Involvement of the ERK/HIF-1α/EMT Pathway in XCL1-Induced Migration of MDA-MB-231 and SK-BR-3 Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
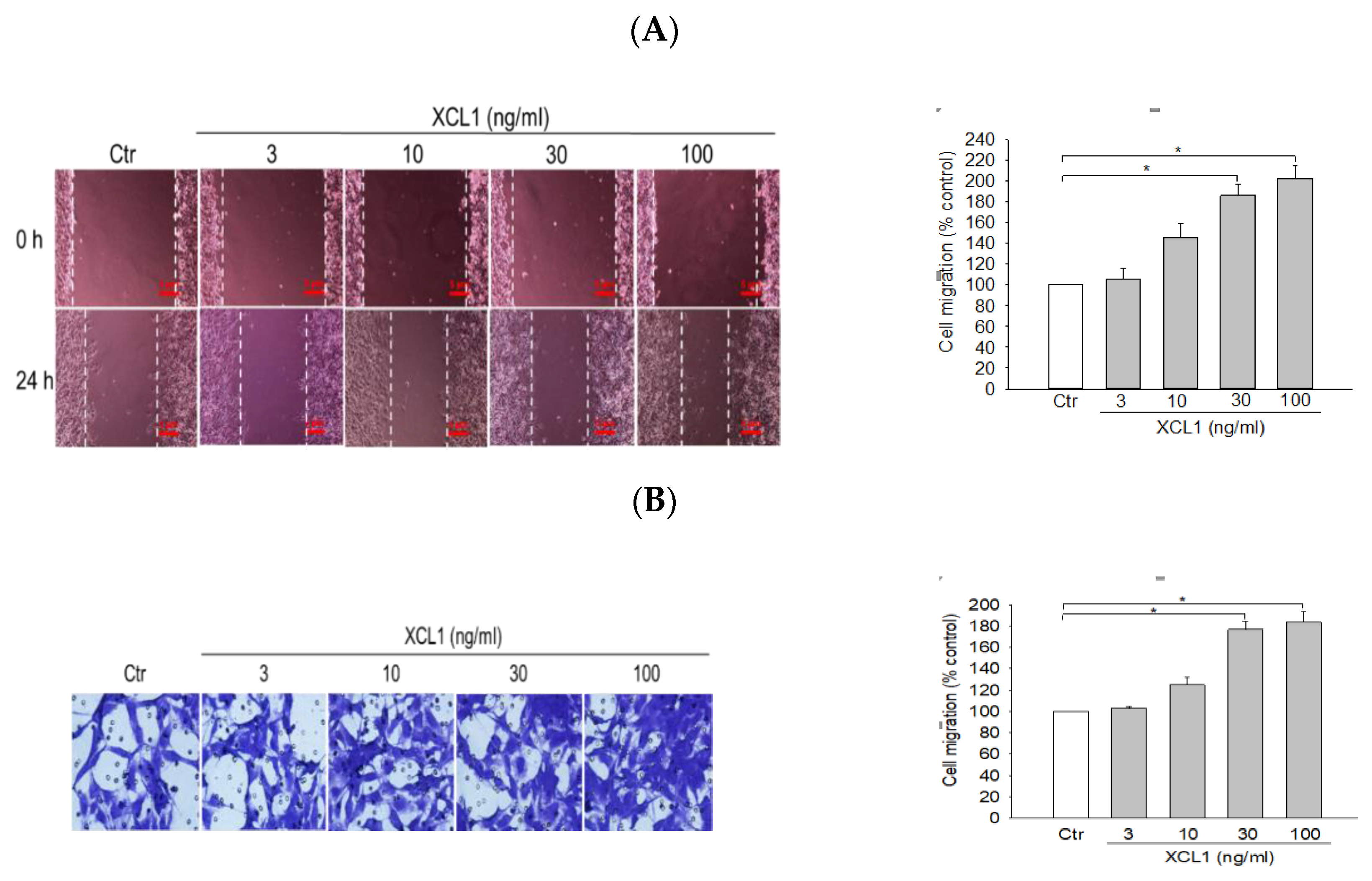
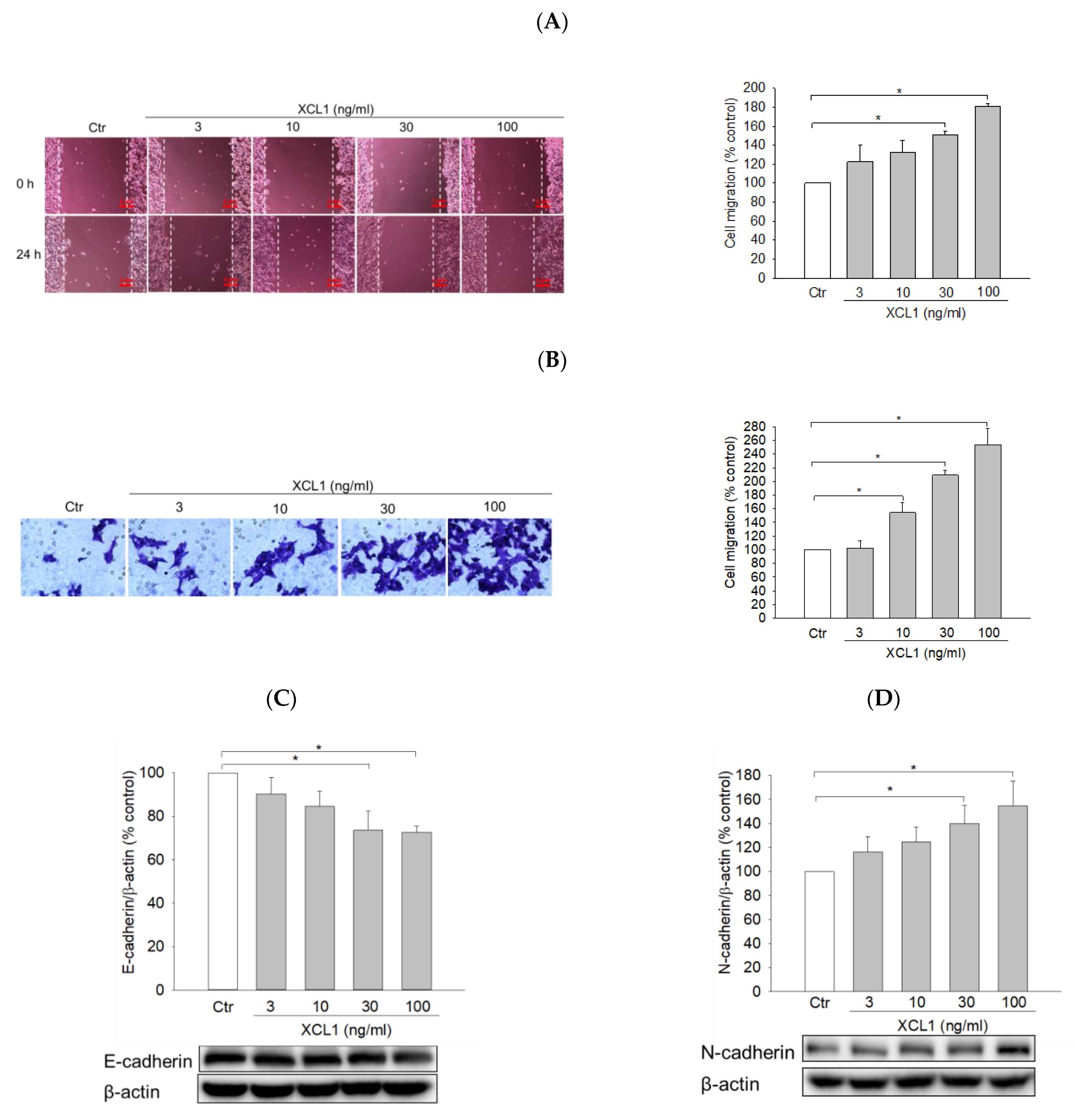
2.1. Effect of XCL1 on MDA-MB-231 Cell Migration
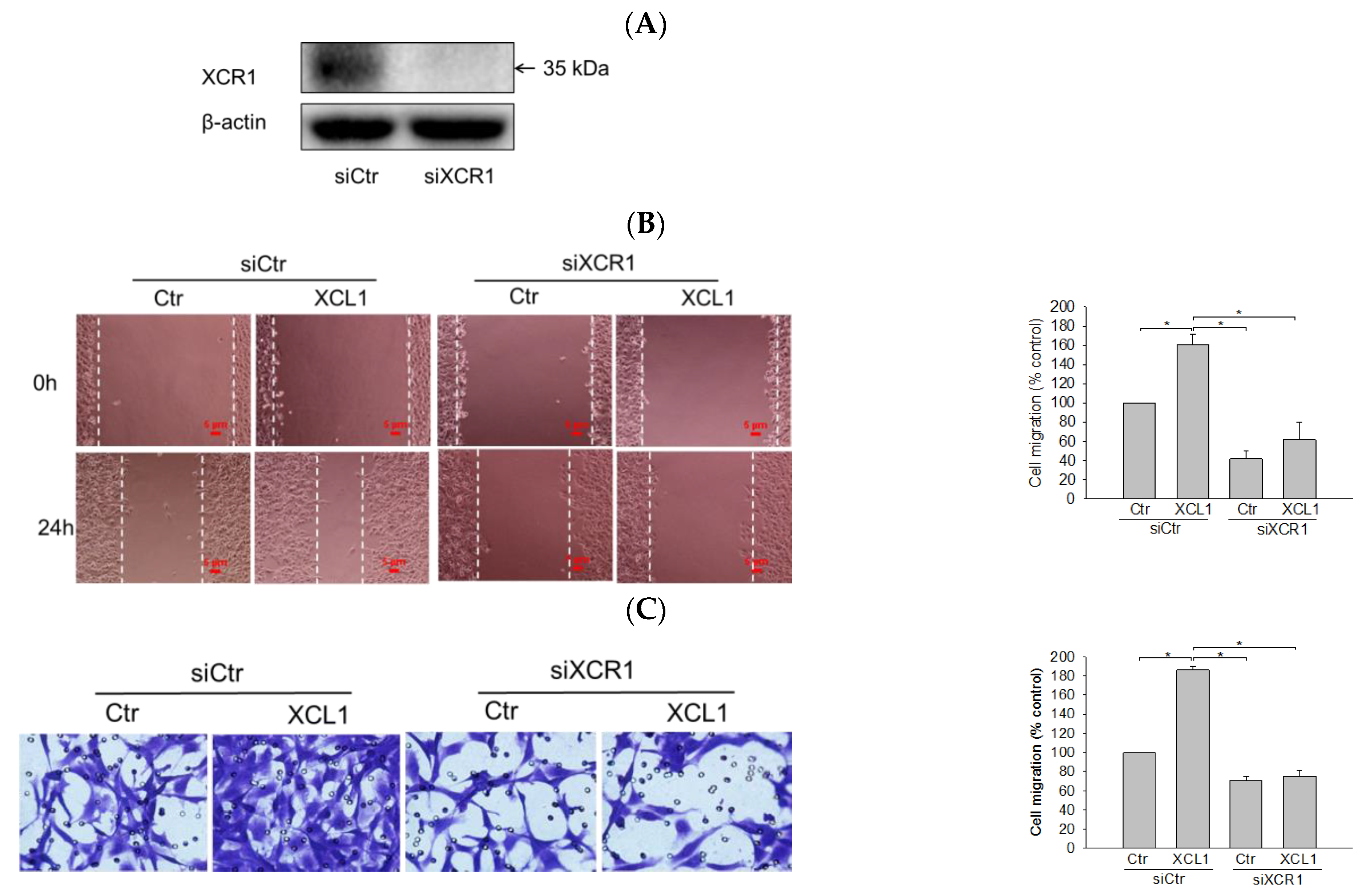
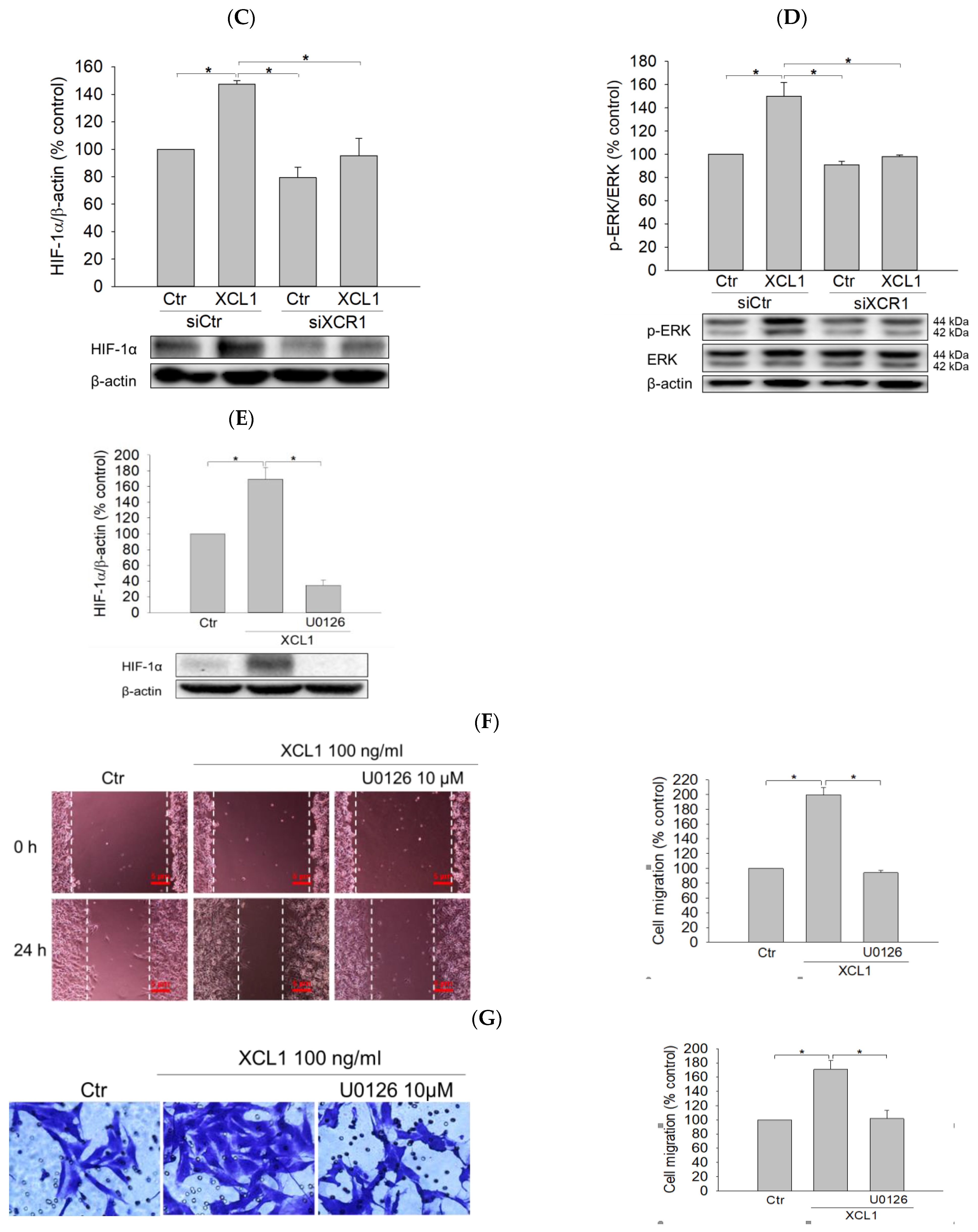
2.2. Effect of XCR1 Knockdown on XCL1-Promoted MDA-MB-231 Cell Migration
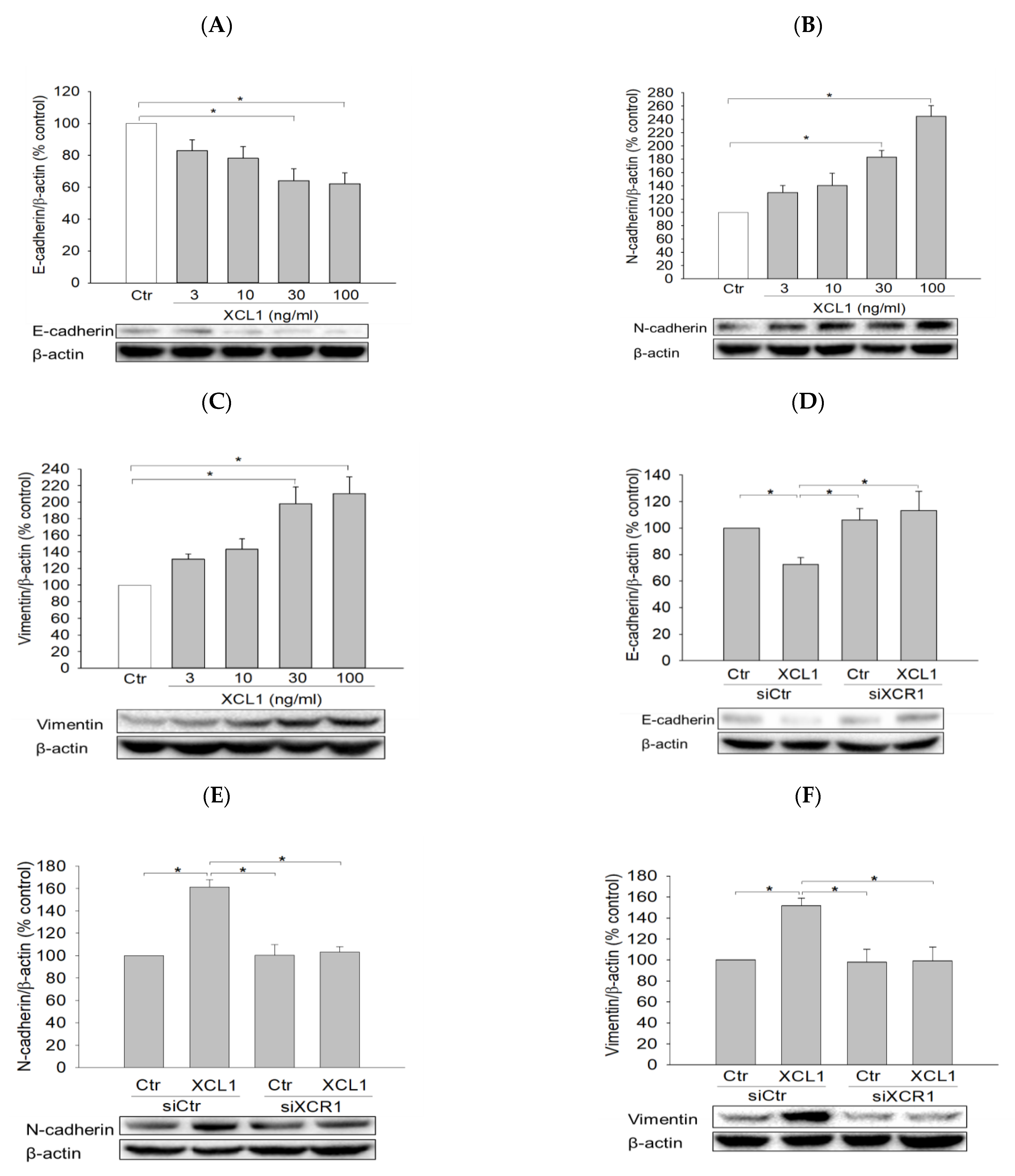
2.3. Effects of XCL1 on EMT in MDA-MB-231 Cells
2.3.1. Effects of XCL1 on the Expression of EMT Markers in MDA-MB-231 Cells
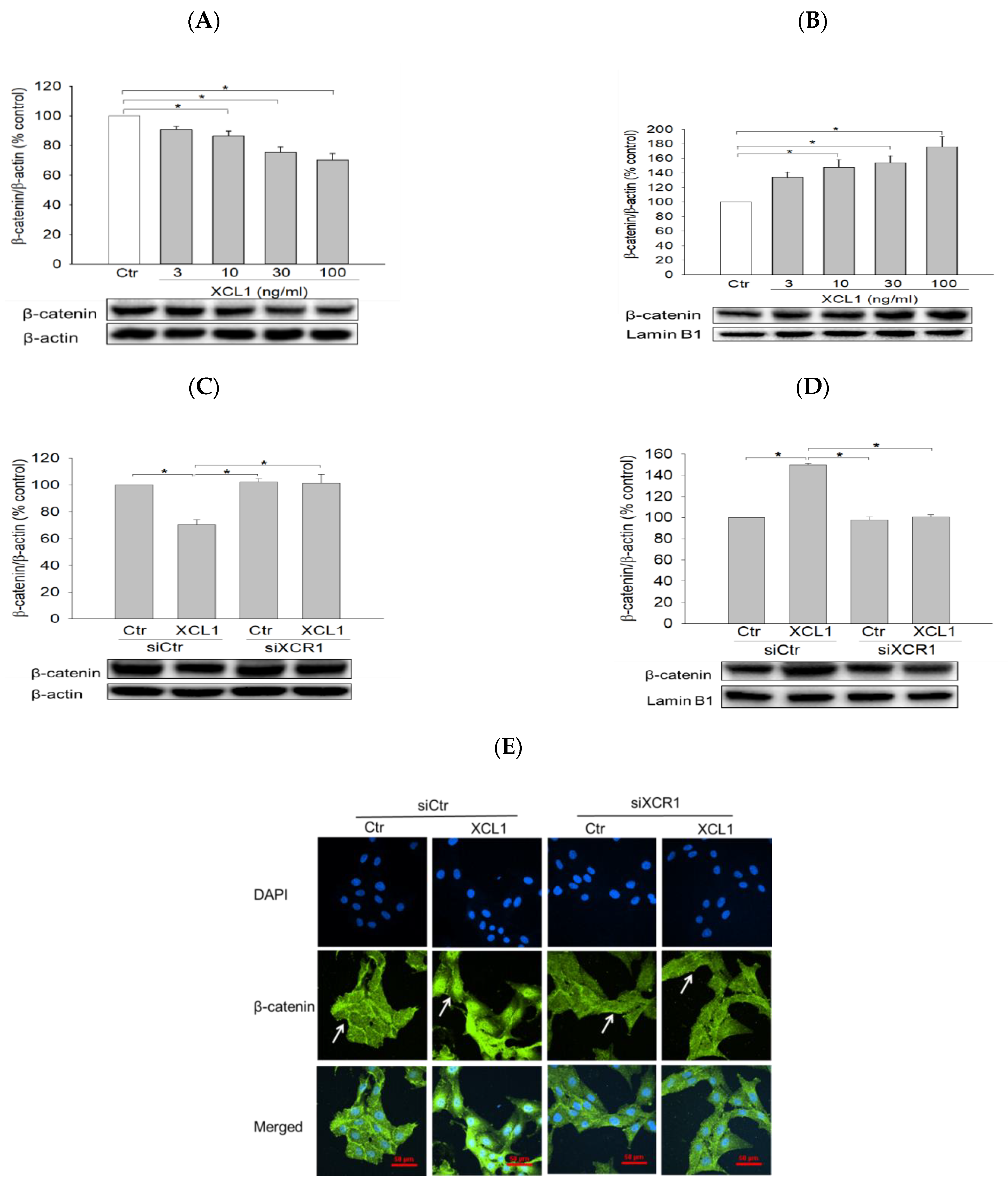
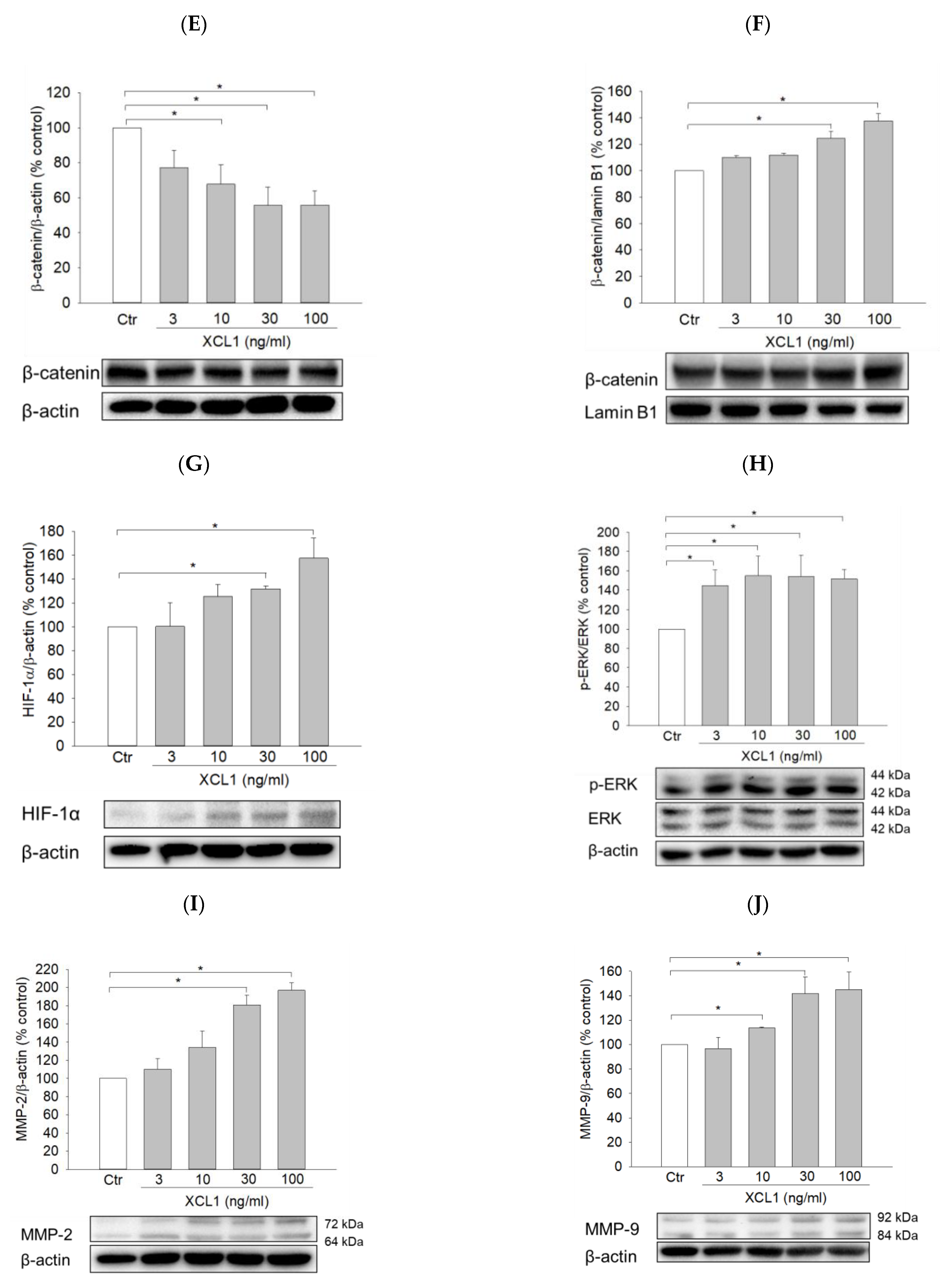
2.3.2. Effect of XCL1 on β-Catenin Nuclear Translocation in MDA-MB-231 Cells
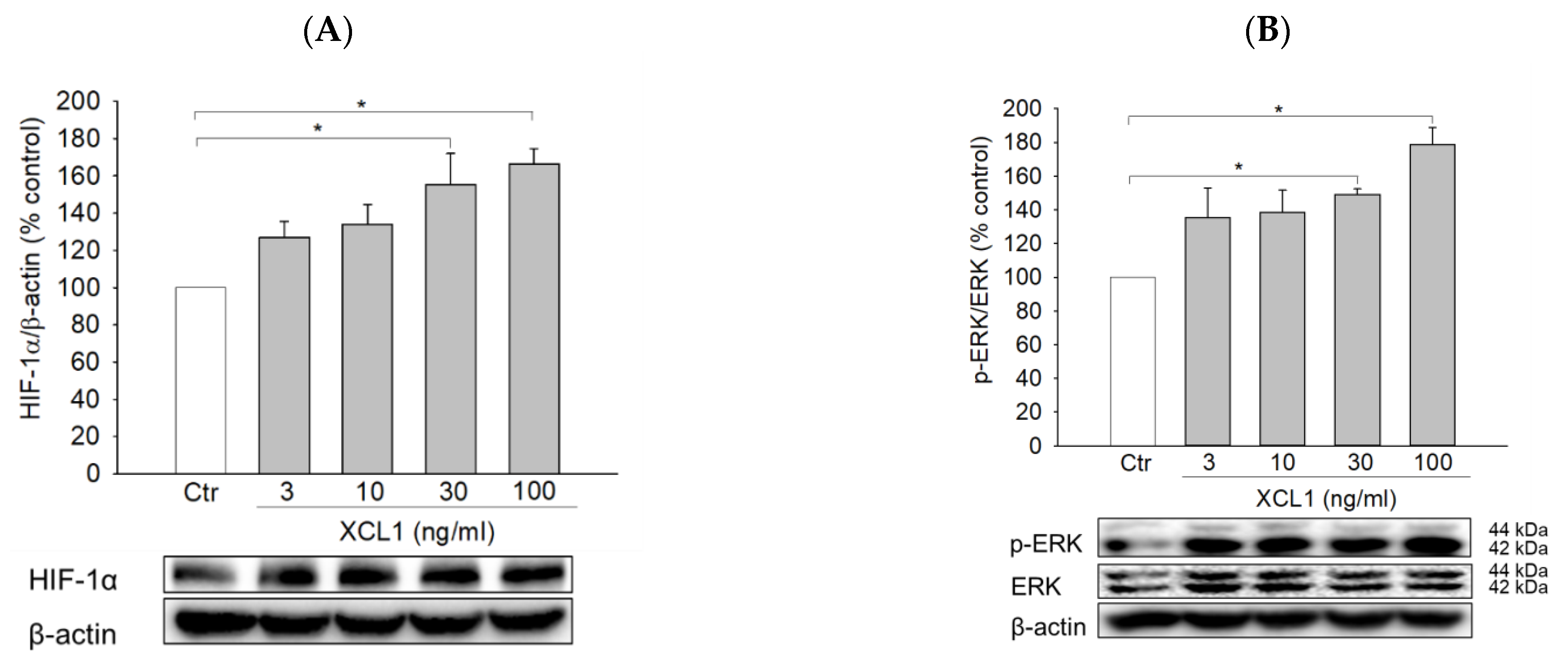
2.4. Effects of XCL1 on ERK/HIF-1α Signaling Pathway in MDA-MB-231 Cells
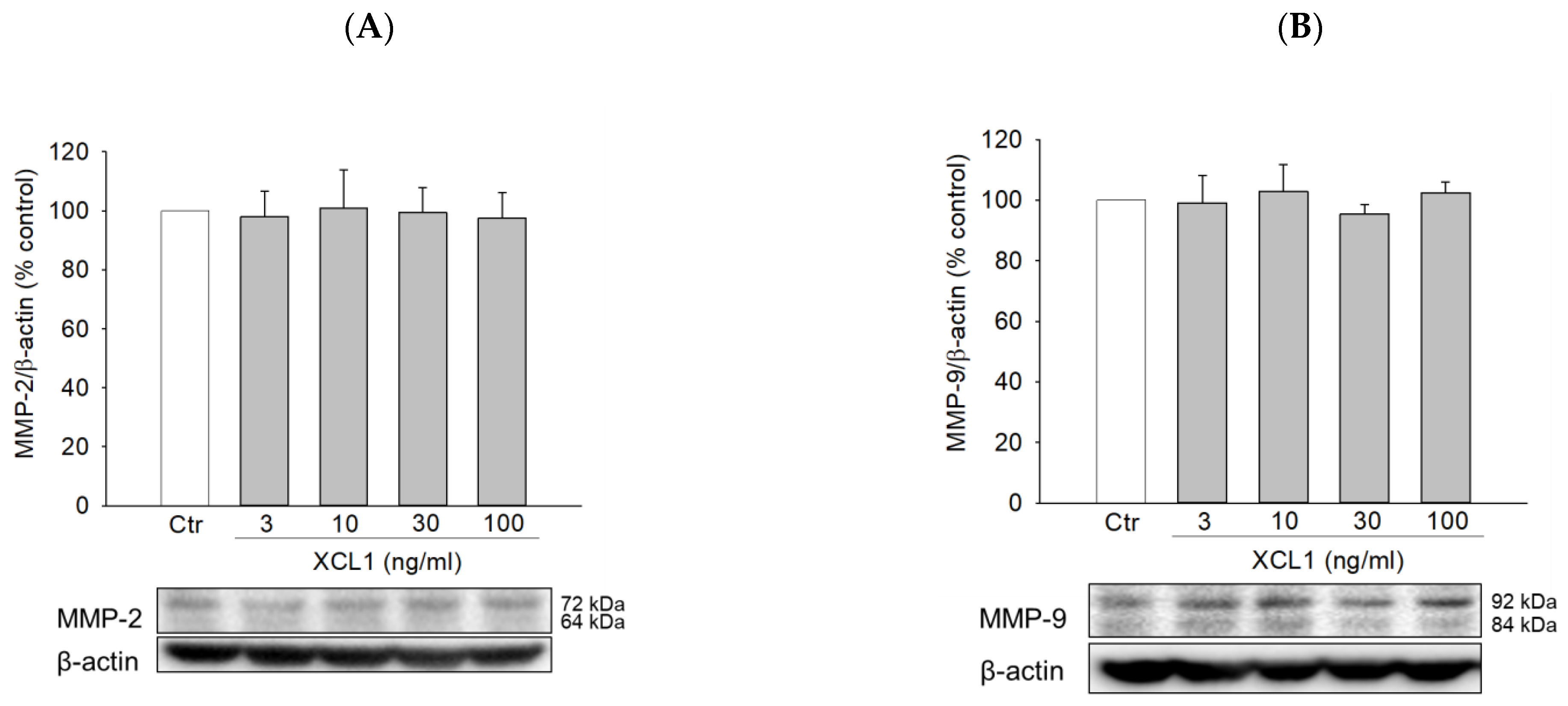
2.5. Effects of XCL1 on the Expression of Matrix Metalloproteinases-2 and -9 in MDA-MB-231 Cells
2.6. Effects of XCL1 on Cell Migration and the Intracellular Signaling Pathway in SK-BR-3 Breast Cancer Cells
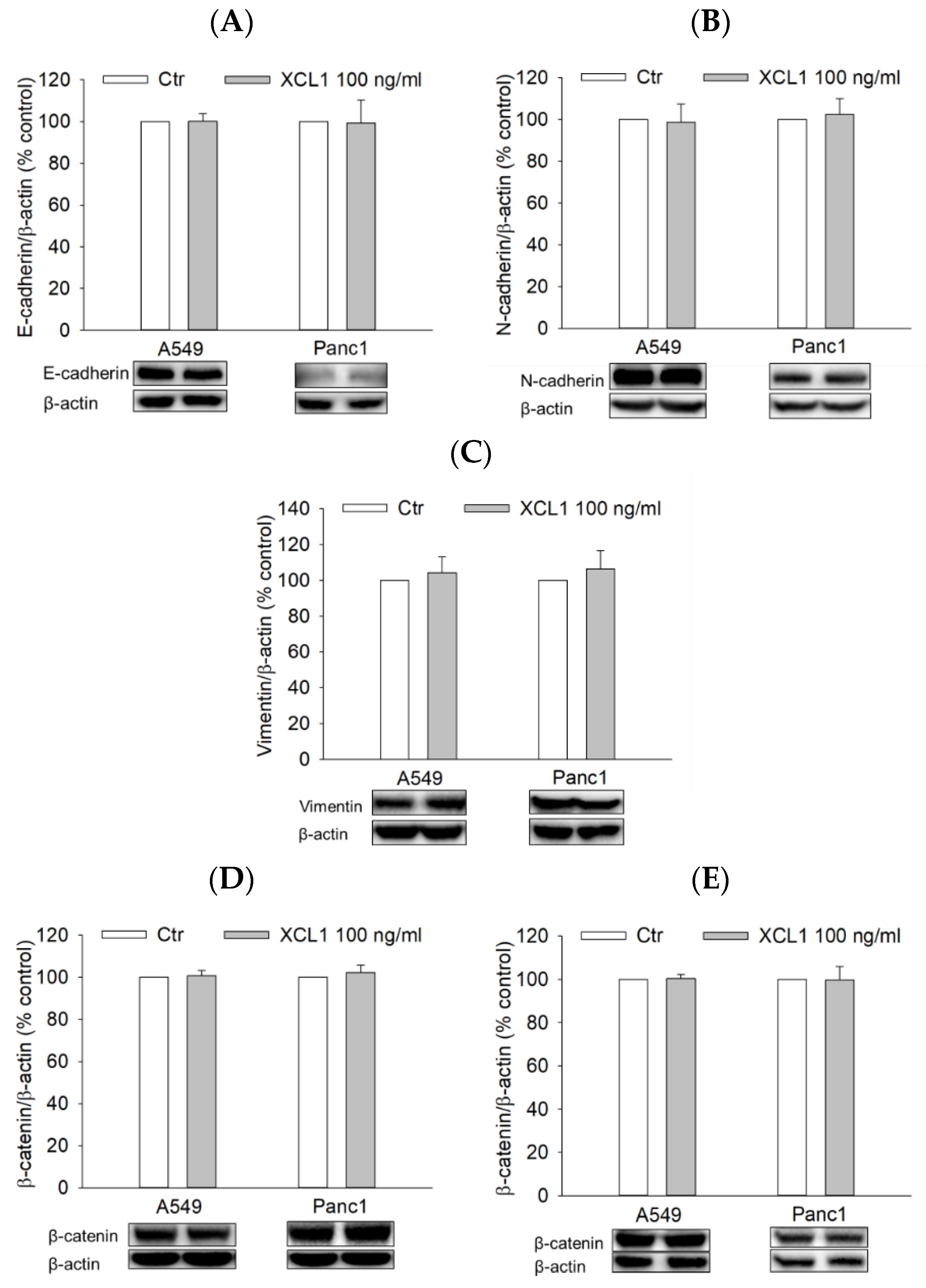
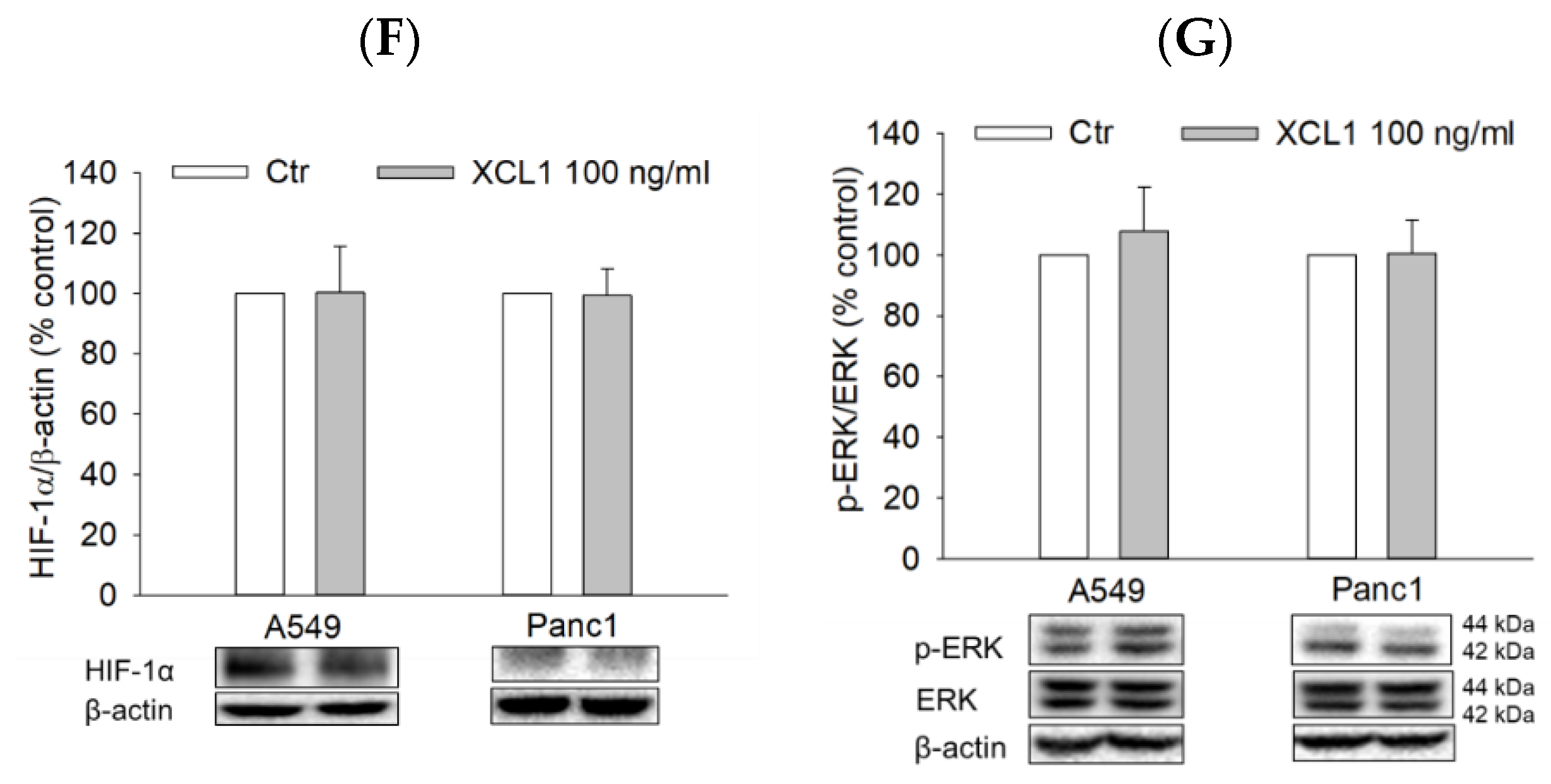
2.7. Effects of XCL1 on the ERK/HIF-1α/EMT Signaling Pathway in A549 NSCLC and Panc1 Pancreatic Cells
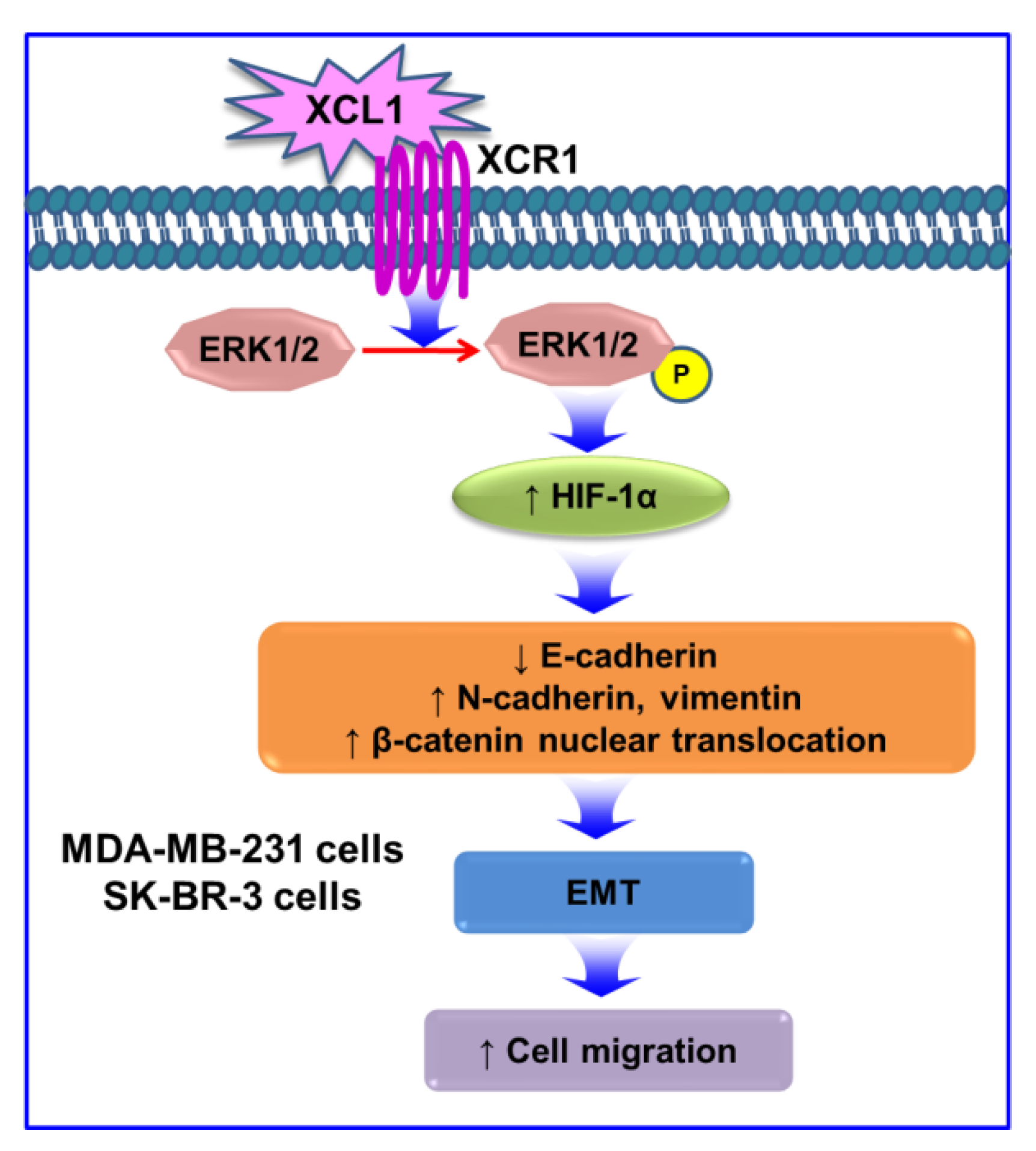
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Line and Culture
4.3. Transfection
4.4. Cell Migration Assays
4.4.1. Wound-Healing Assay
4.4.2. Transwell Migration Assay
4.5. Western Blotting
4.6. Immunocytochemistry
4.7. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| XCL1 | X-C motif chemokine ligand 1 |
| XCR1 | X-C motif chemokine receptor 1 |
| EOC | Epithelial ovarian carcinoma |
| NSCLC | Non-small cell lung carcinoma |
| OSCC | Oral squamous cell carcinoma |
| MAPKs | Mitogen-activated protein kinases |
| TNBC | Triple-negative breast cancers |
| ERK | Extracellular signal-regulated kinase |
| HIF | Hypoxia-inducible factor |
| EMT | Epithelial–mesenchymal transition |
| siRNA | Small interfering RNA |
| siCtr | Small interfering RNA targeting control |
| siXCR1 | Small interfering RNA targeting XCR1 |
| MMP | Matrix metalloproteinase |
| JAK2/STAT3 | Janus tyrosine kinase 2-signal transducer and activator of transcription 3 |
| MEK | Mitogen-activated protein kinase kinase |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yang, Z.; Lu, W.; Chen, Z.; Chen, L.; Han, S.; Wu, X.; Cai, T.; Cai, Y. Chemokines and chemokine receptors: A new strategy for breast cancer therapy. Cancer Med. 2020, 9, 3786–3799. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Cho, J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch. Pharmacal Res. 2013, 36, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Yoshie, O. The Chemokine Superfamily Revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Rooper, L.; Xie, J.; Rayahin, J.; Burdette, J.E.; Kajdacsy-Balla, A.A.; Barbolina, M.V. The Lymphotactin Receptor Is Expressed in Epithelial Ovarian Carcinoma and Contributes to Cell Migration and Proliferation. Mol. Cancer Res. 2012, 10, 1419–1429. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Han, S.; Wu, Z.; Han, Z.; Yan, W.; Liu, T.; Wei, H.; Song, D.; Zhou, W.; Yang, X.; et al. XCR1 promotes cell growth and migration and is correlated with bone metastasis in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2015, 464, 635–641. [Google Scholar] [CrossRef]
- Khurram, S.A.; A Whawell, S.; Bingle, L.; Murdoch, C.; McCabe, B.M.; Farthing, P.M. Functional expression of the chemokine receptor XCR1 on oral epithelial cells. J. Pathol. 2010, 221, 153–163. [Google Scholar] [CrossRef]
- Yang, X.L.; Qi, L.G.; Lin, F.J.; Ou, Z.L. The role of the chemokine receptor XCR1 in breast cancer cells. Breast Cancer Targets Ther. 2017, 9, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Papa, A.; Caruso, D.; Tomao, S.; Rossi, L.; Zaccarelli, E.; Tomao, F. Triple-negative breast cancer: Investigating potential molecular therapeutic target. Expert Opin. Ther. Targets 2014, 19, 55–75. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Hanna, W.M.; Trudeau, M.; Rawlinson, E.; Sun, P.; Narod, S.A. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res. Treat. 2009, 115, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, W.; Wang, L.; Zhu, T.; Zhong, J.; Xie, N. Hypoxia-inducible factor 1 mediates intermittent hypoxia-induced migration of human breast cancer MDA-MB-231 cells. Oncol. Lett. 2017, 14, 7715–7722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xue, D.; Xue, M.; Zhao, J.; Liang, H.; Liu, Y.; Sun, T. Fucoidan inhibits epithelial-to-mesenchymal transition via regulation of the HIF-1α pathway in mammary cancer cells under hypoxia. Oncol. Lett. 2019, 18, 330–338. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, S.; Song, C.; Hu, Z. Paeoniflorin prevents hypoxia-induced epithelial–mesenchymal transition in human breast cancer cells. OncoTargets Ther. 2016, 9, 2511–2518. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Sun, P.; Zhang, Y.; You, X.; Li, P.; Wang, T. Estrogen stimulated migration and invasion of estrogen receptor-negative breast cancer cells involves an ezrin-dependent crosstalk between G protein-coupled receptor 30 and estrogen receptor beta signaling. Steroids 2016, 111, 113–120. [Google Scholar] [CrossRef]
- Park, S.; Koo, J.S.; Kim, M.S.; Park, H.S.; Lee, J.S.; Lee, J.S.; Kim, S.I.; Park, B.-W. Characteristics and outcomes according to molecular subtypes of breast cancer as classified by a panel of four biomarkers using immunohistochemistry. Breast 2012, 21, 50–57. [Google Scholar] [CrossRef]
- Tomaskovic-Crook, E.; Thompson, E.W.; Thiery, J.P. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res. 2009, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Li, J.-Z.; Lu, Y.; Zhang, C.-Y.; Li, Q.; Mao, J.; Li, L. The mechanism between epithelial mesenchymal transition in breast cancer and hypoxia microenvironment. Biomed. Pharmacother. 2016, 80, 393–405. [Google Scholar] [CrossRef]
- O’Toole, S.A.; Beith, J.M.; Millar, E.; West, R.; McLean, A.; Cazet, A.; Swarbrick, A.; Oakes, S.R. Therapeutic targets in triple negative breast cancer. J. Clin. Pathol. 2013, 66, 530–542. [Google Scholar] [CrossRef]
- Curtin, J.C.; Lorenzi, M.V. Drug Discovery Approaches to Target Wnt Signaling in Cancer Stem Cells. Oncotarget 2010, 1, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Prosperi, J.R.; Goss, K.H. A Wnt-ow of opportunity: Targeting the Wnt/beta-catenin pathway in breast cancer. Curr. Drug Targets 2010, 11, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Lin, Y.C.; Rajendran, P.; Thigarajan, V.; Mathew, D.C.; Lin, K.Y.; Way, T.D.; Liao, J.W.; Yang, H.L. Antrodia salmonea suppresses invasion and metastasis in triple-negative breast cancer cells by reversing EMT through the NF-κB and Wnt/β-catenin signaling pathway. Food Chem. Toxicol. 2019, 124, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, Y.-L.; Liang, X.-H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Cheng, C.-W.; Chen, P.-M.; Hsieh, Y.-H.; Weng, C.-C.; Chang, C.-W.; Yao, C.-C.; Hu, L.-Y.; Wu, P.-E.; Shen, C.-Y. Foxo3a-mediated overexpression of microRNA-622 suppresses tumor metastasis by repressing hypoxia-inducible factor-1α in erk-responsive lung cancer. Oncotarget 2015, 6, 44222–44238. [Google Scholar] [CrossRef] [Green Version]
- Whyte, J.; Bergin, O.; Bianchi, A.; McNally, S.; Martin, F. Key signalling nodes in mammary gland development and cancer. Mitogen-activated protein kinase signalling in experimental models of breast cancer progression and in mammary gland development. Breast Cancer Res. 2009, 11, 209. [Google Scholar] [CrossRef]
- Hulit, J.; Suyama, K.; Chung, S.; Keren, R.; Agiostratidou, G.; Shan, W.; Dong, X.; Williams, T.M.; Lisanti, M.P.; Knudsen, K.; et al. N-Cadherin Signaling Potentiates Mammary Tumor Metastasis via Enhanced Extracellular Signal-Regulated Kinase Activation. Cancer Res. 2007, 67, 3106–3116. [Google Scholar] [CrossRef] [Green Version]
- Hatzidaki, E.; Parsonidis, P.; Apostolou, P.; Daikopoulou, V.; Papasotiriou, I. Novel small molecule decreases cell proliferation, migration, clone formation, and gene expression through ERK inhibition in MCF-7 and MDA-MB-231 breast cancer cell lines. Anti-Cancer Drugs 2019, 30, 618–627. [Google Scholar] [CrossRef]
- Teng, Y.; Guo, B.; Mu, X.; Liu, S. KIF26B promotes cell proliferation and migration through the FGF2/ERK signaling pathway in breast cancer. Biomed. Pharmacother. 2018, 108, 766–773. [Google Scholar] [CrossRef]
- He, J.; Xie, N.; Yang, J.; Guan, H.; Chen, W.; Wu, H.; Yuan, Z.; Wang, K.; Li, G.; Sun, J.; et al. siRNA-Mediated Suppression of Synuclein γ Inhibits MDA-MB-231 Cell Migration and Proliferation by Downregulating the Phosphorylation of AKT and ERK. J. Breast Cancer 2014, 17, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zardavas, D.; Baselga, J.; Piccart-Gebhart, M. Emerging targeted agents in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2013, 10, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Bauvois, B. New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: Outside-in signaling and relationship to tumor progression. Biochim. Biophys. Acta (BBA)–Bioenergy 2012, 1825, 29–36. [Google Scholar] [CrossRef]
- John, A.; Tuszynski, G. The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol. Oncol. Res. 2001, 7, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Alaseem, A.; Alhazzani, K.; Dondapati, P.; Alobid, S.; Bishayee, A.; Rathinavelu, A. Matrix Metalloproteinases: A challenging paradigm of cancer management. Semin. Cancer Biol. 2019, 56, 100–115. [Google Scholar] [CrossRef]
- Ko, J.; Yang, M.H.; Baek, S.H.; Nam, D.; Jung, S.H.; Ahn, K.S. Theacrine attenuates epithelial mesenchymal transition in human breast cancer MDA-MB-231 cells. Phytotherapy Res. 2019, 33, 1934–1942. [Google Scholar] [CrossRef]
- Yu, J.-K.; Yue, C.-H.; Pan, Y.-R.; Chiu, Y.-W.; Liu, J.-Y.; Lin, K.-I.; Lee, C.-J. Isochlorogenic Acid C Reverses Epithelial-Mesenchymal Transition via Down-regulation of EGFR Pathway in MDA-MB-231 cells. Anticancer. Res. 2018, 38, 2127–2135. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta. Anat 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Araki, K.; Shimura, T.; Suzuki, H.; Tsutsumi, S.; Wada, W.; Yajima, T.; Kobayahi, T.; Kubo, N.; Kuwano, H. E/N-cadherin switch mediates cancer progression via TGF-β-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br. J. Cancer 2011, 105, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowin, P.; Rowlands, T.M.; Hatsell, S.J. Cadherins and catenins in breast cancer. Curr. Opin. Cell Biol. 2005, 17, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.L.; Kim, A.C.; Hens, J. The role and function of cadherins in the mammary gland. Breast Cancer Res. 2012, 14, 203. [Google Scholar] [CrossRef] [Green Version]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [Green Version]
- González-Moles, M.; Ruiz-Avila, I.; Gil-Montoya, J.; Plaza-Campillo, J.; Scully, C. β-Catenin in oral cancer: An update on current knowledge. Oral Oncol. 2014, 50, 818–824. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Humphries, B.; Brien, R.; Gibbons, A.E.; Chen, Y.-T.; Qyli, T.; Haley, H.R.; Pirone, M.E.; Chiang, B.; Xiao, A.; et al. Functional Isolation of Tumor-Initiating Cells using Microfluidic-Based Migration Identifies Phosphatidylserine Decarboxylase as a Key Regulator. Sci. Rep. 2018, 8, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Wong, C.C.-L.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Khoo, U.-S.; Ng, I.O.-L.; Wirtz, D.; et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.-L.; Zhang, H.; Gilkes, D.M.; Chen, J.; Wei, H.; Chaturvedi, P.; Hubbi, M.E.; Semenza, G.L. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J. Mol. Med. 2012, 90, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Bos, R.; Van Der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; Van Diest, P.J.; Van Der Wall, E. Levels of hypoxia-inducible factor-1? independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 2003, 97, 1573–1581. [Google Scholar] [CrossRef]
- Dales, J.-P.; Garcia, S.; Meunier-Carpentier, S.; Andrac-Meyer, L.; Haddad, O.; Lavaut, M.-N.; Allasia, C.; Bonnier, P.; Charpin, C. Overexpression of hypoxia-inducible factor HIF-1α predicts early relapse in breast cancer: Retrospective study in a series of 745 patients. Int. J. Cancer 2005, 116, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Sang, N.; Stiehl, D.P.; Bohensky, J.; Leshchinsky, I.; Srinivas, V.; Caro, J. MAPK Signaling Up-regulates the Activity of Hypoxia-inducible Factors by Its Effects on p300. J. Biol. Chem. 2003, 278, 14013–14019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like Growth Factor 1 Induces Hypoxia-inducible Factor 1-mediated Vascular Endothelial Growth Factor Expression, Which is Dependent on MAP Kinase and Phosphatidylinositol 3-Kinase Signaling in Colon Cancer Cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [Green Version]
- Agani, F. Oxygen-independent Regulation of HIF-1: Novel Involvement of PI3K/ AKT/mTOR Pathway in Cancer. Curr. Cancer Drug Targets 2013, 13, 245–251. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Sahoo, S.; Brien, R.; Jung, S.; Humphries, B.; Lee, W.; Cheng, Y.-H.; Zhang, Z.; Luker, K.E.; Wicha, M.S.; et al. Single-cell RNA-sequencing of migratory breast cancer cells: Discovering genes associated with cancer metastasis. Analyst 2019, 144, 7296–7309. [Google Scholar] [CrossRef]
- Li, Q.; Mattingly, R.R. Restoration of E-cadherin Cell-Cell Junctions Requires Both Expression of E-cadherin and Suppression of ERK MAP Kinase Activation in Ras-Transformed Breast Epithelial Cells. Neoplasia 2008, 10, 1444–1458. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grünert, S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M.; Liu, P.; Hayashi, N.; Lluch, A.; Ferrer-Lozano, J.; Hortobágyi, G.N. High ERK Protein Expression Levels Correlate with Shorter Survival in Triple-Negative Breast Cancer Patients. Oncologist 2012, 17, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.L.; Hou, M.F.; Kuo, P.L.; Huang, Y.F.; Tsai, E.M. Breast tumor-associated osteoblast-derived CXCL5 increases cancer progression by ERK/MSK1/Elk-1/snail signaling pathway. Oncogene 2013, 32, 4436–4447. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-M.; Tian, F.-J.; Zeng, W.-H.; Ma, X.-L.; Ren, J.-B.; Lin, Y. XCL1-XCR1 pathway promotes trophoblast invasion at maternal-fetal interface by inducing MMP-2/MMP-9 activity. Am. J. Reprod. Immunol. 2018, 80, e12990. [Google Scholar] [CrossRef]
- Ren, W.; Liu, Y.; Wan, S.; Fei, C.; Wang, W.; Chen, Y.; Zhang, Z.; Wang, T.; Wang, J.; Zhou, L.; et al. BMP9 Inhibits Proliferation and Metastasis of HER2-Positive SK-BR-3 Breast Cancer Cells through ERK1/2 and PI3K/AKT Pathways. PLoS ONE 2014, 9, e96816. [Google Scholar] [CrossRef] [PubMed]
- Cairns, C.M.; Gordon, J.R.; Li, F.; Baca-Estrada, M.E.; Moyana, T.; Xiang, J. Lymphotactin Expression by Engineered Myeloma Cells Drives Tumor Regression: Mediation by CD4+and CD8+T Cells and Neutrophils Expressing XCR1 Receptor. J. Immunol. 2001, 167, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, A.J.; Saylor, V.; Figueroa, D.; Mizoue, L.; Xu, Y.; Menon, S.; Abrams, J.; Handel, T.; Zlotnik, A. Lymphotactin is produced by NK cells and attracts both NK cells and T cells in vivo. J. Immunol. 1997, 158, 1533–1540. [Google Scholar] [PubMed]
- Xia, D.J.; Zhang, W.J.; Zheng, S.; Wang, J.; Pan, J.P.; Wang, Q.; Zhang, L.H.; Hamada, H.; Cao, X. Lymphotactin cotransfection enhances the therapeutic efficacy of dendritic cells genetically modified with melanoma antigen gp100. Gene Ther. 2002, 9, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Aguirre, C.H.; Flores-Jiménez, J.A.; Alatorre-Ricardo, J.; Cantú-Rodríguez, O.G.; Rosas-Taraco, A.; Salazar-Riojas, R.; Jaime-Pérez, J.C.; Sánchez-Cárdenas, M.; López-Silva, L.; Martínez-Castilla, A.M. The prognostic significance of serum XCL1 concentration in patients with acute lymphoblastic leukemia: A pilot study. Ann. Hematol. 2017, 96, 2015–2024. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, Y.; Shi, M.; Zhang, Q.; Wang, Q.; Chen, X. Deferoxamine promotes MDA-MB-231 cell migration and invasion through increased ROS-dependent HIF-1α accumulation. Cell Physiol. Biochem. 2014, 33, 1036–1046. [Google Scholar] [CrossRef]
- Dastjerdi, M.N.; Hashemibeni, B.; Kazemi, M.; Salehi, M.; Babazadeh, Z. Comparison of the anti-cancer effect of Disulfiram and 5-Aza-CdR on pancreatic cancer cell line PANC-1. Adv. Biomed. Res. 2014, 3, 156. [Google Scholar] [CrossRef]
- Tan, W.-J.; Tan, Q.-Y.; Wang, T.; Lian, M.; Zhang, L.; Cheng, Z.-S. Calpain 1 regulates TGF-β1-induced epithelial-mesenchymal transition in human lung epithelial cells via PI3K/Akt signaling pathway. Am. J. Transl. Res. 2017, 9, 1402–1409. [Google Scholar]
- Do, H.T.T.; Bui, B.P.; Sim, S.; Jung, J.K.; Lee, H.; Cho, J. Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. Int. J. Mol. Sci. 2020, 21, 2319. [Google Scholar] [CrossRef] [Green Version]
- Bui, B.P.; Oh, Y.; Lee, H.; Cho, J. Inhibition of inflammatory mediators and cell migration by 1,2,3,4-tetrahydroquinoline derivatives in LPS-stimulated BV2 microglial cells via suppression of NF-κB and JNK pathway. Int. Immunopharmacol. 2020, 80, 106231. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Bose, S.; Kim, Y.-M.; Chin, Y.-W.; Cho, J. The ethyl acetate fraction from Physalis alkekengi inhibits LPS-induced pro-inflammatory mediators in BV2 cells and inflammatory pain in mice. J. Ethnopharmacol. 2016, 181, 26–36. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, H.T.T.; Cho, J. Involvement of the ERK/HIF-1α/EMT Pathway in XCL1-Induced Migration of MDA-MB-231 and SK-BR-3 Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 89. https://doi.org/10.3390/ijms22010089
Do HTT, Cho J. Involvement of the ERK/HIF-1α/EMT Pathway in XCL1-Induced Migration of MDA-MB-231 and SK-BR-3 Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(1):89. https://doi.org/10.3390/ijms22010089
Chicago/Turabian StyleDo, Ha Thi Thu, and Jungsook Cho. 2021. "Involvement of the ERK/HIF-1α/EMT Pathway in XCL1-Induced Migration of MDA-MB-231 and SK-BR-3 Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 1: 89. https://doi.org/10.3390/ijms22010089
APA StyleDo, H. T. T., & Cho, J. (2021). Involvement of the ERK/HIF-1α/EMT Pathway in XCL1-Induced Migration of MDA-MB-231 and SK-BR-3 Breast Cancer Cells. International Journal of Molecular Sciences, 22(1), 89. https://doi.org/10.3390/ijms22010089