
Targeting of GLUT5 for Transporter-Mediated Drug-Delivery Is Contingent upon Substrate Hydrophilicity


Abstract
:1. Introduction
2. Results
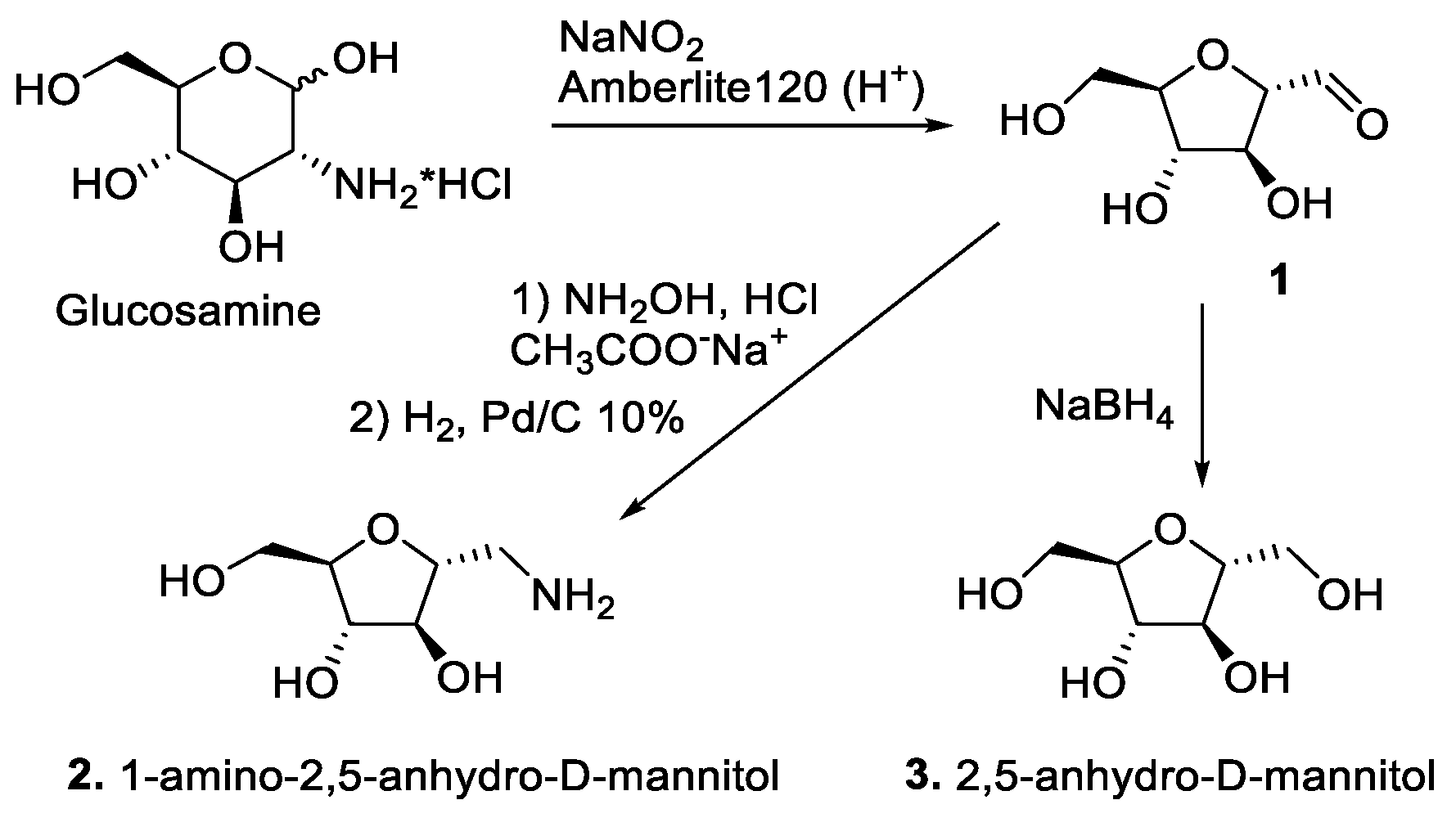
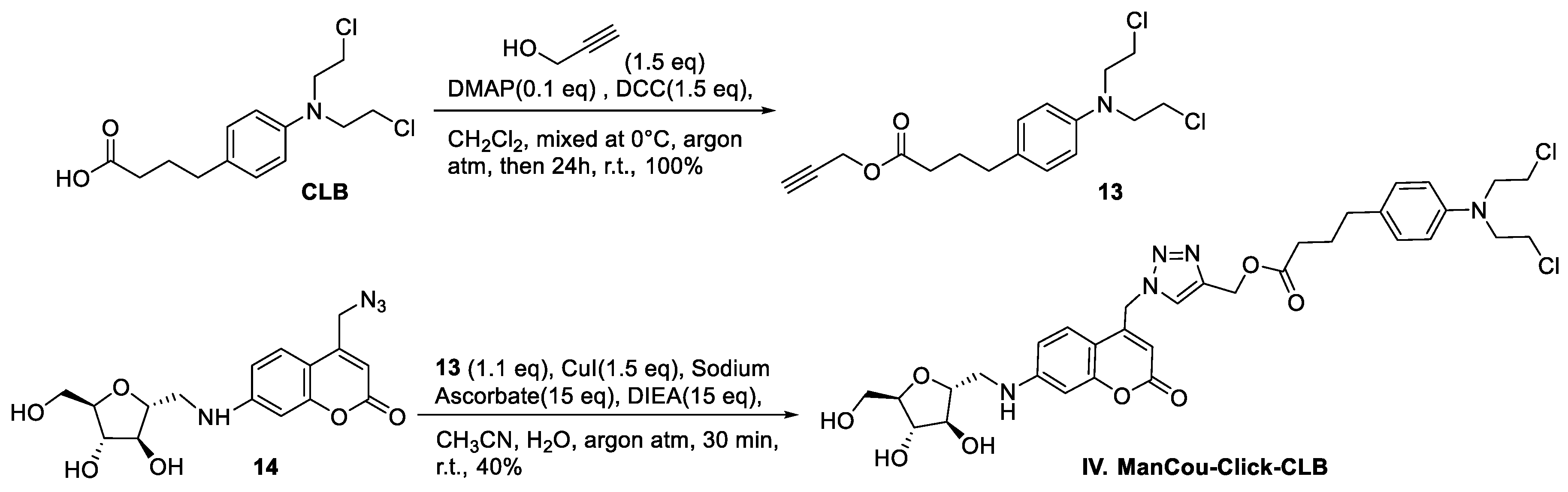
2.1. Synthesis of CLB Conjugates
2.2. Assessing GLUT5 Levels in Cells
2.3. Conjugates I–IV Show GLUT5-Dependent Uptake during Short Incubations
2.4. CLB Conjugates Show Structure-Activity Relationship
2.5. Analysis of CLB Conjugate Hydrophobicity
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Organic Synthesis
4.3. Fluorescence Analysis
4.4. Competitive Inhibition Assay
4.5. Cell Viability Assay
4.6. Determination of Water Solubility via Octanol–Water Buffer Partitioning
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Et Biophys. Acta (BBA) Bioenerg. 2011, 1807, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef] [Green Version]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Tanasova, M.; Fedie, J.R. Molecular Tools for Facilitative Carbohydrate Transporters (Gluts). ChemBioChem 2017, 18, 1774–1788. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, C.; Long, W. Structure of, and functional insight into the GLUT family of membrane transporters. Cell Health Cytoskelet. 2015, 7, 167–183. [Google Scholar] [CrossRef] [Green Version]
- Medina, R.A.; Owen, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef]
- Larson, S.M. Positron Emission Tomography-Based Molecular Imaging in Human Cancer: Exploring the Link between Hypoxia and Accelerated Glucose Metabolism. Clin. Cancer Res. 2004, 10, 2203–2204. [Google Scholar] [CrossRef] [Green Version]
- Machtay, M.; Duan, F.; Siegel, B.A.; Snyder, B.S.; Gorelick, J.J.; Reddin, J.S.; Munden, R.; Johnson, D.W.; Wilf, L.H.; DeNittis, A.; et al. Prediction of Survival by [18F]Fluorodeoxyglucose Positron Emission Tomography in Patients With Locally Advanced Non–Small-Cell Lung Cancer Undergoing Definitive Chemoradiation Therapy: Results of the ACRIN 6668/RTOG 0235 Trial. J. Clin. Oncol. 2013, 31, 3823–3830. [Google Scholar] [CrossRef] [Green Version]
- Wahl, R.L.; Hutchins, G.D.; Buchsbaum, D.J.; Liebert, M.; Grossman, H.B.; Fisher, S. 18F-2-deoxy-2-fluoro-D-glucose uptake into human tumor xenografts. Feasibility studies for cancer imaging with positron-emission tomography. Cancer 1991, 67, 1544–1550. [Google Scholar] [CrossRef]
- Wuest, M.; Trayner, B.J.; Grant, T.N.; Jans, H.-S.; Mercer, J.R.; Murray, D.; West, F.G.; McEwan, A.J.; Wuest, F.; Cheeseman, C.I. Radiopharmacological evaluation of 6-deoxy-6-[18F]fluoro-d-fructose as a radiotracer for PET imaging of GLUT5 in breast cancer. Nucl. Med. Biol. 2011, 38, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Wen, X.; Jia, Z.; Wu, X.; Guo, W.; Sun, H. Synthesis and Preliminary Evaluation of 1-[18F]Fluoro-1-deoxy-2,5-anhydro-D-mannitol as a PET Radiotracer for Breast Cancer Imaging. Chin. J. Chem. 2013, 31, 1159–1163. [Google Scholar] [CrossRef]
- Carter, K.R.; Kotlyarov, E. Common causes of false positive F18 FDG PET/CT scans in oncology. Braz. Arch. Biol. Technol. 2007, 50, 29–35. [Google Scholar] [CrossRef]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef] [Green Version]
- Granchi, C.; Fortunato, S.; Minutolo, F. Anticancer agents interacting with membrane glucose transporters. MedChemComm 2016, 7, 1716–1729. [Google Scholar] [CrossRef] [Green Version]
- Tanasova, M.; Begoyan, V.V.; Weselinski, L.J. Targeting Sugar Uptake and Metabolism for Cancer Identification and Therapy: An Overview. Curr. Top. Med. Chem. 2018, 18, 467–483. [Google Scholar] [CrossRef]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetol. 2014, 58, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Jeong, D.C.; Pak, K.; Han, M.-E.; Kim, J.-Y.; Liangwen, L.; Kim, H.J.; Kim, T.W.; Hyun, D.W.; Oh, S.-O. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget 2017, 8, 68381–68392. [Google Scholar] [CrossRef] [Green Version]
- McQuade, D.T.; Plutschack, M.B.; Seeberger, P.H. Passive fructose transporters in disease: A molecular overview of their structural specificity. Org. Biomol. Chem. 2013, 11, 4909–4920. [Google Scholar] [CrossRef]
- Pujol-Gimenez, J.; de Heredia, F.P.; Idoate, M.A.; Airley, R.; Lostao, M.P.; Evans, A.R. Could GLUT12 be a Potential Therapeutic Target in Cancer Treatment? A Preliminary Report. J. Cancer 2015, 6, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begoyan, V.V.; Xia, S.; Fedie, J.R.; Kannan, S.; Ferrier, A.; Rao, S.; Tanasova, M.; Weselinski, L.J.J. Multicolor GLUT5-permeable fluorescent probes for fructose transport analysis. Chem. Commun. 2018, 54, 3855–3858. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Begoyan, V.V.; Fedie, J.R.; Xia, S.; Weseliński, Ł.J.; Tanasova, M.; Rao, S. Metabolism-Driven High-Throughput Cancer Identification with GLUT5-Specific Molecular Probes. Biosensors 2018, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.rxlist.com/leukeran-drug.htm (accessed on 6 April 2021).
- Reux, B.; Weber, V.; Galmier, M.-J.; Borel, M.; Madesclaire, M.; Madelmont, J.-C.; Debiton, E.; Coudert, P. Synthesis and cytotoxic properties of new fluorodeoxyglucose-coupled chlorambucil derivatives. Bioorg. Med. Chem. 2008, 16, 5004–5020. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.-W.; Zheng, Y.-C.; Duan, Y.-C.; Wang, M.-M.; Yu, B.; Ren, J.-L.; Ma, J.-L.; Zhang, E.; Liu, H.-M. Synthesis and biological evaluation of coumarin–1,2,3-triazole–dithiocarbamate hybrids as potent LSD1 inhibitors. MedChemComm 2014, 5, 650–654. [Google Scholar] [CrossRef]
- Tiwari, V.K.; Mishra, B.B.; Mishra, K.B.; Mishra, N.; Singh, A.S.; Chen, X. Cu-Catalyzed Click Reaction in Carbohydrate Chemistry. Chem. Rev. 2016, 116, 3086–3240. [Google Scholar] [CrossRef] [PubMed]
- Marik, J.; Sutcliffe, J.L. Click for PET: Rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 6681–6684. [Google Scholar] [CrossRef]
- Oecd Guideline for the Testing of Chemicals. Partition Coefficient (N-Octanol/Water): Shake Flask Method. Roecd, 1995, 107.
- Levi, J.; Cheng, Z.; Gheysens, O.; Patel, M.; Chan, C.T.; Wang, Y.; Namavari, M.; Gambhir, S.S. Fluorescent Fructose Derivatives for Imaging Breast Cancer Cells. Bioconjug. Chem. 2007, 18, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Tanasova, M.; Plutschack, M.; Muroski, M.E.; Sturla, S.J.; Strouse, G.F.; McQuade, D.T. Fluorescent THF-Based Fructose Analogue Exhibits Fructose-Dependent Uptake. ChemBioChem 2013, 14, 1263–1270. [Google Scholar] [CrossRef]
- Maly, D.J.; Leonetti, F.; Backes, B.J.; Dauber, D.S.; Harris, J.L.; Craik, C.S.; Ellman, J.A. Expedient Solid-Phase Synthesis of Fluorogenic Protease Substrates Using the 7-Amino-4-carbamoylmethylcoumarin (ACC) Fluorophore. J. Org. Chem. 2002, 67, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Reszka, P.; Schulz, R.; Methling, K.; Lalk, M.; Bednarski, P.J. Synthesis, Enzymatic Evaluation, and Docking Studies of Fluorogenic Caspase 8 Tetrapeptide Substrates. ChemMedChem 2010, 5, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; La Ferla, B. Synthesis of C-Glycoconjugates from Readily Available Unprotected C-Allyl Glycosides by Chemoselective Ligation. J. Carbohydr. Chem. 2008, 27, 203–213. [Google Scholar] [CrossRef]
- Clavel, C.M.; Zava, O.; Schmitt, F.; Kenzaoui, B.H.; Nazarov, A.A.; Juillerat-Jeanneret, L.; Dyson, P.J. Thermoresponsive Chlorambucil Derivatives for Tumour Targeting. Angew. Chem. Int. Ed. 2011, 50, 7124–7127. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCF7 | MDA-MB-231 | 184B5 | |
|---|---|---|---|
| 24 h incubation | |||
| CLB | 145.2 ± 28.7 | 155.9 ± 22.3 | 182.5 ± 33.7 |
| I | 179.4 ± 24.3 | 89.9 ± 17.4 | >900 |
| II | 187.6 ± 34.3 | 194.2 ± 21.9 | 200/6 ± 18.2 |
| III | 490.9 ± 21.8 | 108.4 ± 29.9 | 203.2 ± 33.2 |
| IV | 196.8 ± 11.8 | >500 | 200.4 ± 18.4 |
| 48 h incubation | |||
| CLB | 89.7 ± 12.6 | 114.1 ± 14.4 | 270.4 ± 24.2 |
| I | 85.9 ± 27.1 | 83.6 ± 18.3 | >900 |
| II | 103.2 ± 19.3 | 99.3 ± 11.1 | 380.5 ± 23.7 |
| III | 175.3 ± 10.8 | 104.1 ± 11.7 | 241.5 ± 13.2 |
| IV | 137.6 ± 8.8 | 108.1 ± 15.3 | 350.7 ± 33.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nahrjou, N.; Ghosh, A.; Tanasova, M. Targeting of GLUT5 for Transporter-Mediated Drug-Delivery Is Contingent upon Substrate Hydrophilicity. Int. J. Mol. Sci. 2021, 22, 5073. https://doi.org/10.3390/ijms22105073
Nahrjou N, Ghosh A, Tanasova M. Targeting of GLUT5 for Transporter-Mediated Drug-Delivery Is Contingent upon Substrate Hydrophilicity. International Journal of Molecular Sciences. 2021; 22(10):5073. https://doi.org/10.3390/ijms22105073
Chicago/Turabian StyleNahrjou, Nazanin, Avik Ghosh, and Marina Tanasova. 2021. "Targeting of GLUT5 for Transporter-Mediated Drug-Delivery Is Contingent upon Substrate Hydrophilicity" International Journal of Molecular Sciences 22, no. 10: 5073. https://doi.org/10.3390/ijms22105073
APA StyleNahrjou, N., Ghosh, A., & Tanasova, M. (2021). Targeting of GLUT5 for Transporter-Mediated Drug-Delivery Is Contingent upon Substrate Hydrophilicity. International Journal of Molecular Sciences, 22(10), 5073. https://doi.org/10.3390/ijms22105073