
Involvement of PI3K Pathway in Glioma Cell Resistance to Temozolomide Treatment


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
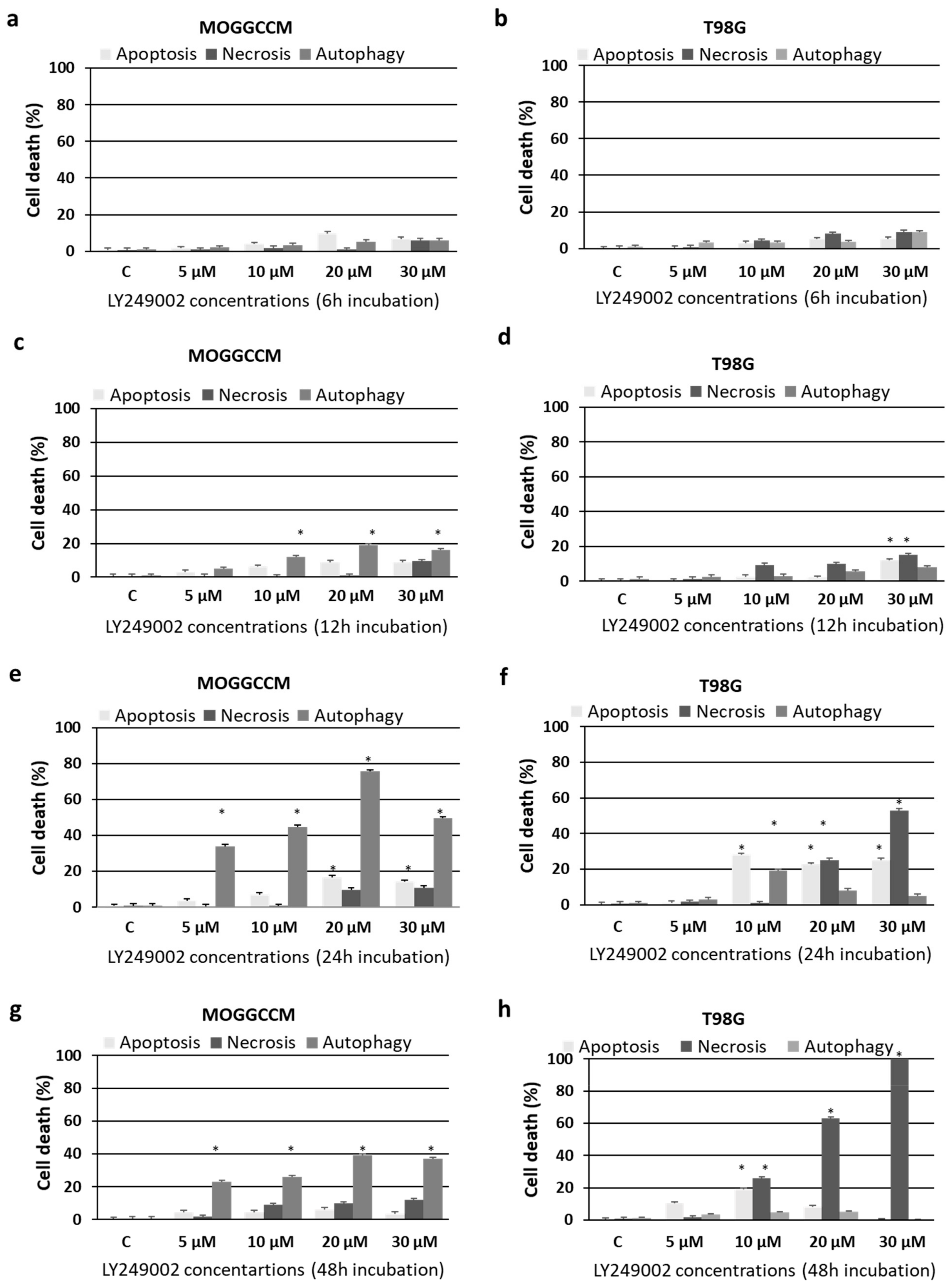
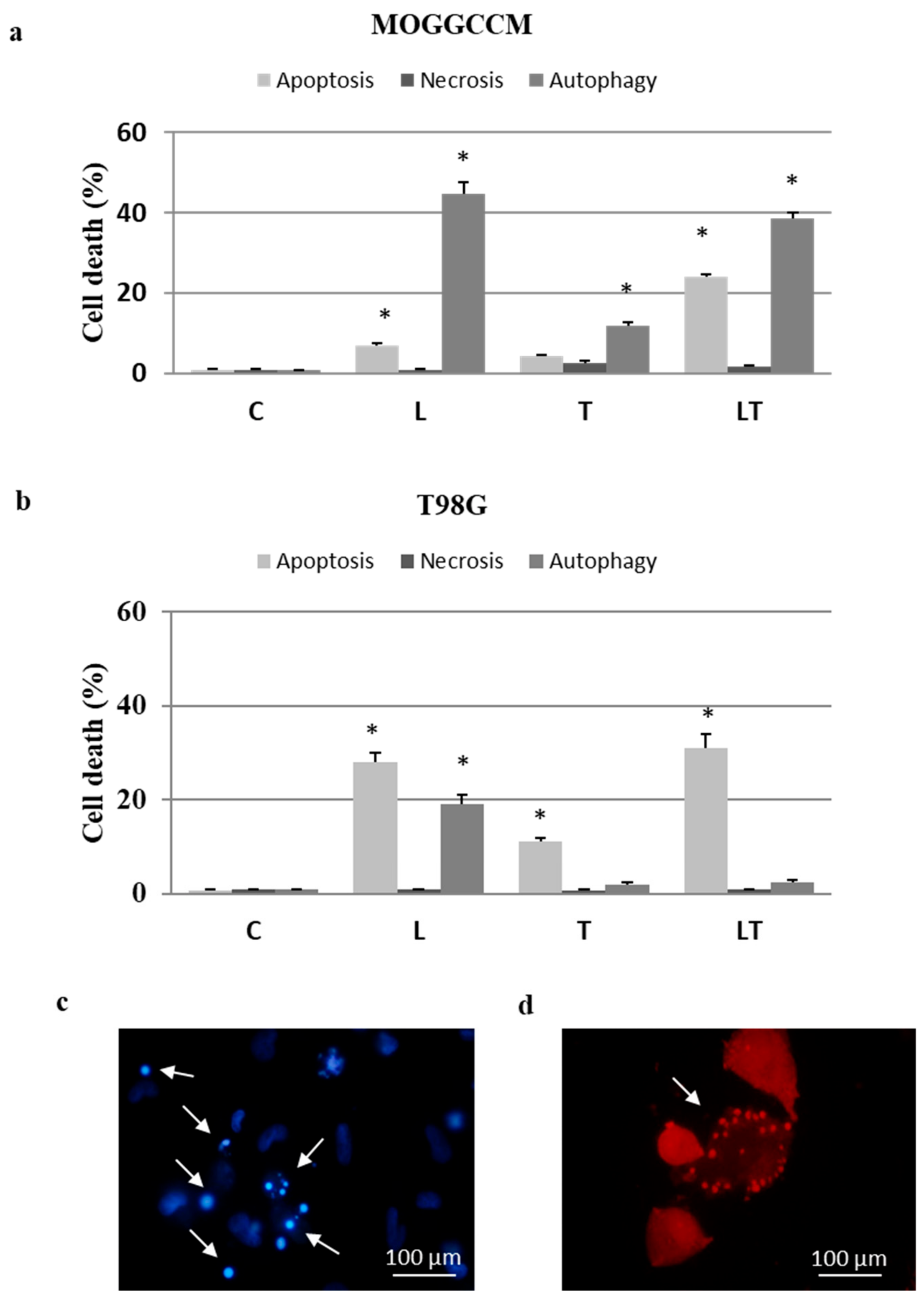
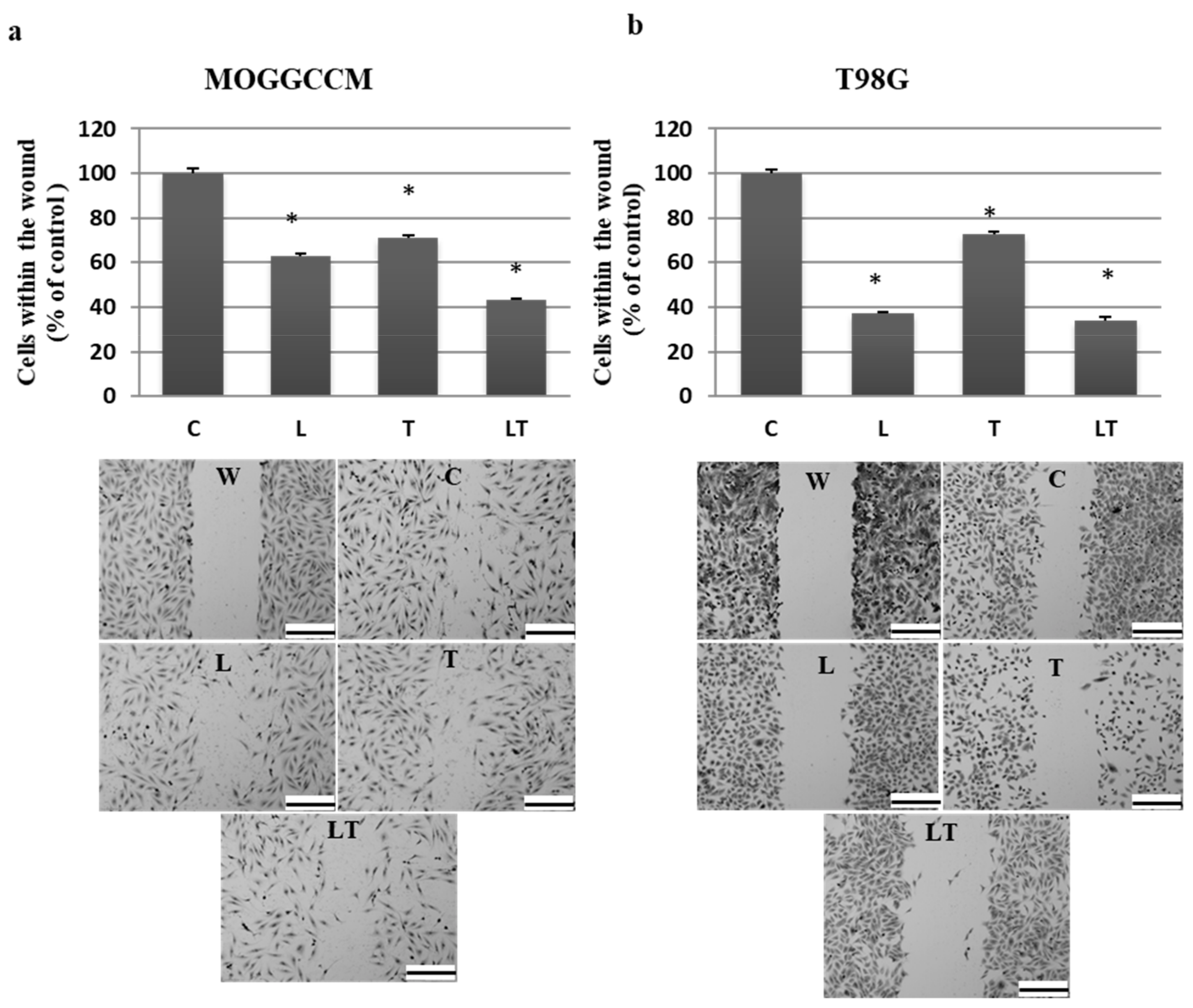
2.1. Cell Death and Migratory Potential after LY294002 and Temozolomide Treatment
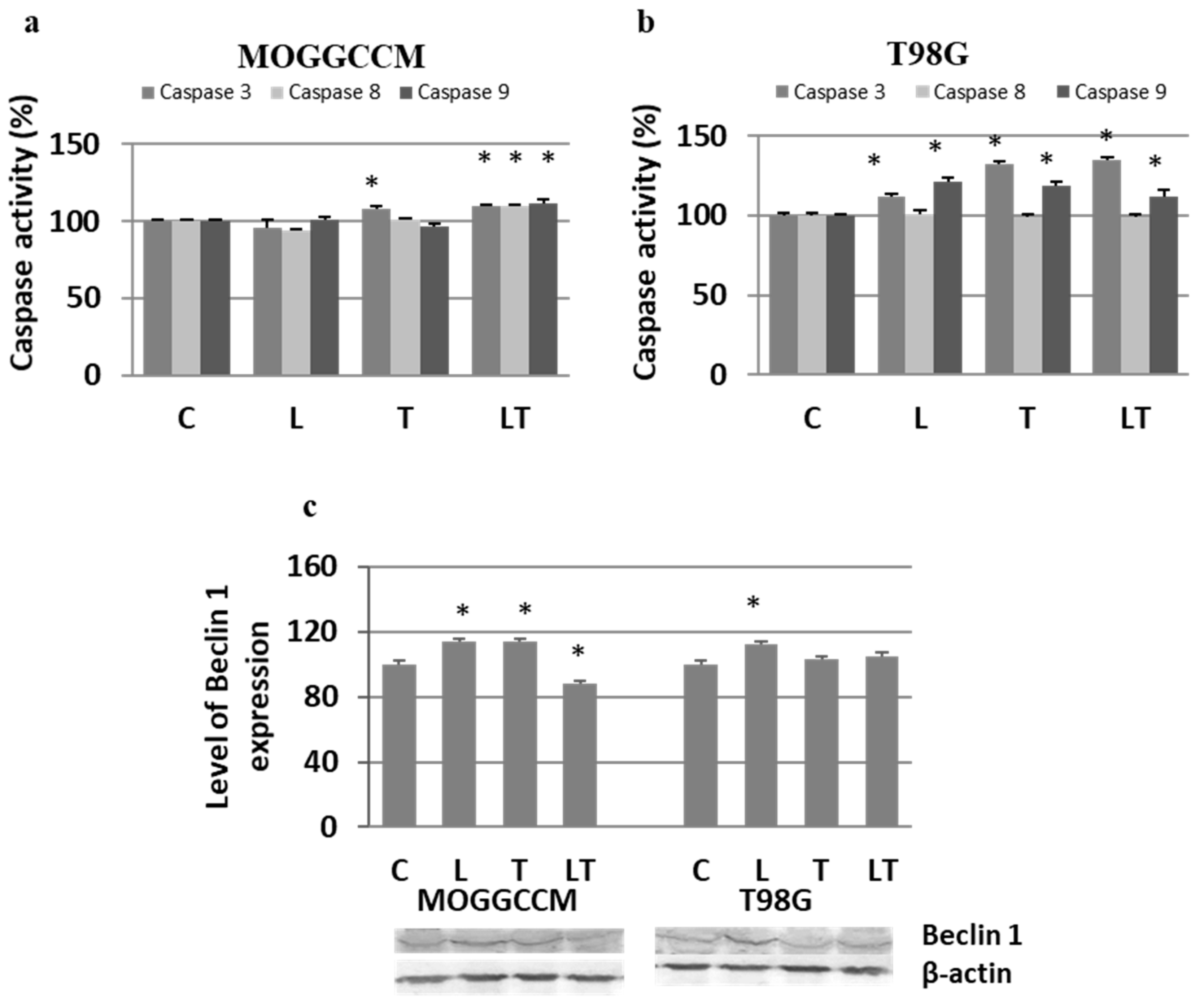
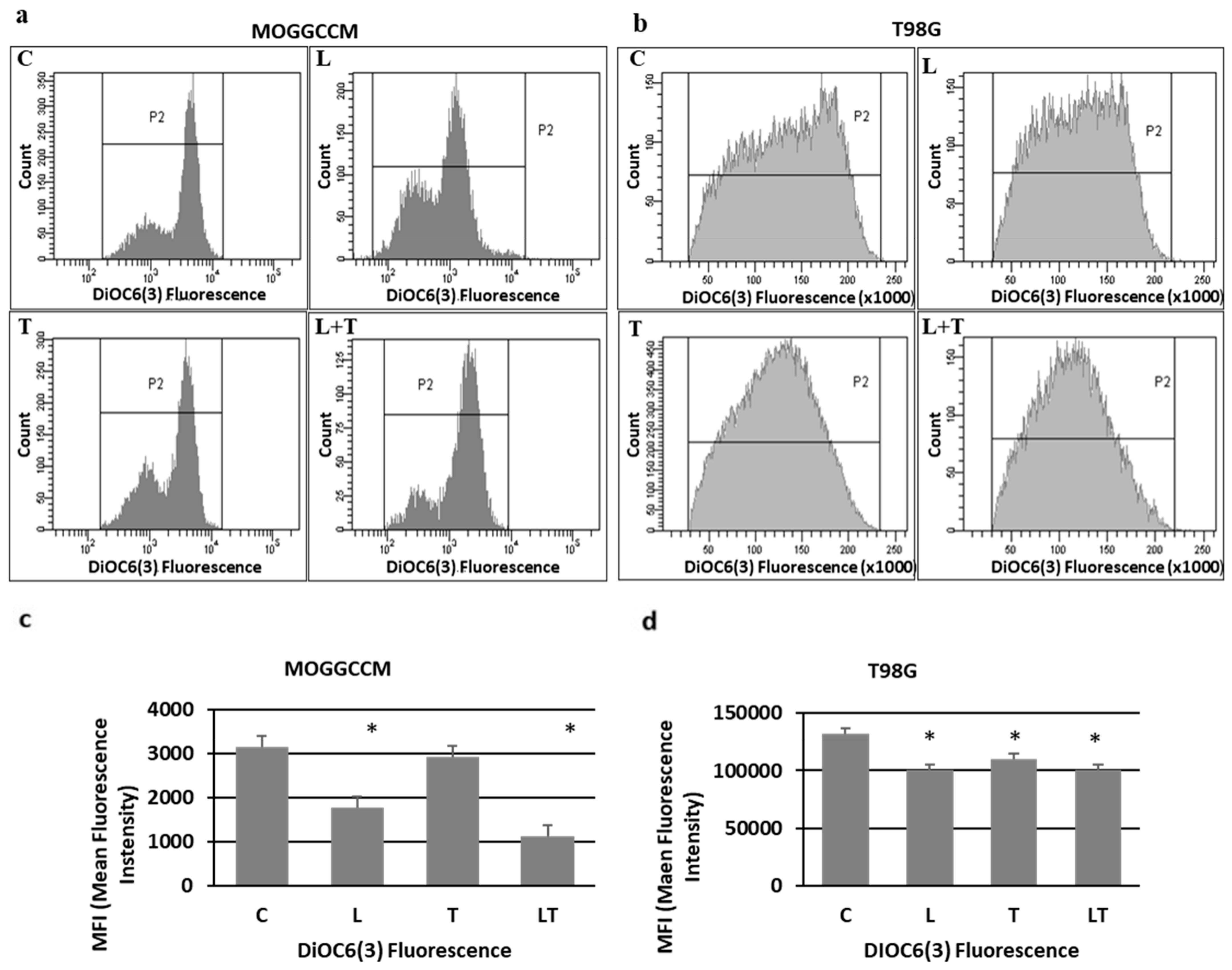
2.2. The Effect of Caspase Activity, Mitochondrial Membrane Potential, and Beclin 1 Expression
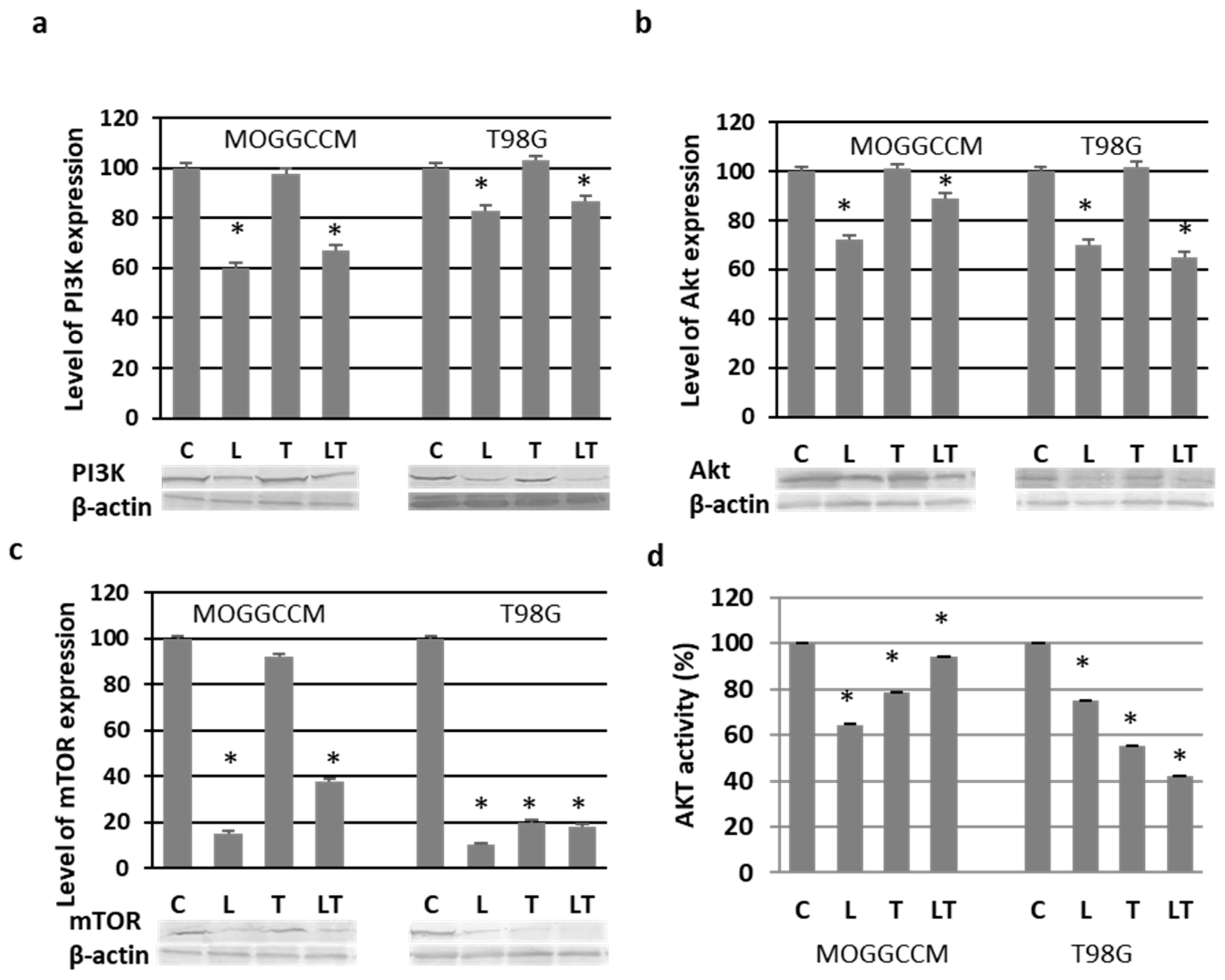
2.3. Effect of LY294002 and Temozolomide on the PI3K/Akt/mTOR Pathway
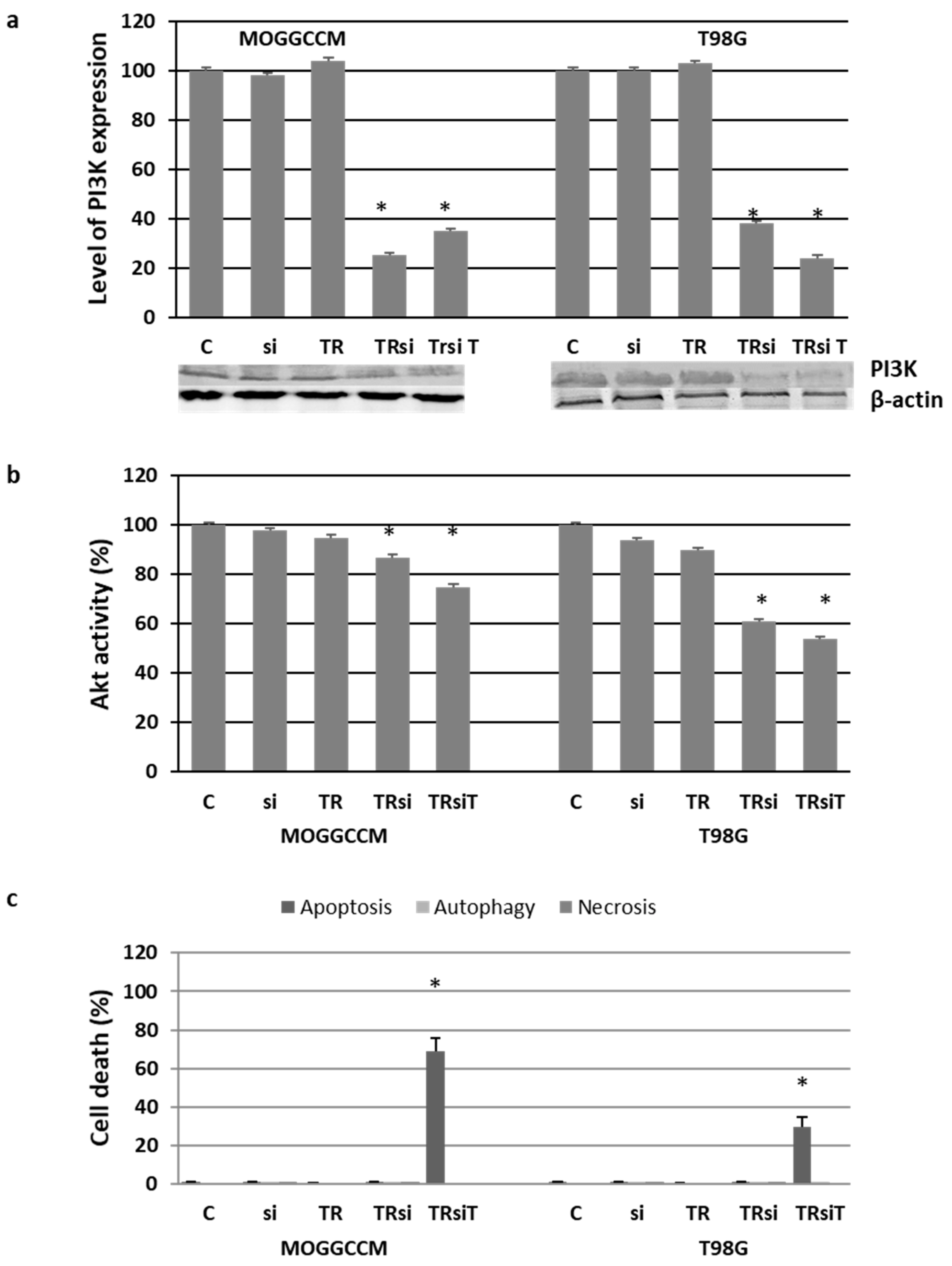
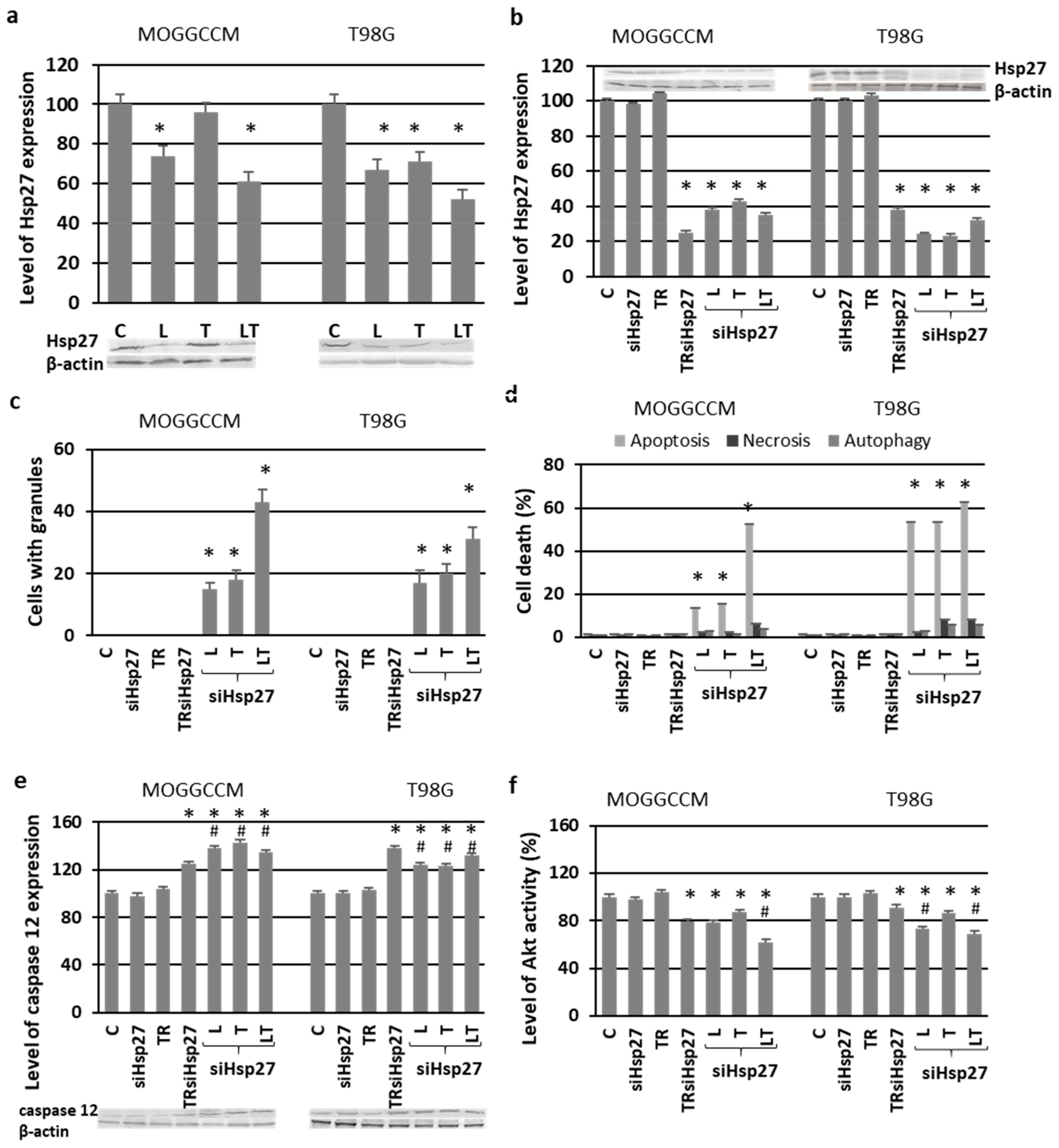
2.4. Blocking the Expression of PI3K
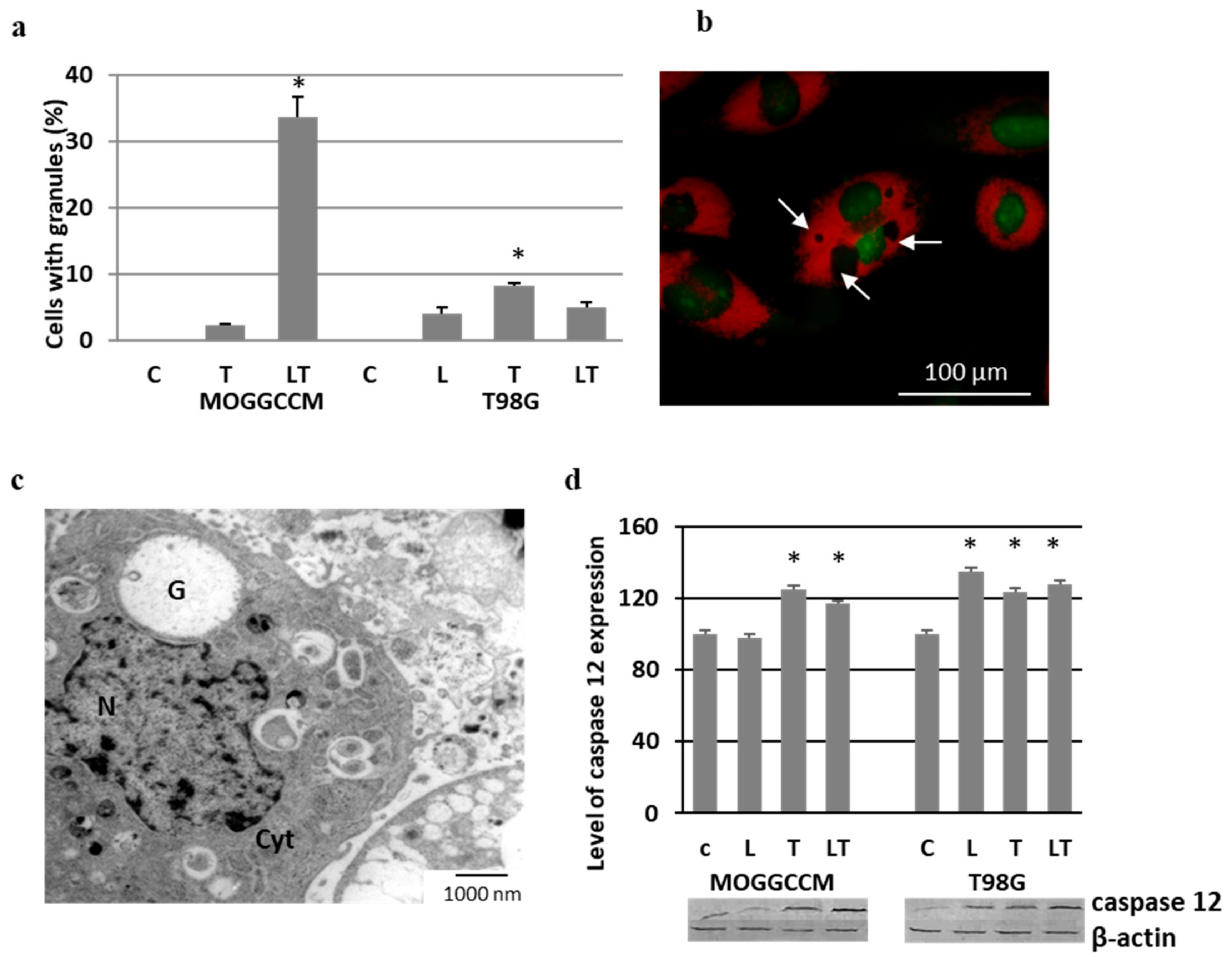
2.5. Effect of LY294002 and Temozolomide on Granule Formation
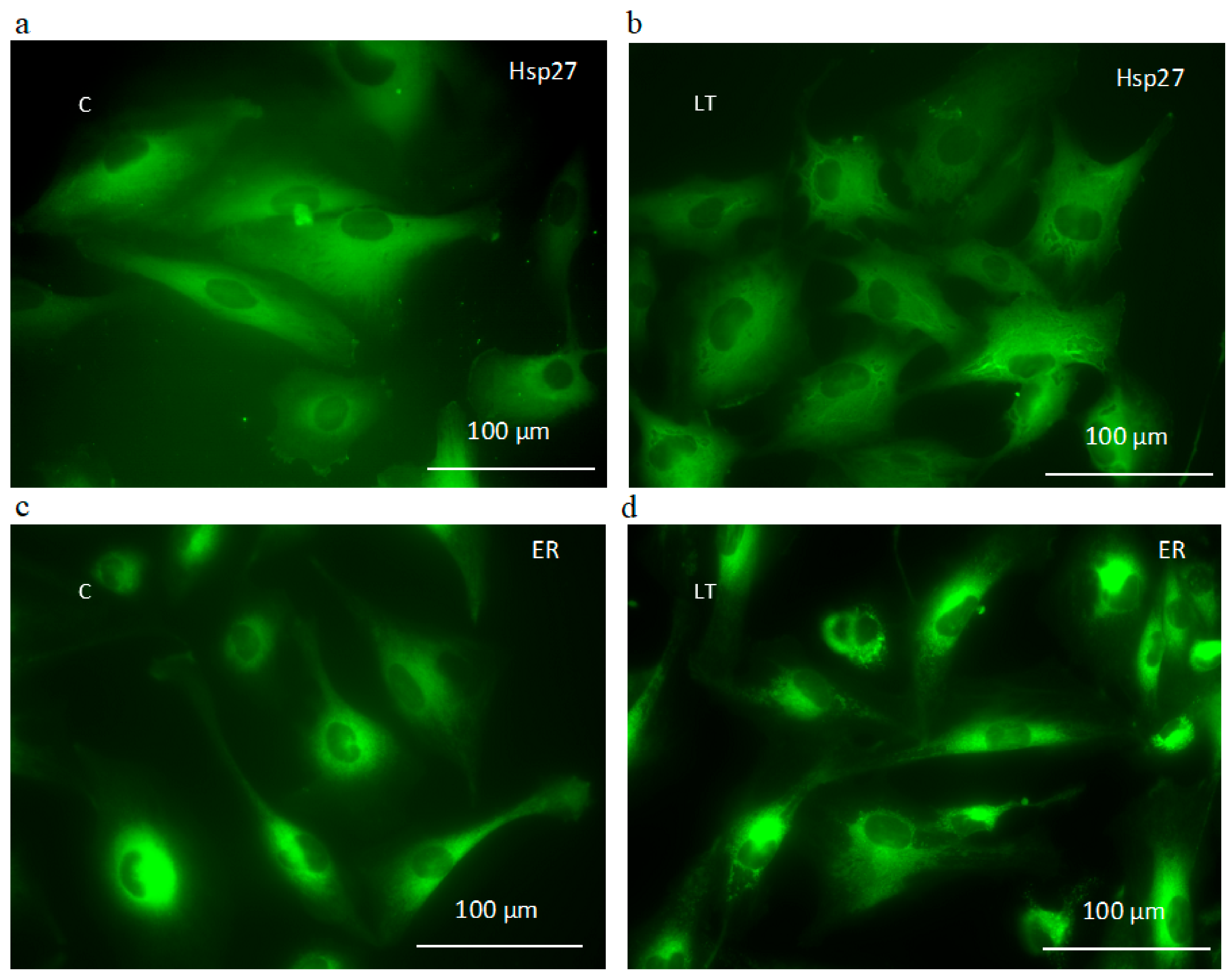
2.6. Involvement of Hsp27 Expression in Glioma Cell Resistance after the LY294002 and Temozolomide Treatment
3. Discussion
4. Materials and Methods
4.1. Cells and Culture Conditions
4.2. Drug Treatment
4.3. Microscopic Detection of Apoptosis, Autophagy, and Necrosis with Fluorochromes
4.4. Detection of Mitochondrial Membrane Potential by Flow Cytometry
4.5. ER Staining
4.6. Cell Migration Test
4.7. Transmission Electron Microscopy
4.8. Immunoblotting
4.9. Akt and Caspase Activity Assay
4.10. Indirect Immunofluorescence
4.11. T98G and MOGGCCM Transfection with siRNA
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Shingu, T.; Holmes, L.; Henry, V.; Wang, Q.; Latha, K.; Gururaj, A.E.; Gibson, L.A.; Doucette, T.; Lang, F.F.; Rao, G.; et al. Suppression of RAF/MEK or PI3K synergizes cytotoxicity of receptor tyrosine kinase inhibitors in glioma tumor-initiating cells. J. Transl. Med. 2016, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 16, 15017. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Cheng, C.K.; Fan, Q.-W.; Weiss, W.A. PI3K Signaling in glioma—Animal models and therapeutic challenges. Brain Pathol. 2009, 19, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.; Kornblum, H.I. Molecular markers in glioma. J. Neurooncol. 2017, 134, 505–512. [Google Scholar] [CrossRef]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Lealhe, J.F.M. PTEN/PI3K/Akt signaling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef]
- Maira, S.-M.; Voliva, C.; Garcia-Echeverria, C. Class IA PI3 kinase: From their biological implication in human cancers to drug discovery. Expert Opin. Ther. Targets 2008, 12, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Echeverria, C. Protein and lipid kinase inhibitors as targeted anticancer agents of the Ras/Raf/MEK and PI3K/PKB pathways. Purinergic Signal. 2009, 5, 117–125. [Google Scholar] [CrossRef]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Fan, X.; Xiong, J.; Zhang, G.; Luo, X.; Li, K.; Jie, Z.; Cao, Y.; Huang, Z.; et al. PRL-3 promotes gastric cancer peritoneal metastasis via the PI3K/AKT signaling pathway in vitro and in vivo. Oncol. Lett. 2018, 15, 9069–9074. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Yamori, T. Phosphatidylinositol 3-kinase inhibitors: Promising drug candidates for cancer therapy. Cancer Sci. 2008, 99, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.-G.; Zhu, B.; Fan, X.-Q.; Liu, H.-H.; Hou, X.; Zhao, K.; Qin, Z.-H. Effects of LY294002 on the invasiveness of human gastric cancer in vivo in nude mice. World J. Gastroenterol. 2009, 15, 5044–5052. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Han, H.; Shi, Z.; Zhang, K.; Liu, Y.; Zheng, Y.; Jiang, T.; Pu, P.; Jiang, C.; Kang, C. LY294002 enhances cytotoxicity of temozolomide in glioma by down-regulation of the PI3K/Akt pathway. Mol. Med. Rep. 2012, 5, 575–579. [Google Scholar] [PubMed]
- Abdallah, M.E.; El-Readi, M.Z.; Althubiti, M.A.; Almaimani, R.A.; Ismail, A.M.; Idris, S.; Refaat, B.; Almalki, W.H.; Babakr, A.T.; Mukhtar, M.H.; et al. Tamoxifen and the PI3K inhibitor: LY294002 synergistically induce apoptosis and cell cycle arrest in breast cancer MCF-7 cells. Molecules 2020, 25, 3355. [Google Scholar] [CrossRef] [PubMed]
- Cabrini, G.; Fabbri, E.; Nigro, C.L.; Dechecchi, M.C.; Gambari, R. Regulation of expression of O6-methylguanine-DNA methyltransferase and the treatment of glioblastoma (Review). Int. J. Oncol. 2015, 47, 417–428. [Google Scholar] [CrossRef]
- Jakubowicz-Gil, J.; Langner, E.; Badziul, D.; Wertel, I.; Rzeski, W. Apoptosis induction in human glioblastoma multiforme T98G cells upon Temozolomide and quercetin treatment. Tumor Biol. 2013, 34, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Jakubowicz-Gil, J.; Langner, E.; Wertel, I.; Piersiak, T.; Rzeski, W. Temozolomide, quercetin and cell death in the MOGGCCM astrocytoma cell line. Chem. Biol. Interact. 2010, 188, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807. [Google Scholar] [CrossRef]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Dowell, J.M.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Wagner, M.; et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin. Cancer Res. 2007, 13, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Noiphithak, R.; Veerasarn, K. Clinical predictors for survival and treatment outcome of high-grade glioma in Prasat Neurological Institute. Asian J. Neurosurg. 2017, 12, 28–33. [Google Scholar] [CrossRef]
- Guillard, G.S.; Clarke, P.A.; Poele, R.T.; Mohri, Z.; Bjerke, L.; Valenti, M.; Raynaud, F.; Eccles, S.A.; Workman, P. Molecular pharmacology of phosphatidylinositol 3-kinase inhibition in human glioma. Cell Cycle 2009, 8, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-H.; Huang, Y.-F.; Chen, C.-C.; Huang, C.-Y.; Chou, C.-Y. Comparing PI3K/Akt inhibitors used in ovarian cancer treatment. Front. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, J.; Shao, W.; Wu, C.; Chen, Z.; To, S.T.; Li, W. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Opel, D.; Westhoff, M.A.; Bender, A.; Braun, V.; Debatin, K.M.; Fulda, S. Phosphatidylinositol 3-kinase inhibition broadly sensitizes glioblastoma cell to death receptor- and drug-induced apoptosis. Cancer Res. 2008, 68, 6271–6280. [Google Scholar] [CrossRef] [PubMed]
- Roca-Agujetas, V.; de Dios, C.; Lestón, L.; Mari, M.; Morales, A.; Colell, A. Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid. Med. Cell. Longev. 2019, 3809308. [Google Scholar] [CrossRef]
- Condello, M.; Pellegrini, E.; Caraglia, M.; Meschini, S. Targeting autophagy to overcome human diseases. Int. J. Mol. Sci. 2019, 20, 725. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Salazar-Ramirez, K.; Molinares-Rodriguez, J.; Bolivar-Gonzalez, S. Molecular mechanisms of autophagy and its role in cancer development. Rev. Fac. Med. 2016, 64, 529–535. [Google Scholar] [CrossRef]
- Kaza, N.; Kohli, L.; Roth, K.A. Autophagy in brain tumors: A new target for therapeutic intervention. Brain Pathol. 2012, 22, 89–98. [Google Scholar] [CrossRef]
- Feng, F.; Zhang, M.; Yang, C.; Heng, X.; Wu, X. The dual roles of autophagy in gliomagenesis and clinical therapy strategies based on autophagic regulation mechanisms. Biomed. Pharmacother. 2019, 120, 109441. [Google Scholar] [CrossRef]
- Li, H.; Jin, X.; Zhang, Z.; Xing, Y.; Kong, X. Inhibition of autophagy enhances apoptosis induced by the PI3K/AKT/mTOR inhibitor NVP-BEZ235 in renal cell carcinoma cells. Cell Biochem. Funct. 2013, 31, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Muilenburg, D.; Parsons, C.; Coates, J.; Virudachalam, S.; Bold, R.J. Role of autophagy in apoptotic regulation by Akt in pancreatic cancer. Anticancer Res. 2014, 34, 631–637. [Google Scholar] [PubMed]
- Tomar, V.S.; Patil, V.; Somasundarm, K. Temozolomide induces activation of Wnt/β-catenin signaling in glioma cells via PI3K/Akt pathway: Implications in glioma therapy. Cell Biol. Toxicol. 2020, 36, 273–278. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.G.; Peng, S.; Sereduk, C.P.; Sodoma, A.M.; Kitange, G.J.; Loftus, J.C.; Sarkaria, J.N.; Tran, N.L. Inhibition of phosphatidylinositol 3-kinase by PX-866 suppresses temozolomide-induced autophagy and promotes apoptosis in glioblastoma cells. Mol. Med. 2019, 25, 49. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Deepti, A.; Deegan, S.; Lisbona, F.; Hetz, C.; Samali, A. Hsp72 protects cells from ER stress-induced apoptosis via enhancement of ire1α-xbp1 signaling through a physical interaction. PLoS Biol. 2010, 8, e1000410. [Google Scholar] [CrossRef]
- Schultz, C.R.; Golembieski, W.A.; King, D.A.; Brown, S.L.; Brodie, C.; Rempel, S.A. Inhibition of HSP27 alone or in combination with pAKT inhibition as therapeutic approaches to target SPARC-induced glioma cell survival. Mol. Cancer 2012, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Rane, M.J.; Pan, Y.; Singh, S.; Powell, D.W.; Wu, R.; Cummins, T.; Chen, Q.; McLeish, K.R.; Klein, J.B. Heat shock protein 27 controls apoptosis by regulating Akt activation. J. Biol. Chem. 2003, 278, 27828–27835. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, J.; Sang, D.; Lan, Q. Phosphorylation of AKT induced by phosphorylated Hsp27 confers the apoptosis-resistance in t-AUCB-treated glioblastoma cells in vitro. J. Neurooncol. 2015, 121, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zając, A.; Sumorek-Wiadro, J.; Langner, E.; Wertel, I.; Maciejczyk, A.; Pawlikowska-Pawlęga, B.; Pawelec, J.; Wasiak, M.; Hułas-Stasiak, M.; Bądziul, D.; et al. Involvement of PI3K Pathway in Glioma Cell Resistance to Temozolomide Treatment. Int. J. Mol. Sci. 2021, 22, 5155. https://doi.org/10.3390/ijms22105155
Zając A, Sumorek-Wiadro J, Langner E, Wertel I, Maciejczyk A, Pawlikowska-Pawlęga B, Pawelec J, Wasiak M, Hułas-Stasiak M, Bądziul D, et al. Involvement of PI3K Pathway in Glioma Cell Resistance to Temozolomide Treatment. International Journal of Molecular Sciences. 2021; 22(10):5155. https://doi.org/10.3390/ijms22105155
Chicago/Turabian StyleZając, Adrian, Joanna Sumorek-Wiadro, Ewa Langner, Iwona Wertel, Aleksandra Maciejczyk, Bożena Pawlikowska-Pawlęga, Jarosław Pawelec, Magdalena Wasiak, Monika Hułas-Stasiak, Dorota Bądziul, and et al. 2021. "Involvement of PI3K Pathway in Glioma Cell Resistance to Temozolomide Treatment" International Journal of Molecular Sciences 22, no. 10: 5155. https://doi.org/10.3390/ijms22105155
APA StyleZając, A., Sumorek-Wiadro, J., Langner, E., Wertel, I., Maciejczyk, A., Pawlikowska-Pawlęga, B., Pawelec, J., Wasiak, M., Hułas-Stasiak, M., Bądziul, D., Rzeski, W., Reichert, M., & Jakubowicz-Gil, J. (2021). Involvement of PI3K Pathway in Glioma Cell Resistance to Temozolomide Treatment. International Journal of Molecular Sciences, 22(10), 5155. https://doi.org/10.3390/ijms22105155