1 in remote ischemic postconditioning-induced mitigation of focal cerebral ischemia-reperfusion. Eur. Rev. Med Pharmacol. Sci. 2015, 19, 4866–4871. [Google Scholar] [PubMed]
- Correia, S.C.; Santos, R.X.; Cardoso, S.; Carvalho, C.; Candeias, E.; Duarte, A.I.; Placido, A.I.; Santos, M.S.; Moreira, P.I. Alzheimer disease as a vascular disorder: Where do mitochondria fit? Exp. Gerontol. 2012, 47, 878–886. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Borland, M.K. Differentiated Alzheimer’s disease transmitochondrial cybrid cell lines exhibit reduced organelle movement. Antioxid. Redox Signal. 2005, 7, 1101–1109. [Google Scholar] [CrossRef]
- Thomas, L.W.; Ashcroft, M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell. Mol. Life Sci. CMLS 2019, 76, 1759–1777. [Google Scholar] [CrossRef]
- Lin, M.Y.; Sheng, Z.H. Regulation of mitochondrial transport in neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Y.; Cheng, X.T.; Tammineni, P.; Xie, Y.; Zhou, B.; Cai, Q.; Sheng, Z.H. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 2017, 94, 595–610.e6. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol. Ther. 2019, 195, 186–198. [Google Scholar] [CrossRef]
- Tang, J.; Oliveros, A.; Jang, M.H. Dysfunctional Mitochondrial Bioenergetics and Synaptic Degeneration in Alzheimer Disease. Int. Neurourol. J. 2019, 23, S5–10. [Google Scholar] [CrossRef]
- Rato, L.; Alves, M.G.; Dias, T.R.; Lopes, G.; Cavaco, J.E.; Socorro, S.; Oliveira, P.F. High-energy diets may induce a pre-diabetic state altering testicular glycolytic metabolic profile and male reproductive parameters. Andrology 2013, 1, 495–504. [Google Scholar] [CrossRef]
- Calvo-Flores Guzman, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef]
- Conway, M.E. Alzheimer’s disease: Targeting the glutamatergic system. Biogerontology 2020, 21, 257–274. [Google Scholar] [CrossRef]
- Tiwari, V.; Patel, A.B. Impaired glutamatergic and GABAergic function at early age in AbetaPPswe-PS1dE9 mice: Implications for Alzheimer’s disease. J. Alzheimers Dis. JAD 2012, 28, 765–769. [Google Scholar] [CrossRef]
- Clark, J.K.; Furgerson, M.; Crystal, J.D.; Fechheimer, M.; Furukawa, R.; Wagner, J.J. Alterations in synaptic plasticity coincide with deficits in spatial working memory in presymptomatic 3xTg-AD mice. Neurobiol. Learn. Mem. 2015, 125, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, Z.; Sun, S.; Duan, Y.; Shi, J.; Qi, Z.; Meng, R.; Sun, Y.; Zeng, X.; Chui, D.; et al. Effects of hypoxic preconditioning on synaptic ultrastructure in mice. Synapse 2015, 69, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.W.; Yang, Y.R.; Sun, S.H.; Liang, K.C.; Wang, R.Y. Post ischemia intermittent hypoxia induces hippocampal neurogenesis and synaptic alterations and alleviates long-term memory impairment. J. Cereb. Blood Flow Metab. 2013, 33, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Biswal, S.; Das, D.; Barhwal, K.; Kumar, A.; Nag, T.C.; Thakur, M.K.; Hota, S.K.; Kumar, B. Epigenetic Regulation of SNAP25 Prevents Progressive Glutamate Excitotoxicty in Hypoxic CA3 Neurons. Mol. Neurobiol. 2017, 54, 6133–6147. [Google Scholar] [CrossRef]
- Stowe, A.M.; Altay, T.; Freie, A.B.; Gidday, J.M. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann. Neurol. 2011, 69, 975–985. [Google Scholar] [CrossRef]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Open Field Test for Measuring Locomotor Activity and Anxiety-Like Behavior. Methods Mol. Biol. 2019, 1916, 99–103. [Google Scholar] [CrossRef]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Alves, M.G.; Oliveira, P.J.; Carvalho, R.A. Substrate selection in hearts subjected to ischemia/reperfusion: Role of cardioplegic solutions and gender. NMR Biomed. 2011, 24, 1029–1037. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Oliveira, C. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci. Rep. 2001, 21, 789–800. [Google Scholar] [CrossRef]
- Gornall, A.G.; Bardawill, C.J.; David, M.M. Determination of serum proteins by means of the biuret reaction. J. Biol. Chem. 1949, 177, 751–766. [Google Scholar] [PubMed]
- Correia, S.C.; Santos, R.X.; Santos, M.S.; Casadesus, G.; Lamanna, J.C.; Perry, G.; Smith, M.A.; Moreira, P.I. Mitochondrial abnormalities in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Oliveira, P.J. Evaluation of Respiration with Clark-Type Electrode in Isolated Mitochondria and Permeabilized Animal Cells. Methods Mol. Biol. 2018, 1782, 7–29. [Google Scholar] [CrossRef] [PubMed]
- Coore, H.G.; Denton, R.M.; Martin, B.R.; Randle, P.J. Regulation of adipose tissue pyruvate dehydrogenase by insulin and other hormones. Biochem. J. 1971, 125, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Fuke, S.; Kubota-Sakashita, M.; Kasahara, T.; Shigeyoshi, Y.; Kato, T. Regional variation in mitochondrial DNA copy number in mouse brain. Biochim. Biophys. Acta 2011, 1807, 270–274. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | WT IHC | 3×Tg-AD | 3×Tg-AD IHC | ||
|---|---|---|---|---|---|
| Oxidative Phosphorylation System | ΔΨ (−mV) | 220.1 ± 3.16 | 222.3 ± 5.14 | 204.6 ± 3.73 | 221.4 ± 4.65 # |
| Repolarization level (−mV) | 217.0 ± 2.26 | 223.7 ± 4.66 | 205.7 ± 2.7 | 223.6 ± 4.57 ## | |
| Repolarization lag phase (min) | 0.73 ± 0.047 | 0.69 ± 0.042 | 0.85 ± 0.086 | 0.67 ± 0.045 | |
| Mitochondrial respiratory function | State 2 (nAtgO/min/mg) | 39.09 ± 2.31 | 35.22 ± 2.12 | 28.46 ± 2.56 | 43.44 ± 5.47 # |
| RCR | 3.80 ± 0.14 | 3.69 ± 0.36 | 3.26 ± 0.09 * | 3.55 ± 0.17 | |
| ADP/O (nmol ADP/ AtgO/min/mg) | 3.94 ± 0.35 | 4.06 ± 0.36 | 4.04 ± 0.48 | 4.03 ± 0.34 | |
| Oligomycin-inhibited respiration (nAtgO/min/mg) | 32.49 ± 2.32 | 40.77 ± 8.37 | 25.32 ± 1.91 | 36.20 ± 4.67 | |
| FCCP-stimulated respiration (nAtgO/min/mg)) | 101.2 ± 11.02 | 114.0± 23.75 | 69.78 ± 5.82 * | 119.9 ± 16.75 |
| Gene | Sequence (5′–3′) | AT (°C) | Amplificon Size (bp) | C |
|---|---|---|---|---|
| ND1 | Sense: GAG CCC TAC GAG CCG TTG CC | 58 | 271 | 30 |
| Antisense: GCG AATG GTC CTG CGG CGT A | ||||
| β-2MG | Sense: GCG TGG GAG GAG CAT CAG GG | 58 | 264 | 30 |
| Antisense: CTC ATC ACC ACC CCG GGG ACT |
| Antibody | Catalog Number | Supplier | Host Specie | Dilution |
|---|---|---|---|---|
| Aβ (6E10) | SIG-39300 | Covance | Mouse | 1:1000 |
| DRP1 | 611113 | BD Biosciences | Mouse | 1:1000 |
| Dynein | sc-13524 | Santa Cruz Biotechnology | Mouse | 1:500 |
| Fis1 | IMG-5113A | ImGenex | Rabbit | 1:750 |
| KIF-5B | Ab5629 | Abcam | Rabbit | 1:1000 |
| LAMP-1 | 3243 | Cell Signaling | Rabbit | 1:500 |
| LC3B (D11) | 3868 | Cell Signaling | Rabbit | 1:1000 |
| Mnf-1 | sc-50330 | Santa Cruz Biotechnology | Rabbit | 1:1000 |
| Mnf-2 | sc-100560 | Santa Cruz Biotechnology | Mouse | 1:1000 |
| OPA-1 | 612607 | BD Biosciences | Mouse | 1:1000 |
| Parkin | 2132 | Cell Signaling | Rabbit | 1:1000 |
| PINK-1 | sc-517353 | Santa Cruz Biotechnology | Mouse | 1:500 |
| PSD-95 | D27E11 | Cell Signaling | Rabbit | 1:1000 |
| pSer616-DRP1 | 3455 | Cell Signaling | Rabbit | 1:1000 |
| SNAP-25 | S5187 | Sigma Aldrich | Mouse | 1:1000 |
| Synaptotagmin-I | sc-136480 | Santa Cruz Biotechnology | Mouse | 1:1000 |
| Syntaphilin | sc-365606 | Santa Cruz Biotechnology | Mouse | 1:500 |
| Tau (AT8) | Mn1020 | Thermo Scientific | Mouse | 1:750 |
| TFAM | sc-30965 | Santa Cruz Biotechnology | Goat | 1:750 |
| TOM20 | sc-11415 | Santa Cruz Biotechnology | Rabbit | 1:1000 |
| β-Actin | A5441 | Sigma Aldrich | Mouse | 1:5000 |
| Mouse IgG alkaline phosphatase conjugate | NIF1316 | Amersham Pharmacia Biotech | Goat | 1:10,000 |
| Rabbit IgG alkaline phosphatase conjugate | NIF1317 | Amersham Pharmacia Biotech | Goat | 1:10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia, S.C.; Machado, N.J.; Alves, M.G.; Oliveira, P.F.; Moreira, P.I. Intermittent Hypoxic Conditioning Rescues Cognition and Mitochondrial Bioenergetic Profile in the Triple Transgenic Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 461. https://doi.org/10.3390/ijms22010461
Correia SC, Machado NJ, Alves MG, Oliveira PF, Moreira PI. Intermittent Hypoxic Conditioning Rescues Cognition and Mitochondrial Bioenergetic Profile in the Triple Transgenic Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(1):461. https://doi.org/10.3390/ijms22010461
Chicago/Turabian StyleCorreia, Sónia C., Nuno J. Machado, Marco G. Alves, Pedro F. Oliveira, and Paula I. Moreira. 2021. "Intermittent Hypoxic Conditioning Rescues Cognition and Mitochondrial Bioenergetic Profile in the Triple Transgenic Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 1: 461. https://doi.org/10.3390/ijms22010461
APA StyleCorreia, S. C., Machado, N. J., Alves, M. G., Oliveira, P. F., & Moreira, P. I. (2021). Intermittent Hypoxic Conditioning Rescues Cognition and Mitochondrial Bioenergetic Profile in the Triple Transgenic Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 22(1), 461. https://doi.org/10.3390/ijms22010461