
Hybrid Cyclobutane/Proline-Containing Peptidomimetics: The Conformational Constraint Influences Their Cell-Penetration Ability

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
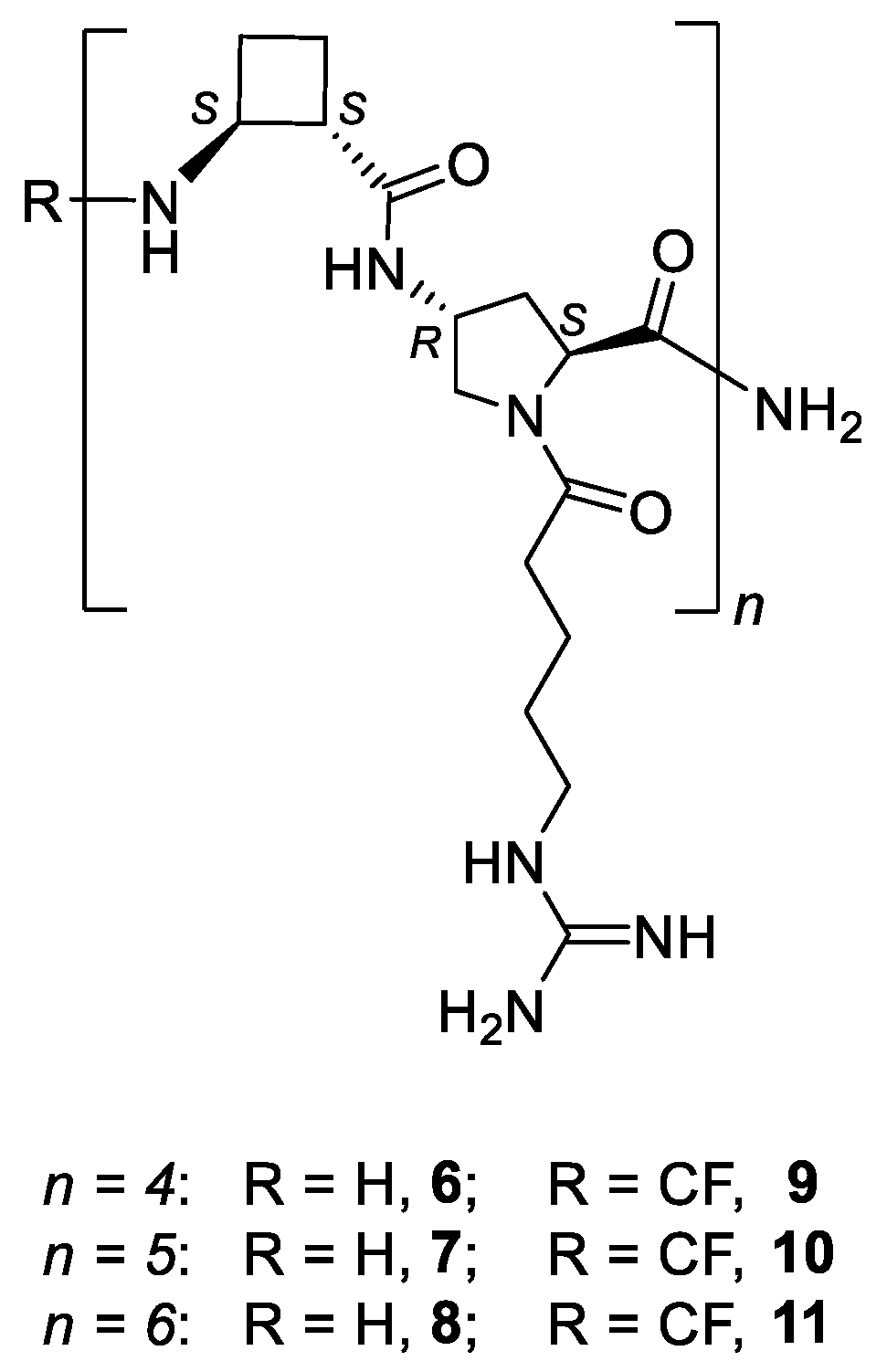
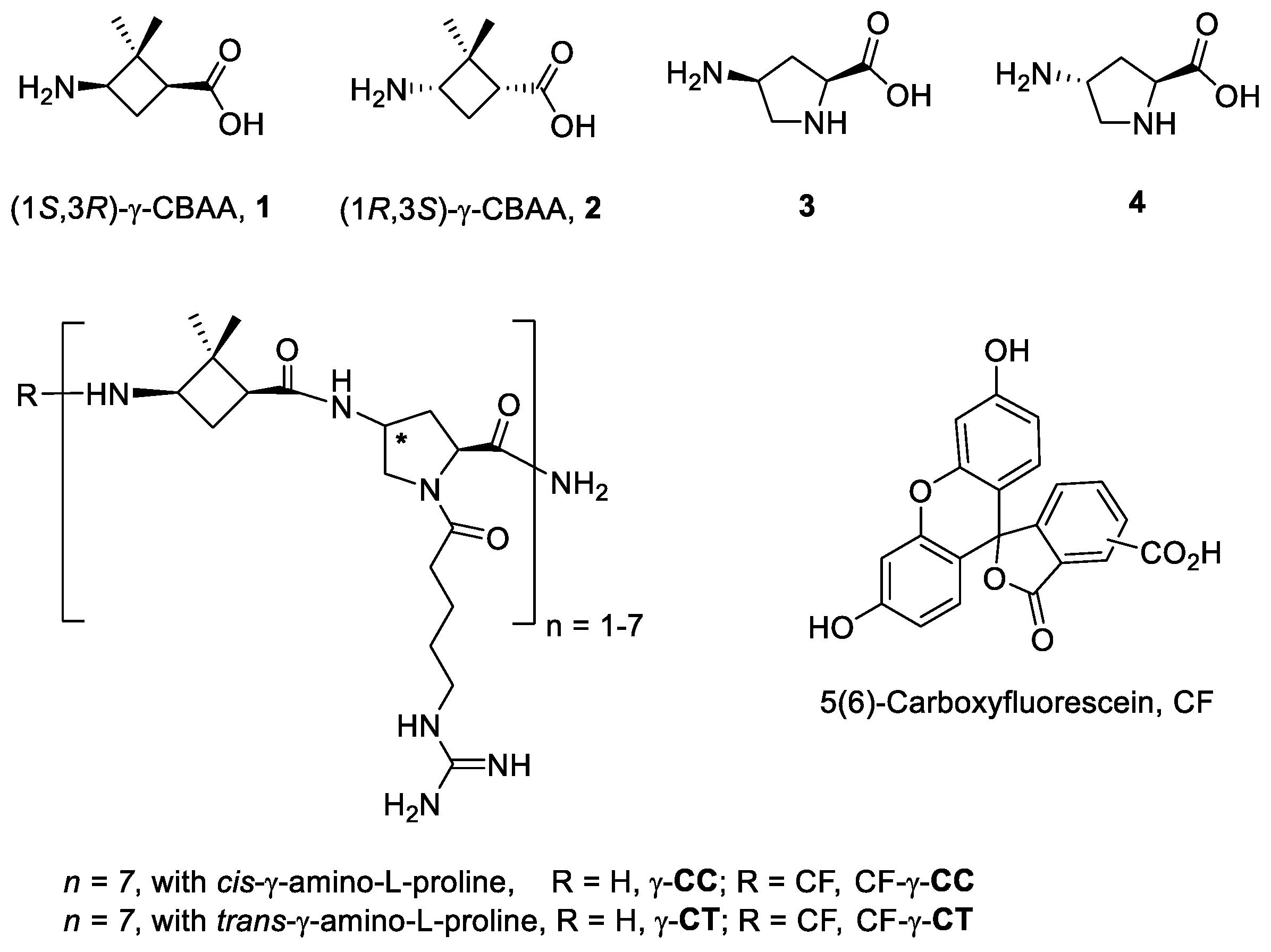
2.1. Synthesis of the Peptides
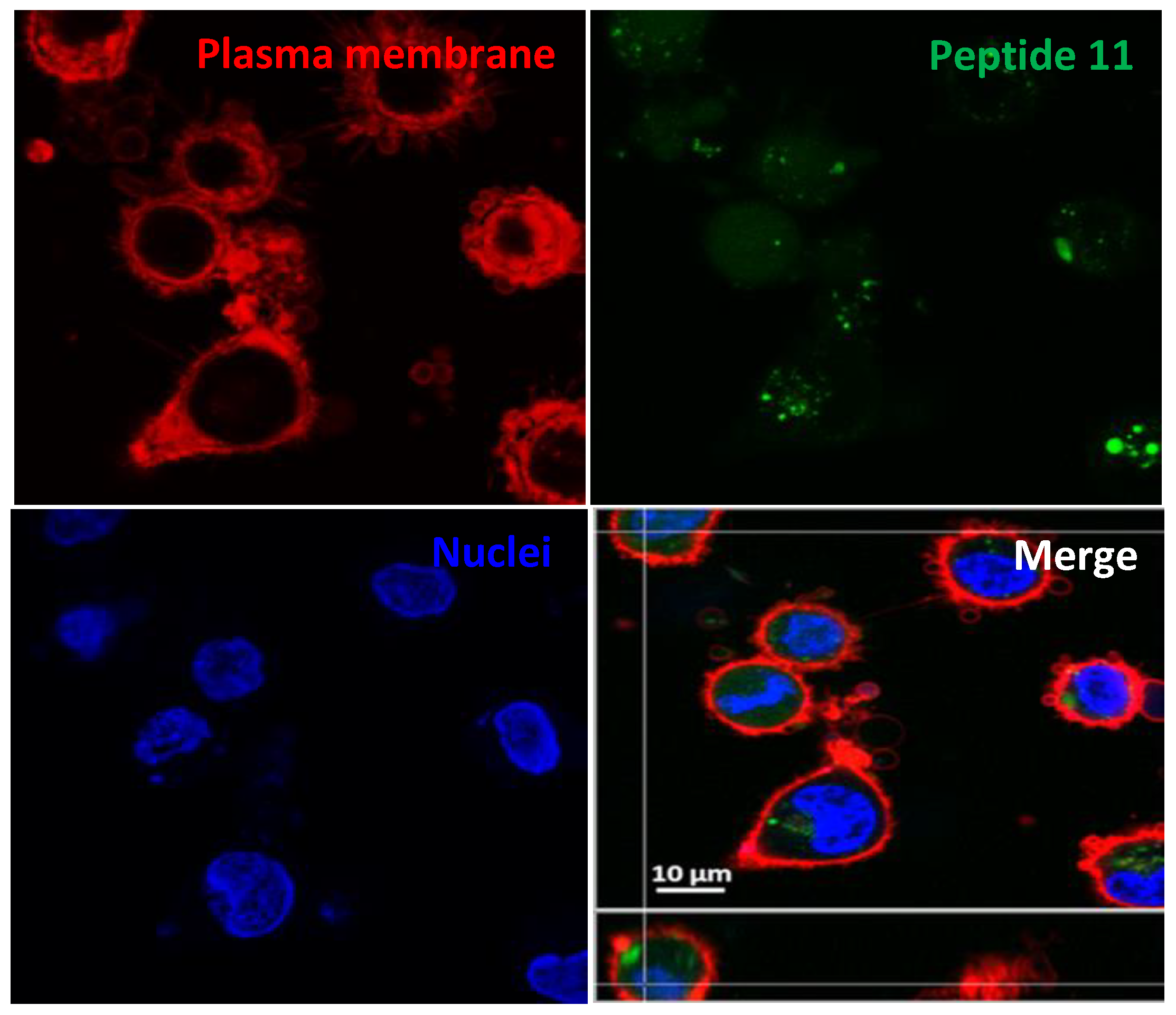
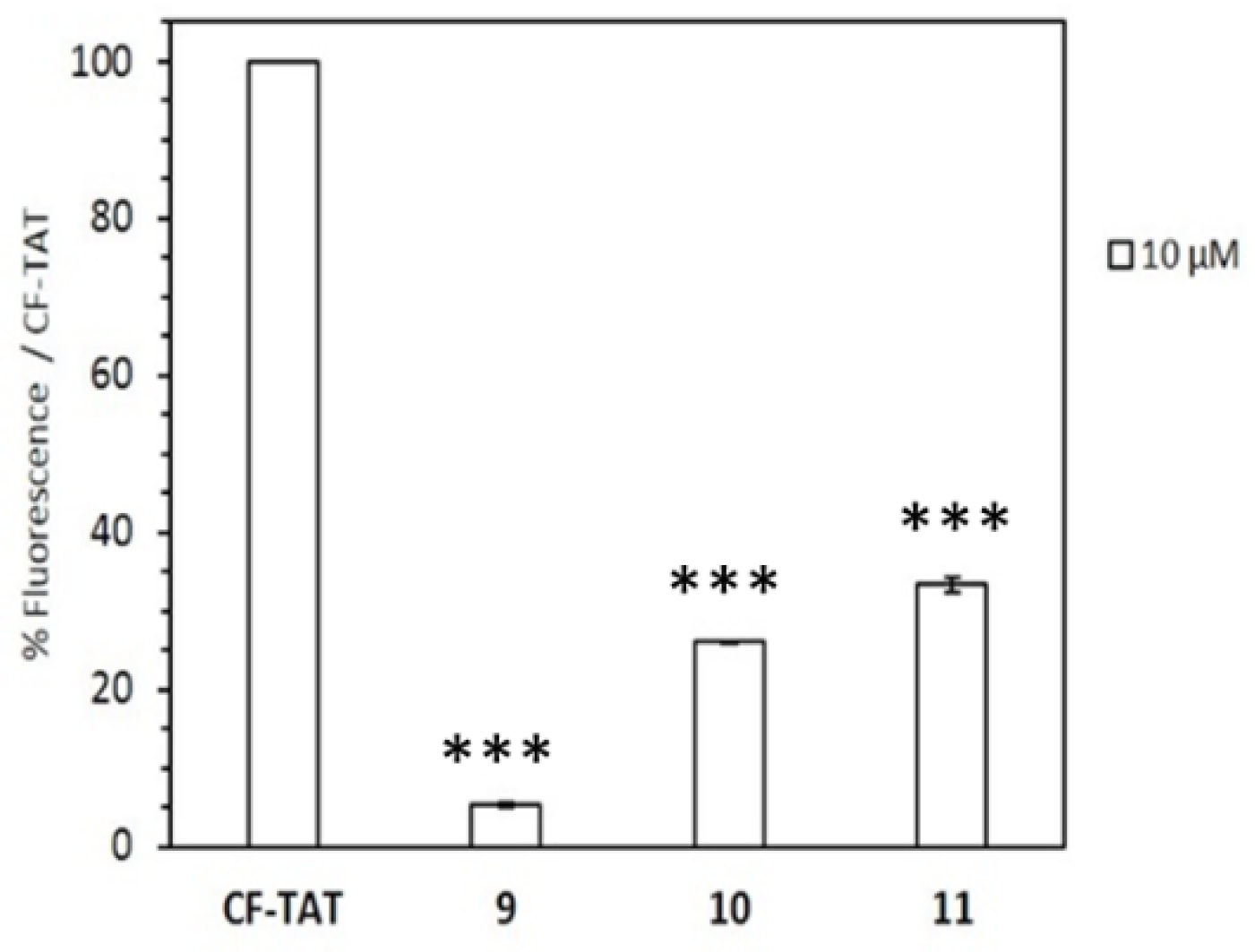
2.2. Cytotoxicity and Cellular Uptake in HeLa Cells
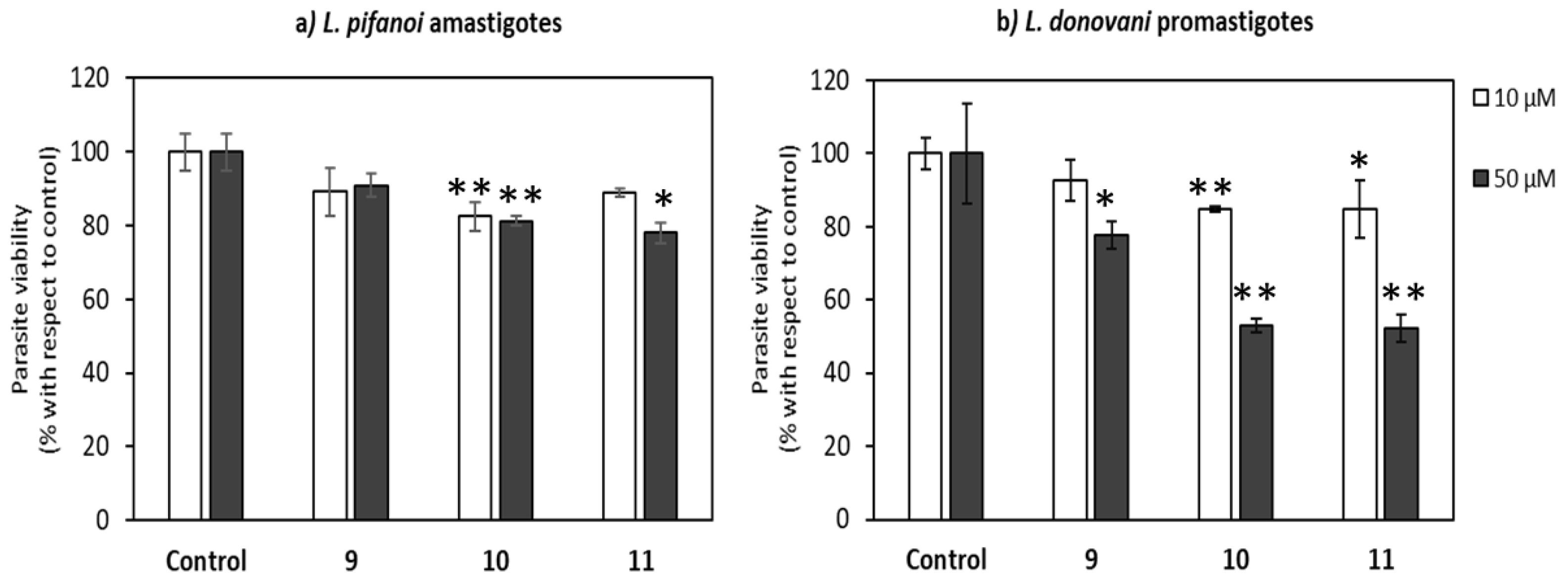
2.3. Uptake, Microbicidal Activity, and Intracellular Location of Peptides on Leishmania Parasites
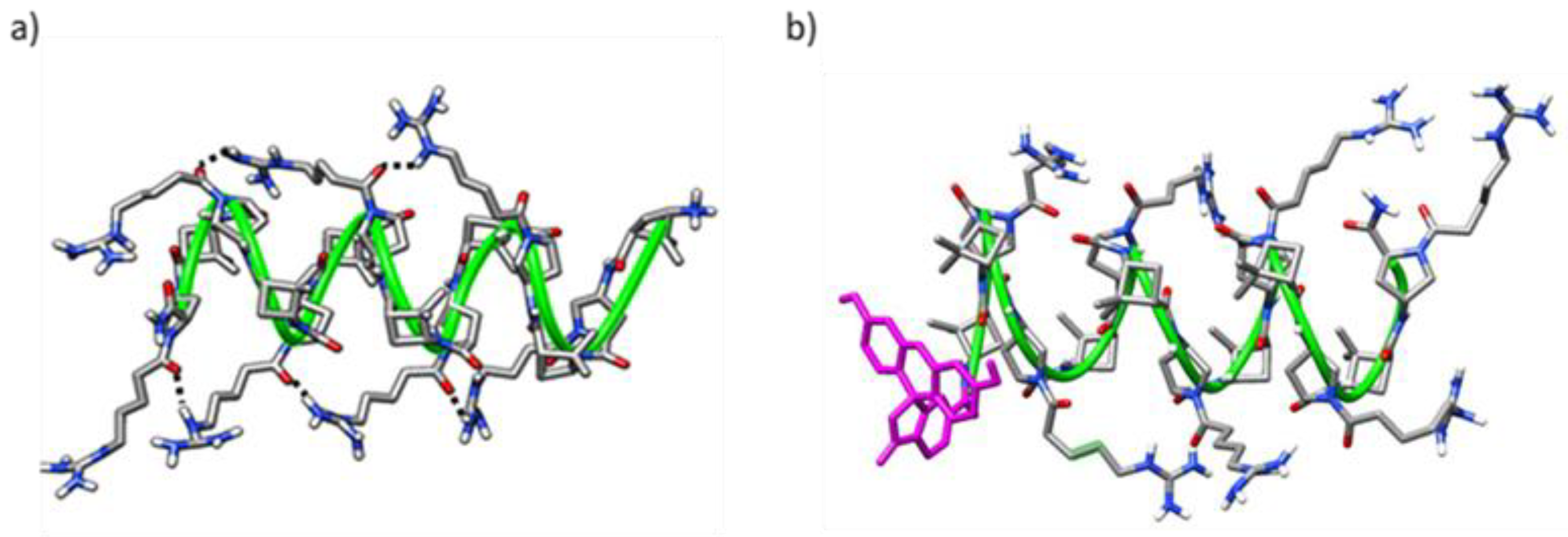
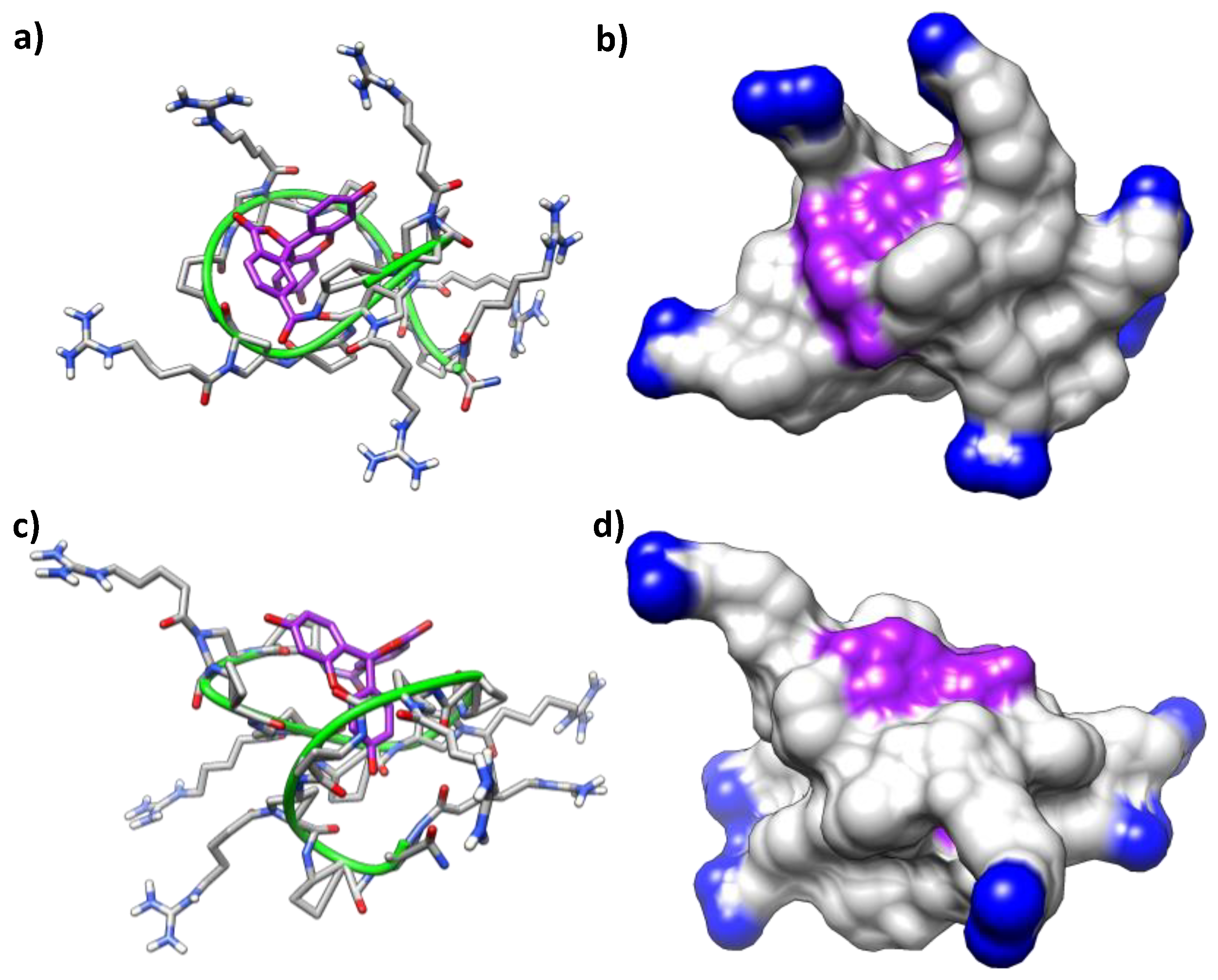
2.4. Molecular Modeling
3. Conclusions
4. Materials and Methods
4.1. Synthesis of the β, γ-Peptides 6–11 and TAT48–57 Peptide
4.2. Peptide Purification
4.3. Peptide Characterization
4.3.1. Octameric Peptide (6)
4.3.2. Decameric Peptide (7)
4.3.3. Dodecameric Peptide (8)
4.3.4. Octameric CF-Peptide (9)
4.3.5. Decameric CF-Peptide (10)
4.3.6. Dodecameric CF-Peptide (11)
4.4. Cellular Viability, Internalization, and Localization Experiments with HeLa Cells
4.4.1. HeLa Cells Culture
4.4.2. Cellular Viability
4.4.3. Peptide Internalization
4.5. Cellular Viability, Internalization, and Localization Experiments with Leishmania Parasites
4.5.1. Cellular Viability
4.5.2. Peptide Uptake for Leishmania donovani Promastigotes
4.5.3. Intracellular Localization of the Internalized Peptides in Leishmania donovani Promastigotes
4.5.4. Statistical Treatment
4.6. Computational Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| Alloc | Allyloxycarbonyl |
| Amp | 4-Aminoproline |
| CBAA | Cyclobutane Amino Acid |
| CF | 5(6)-Carboxyfluorescein |
| CPP | Cell Penetrating Peptide |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DCM | Dichloromethane |
| DDS | Drug Delivery Systems |
| DFT | Density Functional Theory |
| DIC | N,N′-Diisopropylcarbodiimide |
| DIPEA | N-Diisopropylethylamine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl Sulfoxide |
| EDTA | Ethylenediaminetetraacetic Acid |
| ESI | Electrospray Ionization |
| FBS | Fetal Bovine Serum |
| Fmoc | Fluorenylmethyloxycarbonyl |
| Glc | Glucose |
| HBSS | Hanks Buffered Saline Solution |
| HPLC | High-Performance Liquid Chromatography |
| LPG | Lypophosphoglycan |
| MALDI | Matrix Assisted Laser Desorption Ionization |
| MS | Mass Spectroscopy |
| MD | Molecular Dynamics |
| MEM | Minimum Essential Medium |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide |
| NMR | Nuclear Magnetic Resonance |
| PBS | Phosphate-Buffered Saline |
| PCA | Principal Component Analysis |
| PES | Potential Energy Surface |
| PI | Propidium Iodide |
| PyBOP | (7-Azabenzotriazol-1-yloxy)-tripyrrolidinophosphonium Hexafluorophosphate |
| RESP | Restrained Electrostatic Potential |
| RMSD | Root-Mean-Square Deviation |
| RMSF | Root-Mean-Square Fluctuation |
| RP | Reverse Phase |
| SASA | Solvent Accessible Surface Area |
| SD | Standard Deviation |
| SM | Supplementary Materials |
| SPPS | Solid Phase Peptide Synthesis |
| TIS | Triisopropylsilane |
| TOF | Time of Flight |
| UV | Ultraviolet Spectroscopy |
References
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-penetrating Peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Langel, Ü. Cell-Penetrating Peptides in Processes and Applications; CRC Press Pharmacology and Toxicology Series; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar] [CrossRef]
- Lundberg, P.; Langel, Ü. A brief introduction to cell-penetrating peptides. J. Mol. Recognit. 2003, 16, 227–233. [Google Scholar] [CrossRef]
- Kim, G.C.; Cheon, D.H.; Lee, Y. Challenge to overcome current limitations of cell-penetrating peptides. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140604. [Google Scholar] [CrossRef]
- Wu, J.; Li, J.; Wang, H.; Liu, C.B. Mitochondrial-targeted penetrating peptide delivery for cancer therapy. Expert Opin. Drug Deliv. 2021, 15, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Vivès, E.; Schmidt, J.; Pèlegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, E.; Torchillin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, J.; Xu, D. Cell penetrating peptides as noninvasive transmembrane vectors for the development of new functional drug delivery systems. J. Control. Release 2016, 229, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dissanayake, S.; Denny, W.A.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control. Release 2017, 250, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Fominaya, J.; Bravo, J.; Rebollo, A. Strategies to stabilize cell penetrating peptides for in vivo applications. Ther. Deliv. 2015, 6, 1171–1194. [Google Scholar] [CrossRef]
- Berlicki, Ł.; Kaske, M.; Gutiérrez-Abad, R.; Bernhardt, G.; Illa, O.; Ortuño, R.M.; Cabrele, C.; Buschauer, A.; Reiser, O. Replacement of Th32 and Gln34 in the C-terminal neuropeptide Y fragment 25-36 by cis-cyclobutane- and cis-cyclopentane-amino acids shifts selectivity toward the Y4 receptor. J. Med. Chem. 2013, 56, 8422–8431. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Maynard, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Feliciani, F.; Pinnen, F.; Stefanuccia, A.; Costante, R.; Cacciatore, I.; Lucente, G.; Mollica, A. Structure-Activity Relationships of Biphalin Analogs and their Biological Evaluation on Opioid Receptors. Mini-Rev. Med. Chem. 2013, 13, 11–33. [Google Scholar] [CrossRef]
- Mollica, A.; Costante, R.; Stefanucci, A.; Pinnen, F.; Lucente, G.; Fidanzad, S.; Pieretti, S. Antinociceptive profile of potent opioid peptide AM94, a fluorinated analogue of biphalin with non-hydrazine linker. J. Pept. Sci. 2013, 19, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Stefanucci, A.; Pinnen, F.; Feliciani, F.; Cacciatore, I.; Lucente, G.; Mollica, A. Conformationally Constrained Histidines in the Design of Peptidomimetics: Strategies for the χ-Space Control. Int. J. Mol. Sci. 2011, 12, 2853–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar] [CrossRef]
- Dobitz, S.; Aronoff, M.R.; Wennemers, H. Oligoprolines as molecular entities for controlling distance in biological and material sciences. Acc. Chem. Res. 2017, 50, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Potocky, T.B.; Menon, A.K.; Gellman, S.H. Effects of conformational stability and geometry of guanidinium display on cell entry by β-peptides. J. Am. Chem. Soc. 2005, 127, 3686–3687. [Google Scholar] [CrossRef]
- Nagel, Y.A.; Raschle, P.S.; Wennemers, H. Effect of preorganized charge-display on the cell-penetrating properties of cationic peptides. Angew. Chem. Int. Ed. 2017, 56, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zeng, X.; Li, J.; Jiang, Y.; Zhao, H.; Wang, D.; Huang, X.; Li, Z. Achieving enhanced cell penetration of short conformationally constrained peptides through amphiphilicity tuning. Chem. Sci. 2017, 8, 7576–7581. [Google Scholar] [CrossRef] [Green Version]
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.C.; Hackenberger, C.P.R. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. Int. Ed. 2015, 54, 1950–1953. [Google Scholar] [CrossRef]
- Lättig-Tünnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-Apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Abad, R.; Carbajo, D.; Nolis, P.; Acosta-Silva, C.; Cobos, J.A.; Illa, O.; Royo, M.; Ortuño, R.M. Synthesis and structural study of highly constrained hybrid cyclobutane-proline γ,γ-peptides. Amino Acids 2011, 41, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Gorrea, E.; Carbajo, D.; Gutiérrez-Abad, R.; Illa, O.; Branchadell, V.; Royo, M.; Ortuño, R.M. Searching for new cell-penetrating agents: Hybrid cyclobutane–proline γ,γ-peptides. Org. Biomol. Chem. 2012, 10, 4050–4057. [Google Scholar] [CrossRef]
- Farrera-Sinfreu, J.; Giralt, E.; Castel, S.; Albericio, F.; Royo, M. Cell-penetrating cis-γ-amino-L-proline-derived peptides. J. Am. Chem. Soc. 2005, 127, 9459–9468. [Google Scholar] [CrossRef] [PubMed]
- Illa, O.; Olivares, J.A.; Gaztelumendi, N.; Martínez-Castro, L.; Ospina, J.; Abengozar, M.Á.; Sciortino, G.; Maréchal, J.D.; Nogués, C.; Royo, M.; et al. Chiral cyclobutane-containing cell-penetrating peptides as selective vectors for anti-Leishmania drug delivery systems. Int. J. Mol. Sci. 2020, 21, 7502. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.; Gorrea, E.; Da Silva, E.; Nolis, P.; Branchadell, V.; Ortuño, R.M. Prevalence of eight-membered hydrogen-bonded rings in some bis(cyclobutane)-dipeptides including residues with trans stereochemistry. Org. Lett. 2009, 11, 2301–2304. [Google Scholar] [CrossRef]
- Fernandes, C.; Faure, S.; Pereira, E.; Théry, V.; Declerck, V.; Guillot, R.; Aitken, D.J. 12-Helix folding of cyclobutane β-amino acid oligomers. Org. Lett. 2010, 12, 3606–3609. [Google Scholar] [CrossRef]
- Gorrea, E.; Pohl, G.; Nolis, P.; Celis, S.; Burusco, K.; Branchadell, V.; Perczel, A.; Ortuño, R.M. Secondary structure of short β-peptides as the chiral expression of monomeric building units: A rational and predictive model. J. Org. Chem. 2012, 77, 9795–9806. [Google Scholar] [CrossRef] [PubMed]
- Illa, O.; Olivares, J.A.; Nolis, P.; Ortuño, R.M. The relevance of the relative configuration in the folding of hybrid peptides containing β-cyclobutane amino acids and γ-amino-L-proline residues. Tetrahedron 2017, 73, 6286–6295. [Google Scholar] [CrossRef]
- Fernandes, C.; Gauzy, C.; Yang, Y.; Roy, O.; Pereira, E.; Faure, S.; Aitken, D.J. [2+2] Photocycloadditions with chiral uracyl derivatives: Access to all four stereoisomers of 2-aminocyclobutanecarboxylic acid. Synthesis 2007, 2222–2232. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus TAT trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the TAT protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 TAT protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Mosnann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sinclair, A.N.; de Graffenried, C.L. More than Microtubules: The Structure and Function of the Subpellicular Array in Trypanosomatids. Trends Parasitol. 2019, 35, 760–777. [Google Scholar] [CrossRef]
- Halliday, C.; de Castro-Neto, A.; Alcantara, C.L.; Cunha-e-Silva, N.L.; Vaughan, S.; Sunter, J.D. Trypanosomatid Flagellar Pocket from Structure to Function. Trends Parasitol. 2021, 37, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Mottram, J.C.; Coombs, G.H.; Alexander, J. Cysteine peptidases as virulence factors of Leishmania. Curr. Opin. Microbiol. 2004, 7, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Pupkis, M.F.; Tetley, L.; Coombs, G.H. Leishmania mexicana: Amastigote hydrolases in unusual lysosomes. Exp. Parasitol. 1986, 62, 29–39. [Google Scholar] [CrossRef]
- Aguilera, T.A.; Timmers, M.M.; Olson, E.S.; Jiang, T.; Tsien, R.Y. Systemic in vivo distribution of activatable cell penetrating peptides is superior to cell penetrating peptides. Integr. Biol. 2009, 1, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Descoteaux, A.; Turco, S.J. Glycoconjugates in Leishmania infectivity. Biochim. Biophys. Acta Mol. Basis Dis. 1999, 1455, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Pae, J.; Liivamägi, L.; Lubenets, D.; Arukuusk, P.; Langel, Ü.; Pooga, M. Glycosaminoglycans are required for translocation of amphipathic cell-penetrating peptides across membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1860–1867. [Google Scholar] [CrossRef]
- Nakase, I.; Takeuchi, T.; Tanaka, G.; Futaki, S. Methodological and cellular aspects that govern the internalization mechanisms of arginine-rich cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Vazdar, M.; Heyda, J.; Mason, P.E.; Tesei, G.; Allolio, C.; Lund, M.; Jungwirth, P. Arginine “Magic”: Guanidinium Like-Charge Ion Pairing from Aqueous Salts to Cell Penetrating Peptides. Acc. Chem. Res. 2018, 51, 1455–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homans, S.W.; Mehlert, A.; Turco, S.J. Solution Structure of the Lipophosphoglycan of Leishmania donovani. Biochemistry 1992, 31, 654–661. [Google Scholar] [CrossRef]
- Ablan, F.D.O.; Spaller, B.L.; Abdo, K.I.; Almeida, P.F. Charge Distribution Fine-Tunes the Translocation of a-Helical Amphipatic Peptides across Membranes. Biophys. J. 2016, 111, 1738–1749. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Witek, J.; Landrum, G.A.; Riniker, S. Improving Conformer Generation for Small Rings and Macrocycles Based on Distance Geometry and Experimental Torsional-Angle Preferences. J. Chem. Inf. Model. 2020, 60, 2044–2058. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high-performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- Rodríguez-Guerra Pedregal, J.; Alonso-Cotchico, L.; Velasco-Carneros, L.; Maréchal, J.-D. OMMProtocol: A command line application to launch molecular dynamics simulations with OpenMM. ChemRxiv 2018. [Google Scholar] [CrossRef]
- Sciortino, G.; Sánchez-Aparicio, J.-E.; Rodríguez-Guerra Pedregal, J.; Garribba, E.; Maréchal, J.-D. Computational insight into the interaction of oxaliplatin with insulin. Metallomics 2019, 11, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Alemán, R.; Hernández-Castillo, D.; Caballero, J.; Montero-Cabrera, L.A. Quality Threshold Clustering of Molecular Dynamics: A Word of Caution. J. Chem. Inf. Model. 2020, 60, 467–472. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Guanidinium Group | Peptide 8 | CF-Conjugate 11 | ||
|---|---|---|---|---|
| SASA (nm2) | Percentage 1 | SASA (nm2) | Percentage 1 | |
| 1 | 1.119 ± 0.240 | 85.1 ± 18.3% | 1.194 ± 0.186 | 90.8 ± 14.1% |
| 2 | 1.105 ± 0.250 | 84.0 ± 19.0% | 1.143 ± 0.194 | 86.9 ± 14.8% |
| 3 | 1.083 ± 0.257 | 82.4 ± 19.5% | 1.212 ± 0.164 | 92.2 ± 12.5% |
| 4 | 1.147 ± 0.213 | 87.2 ± 16.2% | 1.159 ± 0.201 | 88.1 ± 15.3% |
| 5 | 1.185 ± 0.171 | 90.1 ± 13.0% | 1.154 ± 0.196 | 87.8 ± 14.9% |
| 6 | 1.232 ± 0.151 | 93.7 ± 11.5% | 1.099 ± 0.229 | 83.6 ± 17.4% |
| Average | 1.145 ± 0.084 | 87.1 ± 6.4% | 1.160 ± 0.078 | 88.2 ± 5.9% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illa, O.; Ospina, J.; Sánchez-Aparicio, J.-E.; Pulido, X.; Abengozar, M.Á.; Gaztelumendi, N.; Carbajo, D.; Nogués, C.; Rivas, L.; Maréchal, J.-D.; et al. Hybrid Cyclobutane/Proline-Containing Peptidomimetics: The Conformational Constraint Influences Their Cell-Penetration Ability. Int. J. Mol. Sci. 2021, 22, 5092. https://doi.org/10.3390/ijms22105092
Illa O, Ospina J, Sánchez-Aparicio J-E, Pulido X, Abengozar MÁ, Gaztelumendi N, Carbajo D, Nogués C, Rivas L, Maréchal J-D, et al. Hybrid Cyclobutane/Proline-Containing Peptidomimetics: The Conformational Constraint Influences Their Cell-Penetration Ability. International Journal of Molecular Sciences. 2021; 22(10):5092. https://doi.org/10.3390/ijms22105092
Chicago/Turabian StyleIlla, Ona, Jimena Ospina, José-Emilio Sánchez-Aparicio, Ximena Pulido, María Ángeles Abengozar, Nerea Gaztelumendi, Daniel Carbajo, Carme Nogués, Luis Rivas, Jean-Didier Maréchal, and et al. 2021. "Hybrid Cyclobutane/Proline-Containing Peptidomimetics: The Conformational Constraint Influences Their Cell-Penetration Ability" International Journal of Molecular Sciences 22, no. 10: 5092. https://doi.org/10.3390/ijms22105092