
Role of Matricellular CCN Proteins in Skeletal Muscle: Focus on CCN2/CTGF and Its Regulation by Vasoactive Peptides


{kind=link}
{kind=link}
{kind=link}
Abstract
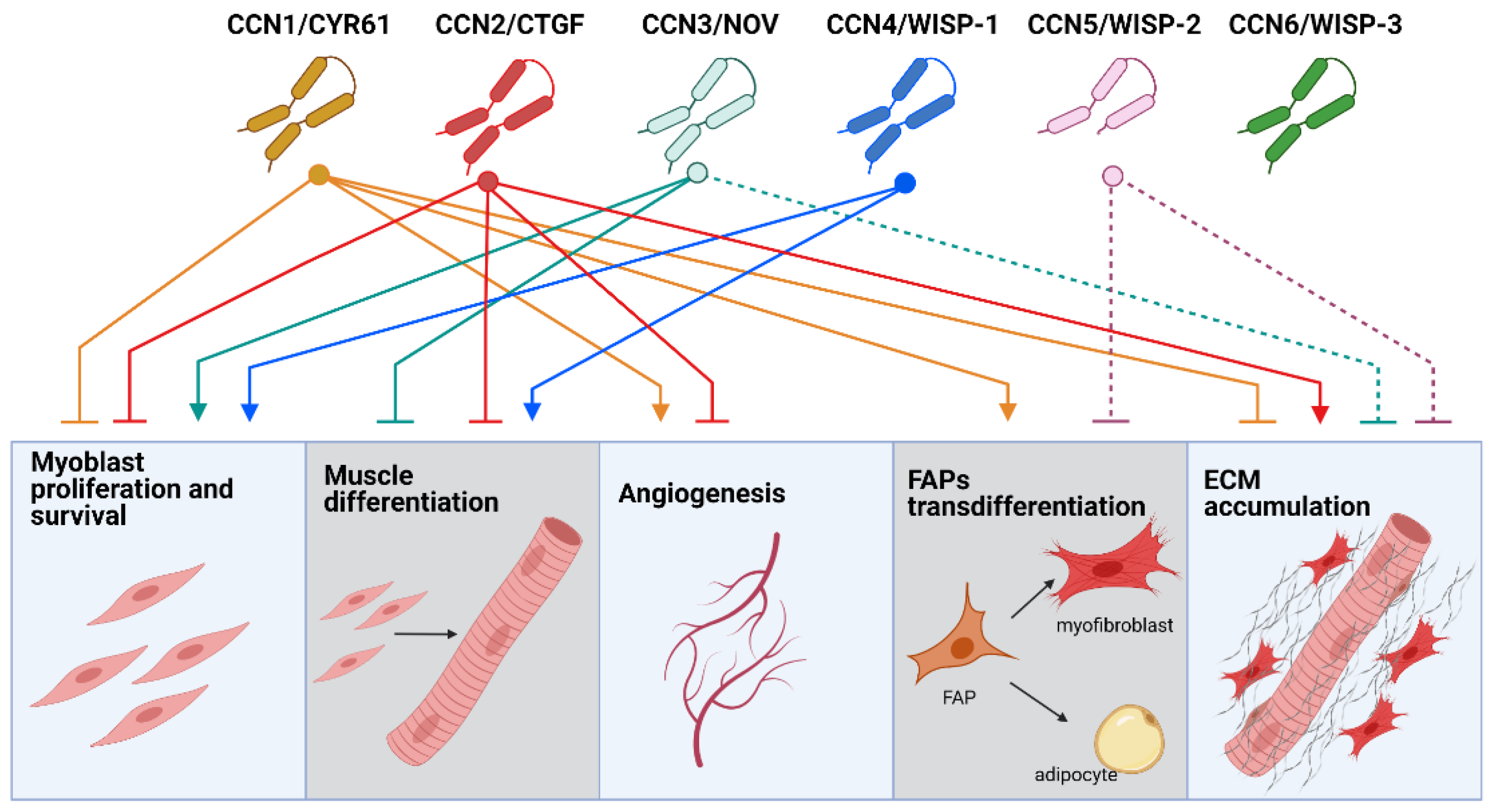
1. The CCN Family of Matricellular Proteins
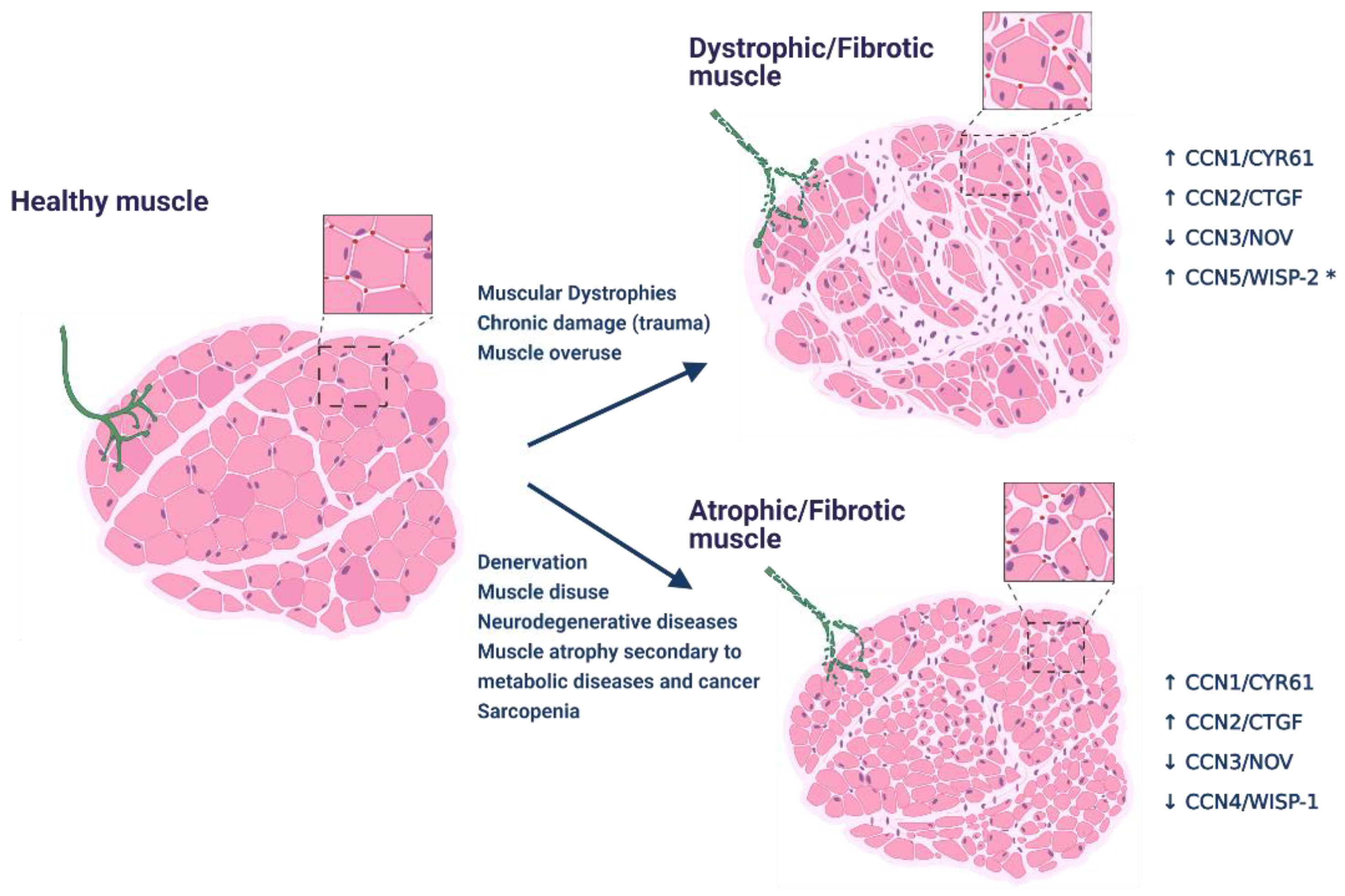
2. CCN Proteins in Skeletal Muscle Function and Disease
2.1. CCNs in Skeletal Muscle Regeneration
2.2. CCNs Role in Skeletal Muscle Atrophy
2.3. CCNs in Angiogenesis and Blood Perfusion of Skeletal Muscle
3. CCN Proteins in Skeletal Muscle Fibrosis
3.1. CCN2/CTGF Drives Skeletal Muscle Fibrosis
3.2. Role of Other CCNs in Skeletal Muscle Fibrosis
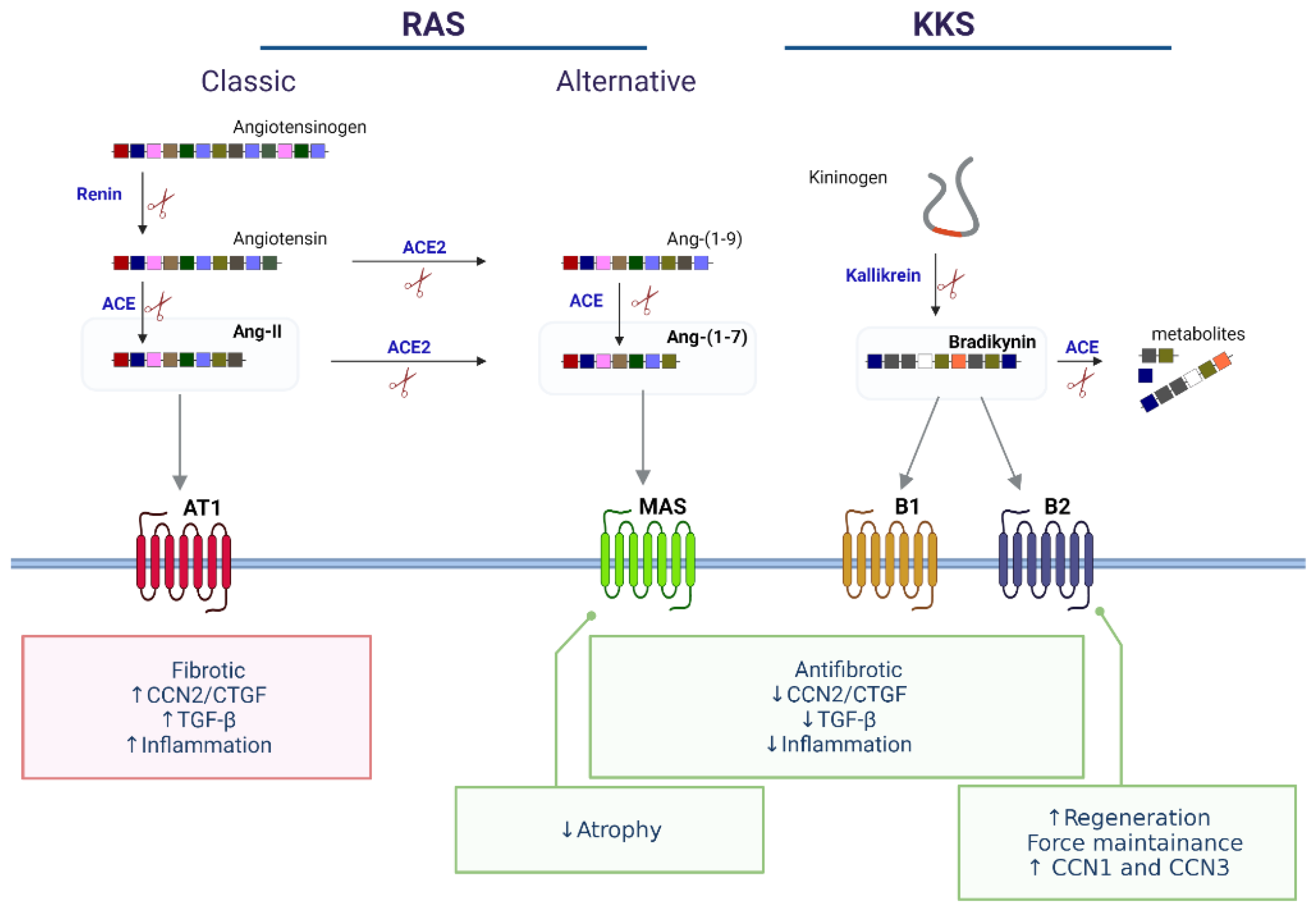
4. Regulation of CCN2/CTGF and Fibrosis in Skeletal Muscle: Role of Vasoactive Peptides
4.1. Vasoactive Peptides Modulate Fibrosis and CCN2/CTGF in Skeletal Muscle
4.2. CCN2/CTGF Is Upregulated by the RAS Classical Axis in Skeletal Muscle and Promotes Fibrosis
4.3. The Non-Classical RAS Axis and KKS Reduce CCN2/CTGF Levels in Models of Skeletal Muscle Fibrosis
4.4. Vasoactive Peptides and Other CCN Proteins
5. Summary and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119 Pt 23, 4803–4810. [Google Scholar] [CrossRef]
- Chen, C.-C.; Lau, L.F. Functions and mechanisms of action of CCN matricellular proteins. Int. J. Biochem. Cell Biol. 2009, 41, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Kubota, S.; Takigawa, M. The CCN family acting throughout the body: Recent research developments. BioMol. Concepts 2013, 4, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B.; Tweedie, S.; Bruford, E. The official unified nomenclature adopted by the HGNC calls for the use of the acronyms, CCN1-6, and discontinuation in the use of CYR61, CTGF, NOV and WISP 1-3 respectively. J. Cell Commun. Signal. 2018, 12, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B. The concept of the CCN protein family revisited: A centralized coordination network. J. Cell Commun. Signal. 2018, 12, 3–12. [Google Scholar] [CrossRef]
- Takigawa, M. The CCN proteins: An overview. Methods Mol. Biol. 2017, 1489, 1–8. [Google Scholar]
- Kaasbøll, O.J.; Gadicherla, A.K.; Wang, J.H.; Monsen, V.T.; Hagelin, E.M.V.; Dong, M.Q.; Attramadal, H. Connective tissue growth factor (CCN2) is a matricellular preproprotein controlled by proteolytic activation. J. Biol. Chem. 2018, 293, 17953–17970. [Google Scholar] [CrossRef]
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; Van Den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.F. Cell surface receptors for CCN proteins. J. Cell Commun. Signal. 2016, 10, 121–127. [Google Scholar] [CrossRef]
- Li, J.; Ye, L.; Owen, S.; Weeks, H.P.; Zhang, Z.; Jiang, W.G. Emerging role of CCN family proteins in tumorigenesis and cancer metastasis (Review). Int. J. Mol. Med. 2015, 36, 1451–1463. [Google Scholar] [CrossRef]
- Kim, H.; Son, S.; Shin, I. Role of the CCN protein family in cancer. BMB Rep. 2018, 51, 486–492. [Google Scholar] [CrossRef]
- Kular, L.; Pakradouni, J.; Kitabgi, P.; Laurent, M.; Martinerie, C. The CCN family: A new class of inflammation modulators? Biochimie 2011, 93, 377–388. [Google Scholar] [CrossRef]
- Wei, Y.; Peng, L.; Li, Y.; Zhang, N.; Shang, K.; Duan, L.; Zhong, J.; Chen, J. Higher serum CCN3 is associated with disease activity and inflammatory markers in rheumatoid arthritis. J. Immunol. Res. 2020, 2020, 3891425. [Google Scholar] [CrossRef]
- Bassyouni, I.H.; Mohammed, W.H.S.; Taha, F.M.; El Refai, R.M. Clinical significance of CCN2/connective tissue growth factor in Behçet’s disease patients. Int. J. Rheum. Dis. 2019, 22, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Winterhager, E.; Gellhaus, A. The role of the CCN family of proteins in female reproduction. Cell Mol. Life Sci. 2014, 71, 2299–2311. [Google Scholar] [CrossRef] [PubMed]
- Grünberg, J.R.; Elvin, J.; Paul, A.; Hedjazifar, S.; Hammarstedt, A.; Smith, U. CCN5/WISP2 and metabolic diseases. J. Cell Commun. Signal. 2018, 12, 309–318. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, H.; Liu, X. Emerging role of CCN family proteins in fibrosis. J. Cell. Physiol. 2021, 236, 4195–4206. [Google Scholar] [CrossRef] [PubMed]
- Díez, J.; González, A.; Ravassa, S. Understanding the role of CCN matricellular proteins in myocardial fibrosis. J. Am. Coll. Cardiol. 2016, 67, 1569–1571. [Google Scholar] [CrossRef]
- Leask, A. CCN2 in skin fibrosis. Methods Miol. Biol. 2017, 1489, 417–421. [Google Scholar]
- Yin, Q.; Liu, H. Connective Tissue Growth Factor and Renal Fibrosis; Springer: Singapore, 2019; pp. 365–380. [Google Scholar]
- Gonzalez, D.; Brandan, E. CTGF/CCN2 from skeletal muscle to nervous system: Impact on neurodegenerative diseases. Mol. Neurobiol. 2019, 56, 5911–5916. [Google Scholar] [CrossRef]
- Rebolledo, D.L.; Lipson, K.E.; Brandan, E. Driving fibrosis in neuromuscular diseases: Role and regulation of connective tissue growth factor (CCN2/CTGF). Matrix Biol. Plus 2021, in press. [Google Scholar] [CrossRef]
- Malik, A.R.; Liszewska, E.; Jaworski, J. Matricellular proteins of the CYR61/CTGF/NOV (CCN) family and the nervous system. Front. Cell. Neurosci. 2015, 9, 237. [Google Scholar] [CrossRef] [PubMed]
- Chaqour, B. Regulating the regulators of angiogenesis by CCN1 and taking it up a Notch. J. Cell Commun. Signal. 2016, 10, 259–261. [Google Scholar] [CrossRef]
- Grote, K.; Salguero, G.; Ballmaier, M.; Dangers, M.; Drexler, H.; Schieffer, B. The angiogenic factor CCN1 promotes adhesion and migration of circulating CD34+ progenitor cells: Potential role in angiogenesis and endothelial regeneration. Blood 2007, 110, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, M. CCN2: A master regulator of the genesis of bone and cartilage. J. Cell Commun. Signal. 2013, 7, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenes Tissue Repair 2012, 5 (Suppl. 1), S24. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, N.; Chu, H.Y.; Yu, Y.; Zhang, Z.K.; Zhang, G.; Zhang, B.T. Connective tissue growth factor: From molecular understandings to drug discovery. Front. Cell Dev. Biol. 2020, 8, 593269. [Google Scholar] [CrossRef]
- Leask, A. CCN2: A novel, specific and valid target for anti-fibrotic drug intervention. Expert Opin. Ther. Targets 2013, 17, 1067–1071. [Google Scholar] [CrossRef]
- Kubota, S.; Takigawa, M. Cellular and molecular actions of CCN2/CTGF and its role under physiological and pathological conditions. Clin. Sci. 2015, 128, 181–196. [Google Scholar] [CrossRef]
- Lin, C.G.; Leu, S.J.; Chen, N.; Tebeau, C.M.; Lin, S.X.; Yeung, C.Y.; Lau, L.F. CCN3 (NOV) is a novel angiogenic regulator of the CCN protein family. J. Biol. Chem. 2003, 278, 24200–24208. [Google Scholar] [CrossRef]
- Liu, J.L.; Kaddour, N.; Chowdhury, S.; Li, Q.; Gao, Z.H. Role of CCN5 (WNT1 inducible signaling pathway protein 2) in pancreatic islets. J. Diabetes 2017, 9, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Mirr, M.; Owecki, M. An Update to the WISP-1/CCN4 role in obesity, insulin resistance and diabetes. Medicina 2021, 57, 100. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Jia, S.; Ji, K.; Jiang, W.G. Wnt1 inducible signalling pathway protein-2 (WISP-2/CCN5): Roles and regulation in human cancers (review). Oncol. Rep. 2014, 31, 533–539. [Google Scholar] [CrossRef]
- Jia, Q.; Xu, B.; Zhang, Y.; Ali, A.; Liao, X. CCN Family Proteins in Cancer: Insight Into Their Structures and Coordination Role in Tumor Microenvironment. Front. Genet. 2021, 12, 649387. [Google Scholar] [CrossRef]
- Nivison, M.P.; Meier, K.E. The role of CCN4/WISP-1 in the cancerous phenotype. Cancer Manag. Res. 2018, 10, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. CCN6: A modulator of breast cancer progression. J. Cell Commun. Signal. 2016, 10, 163–164. [Google Scholar] [CrossRef]
- McMullen, E.R.; Zoumberos, N.A.; Kleer, C.G. Metaplastic breast carcinoma: Update on histopathology and molecular alterations. Arch. Pathol. Lab. Med. 2019, 143, 1492–1496. [Google Scholar] [CrossRef]
- Tran, M.N.; Kleer, C.G. Matricellular CCN6 (WISP3) protein: A tumor suppressor for mammary metaplastic carcinomas. J. Cell Commun. Signal. 2018, 12, 13–19. [Google Scholar] [CrossRef]
- Tamura, I.; Rosenbloom, J.; Macarak, E.; Chaqour, B. Regulation of Cyr61 gene expression by mechanical stretch through multiple signaling pathways. Am. J. Physiol. Cell Physiol. 2001, 281, C1524–C1532. [Google Scholar] [CrossRef]
- Schild, C.; Trueb, B. Three members of the connective tissue growth factor family CCN are differentially regulated by mechanical stress. Biochim. Biophys. Acta 2004, 1691, 33–40. [Google Scholar] [CrossRef]
- Sabaratnam, R.; Pedersen, A.J.T.; Kristensen, J.M.; Handberg, A.; Wojtaszewski, J.F.P.; Højlund, K. Intact regulation of muscle expression and circulating levels of myokines in response to exercise in patients with type 2 diabetes. Physiol. Rep. 2018, 6, e13723. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, R.; Kyröläinen, H.; Selänne, H.; Komi, P.V.; Kainulainen, H.; Vihko, V. A single bout of exercise with high mechanical loading induces the expression of Cyr61/CCN1 and CTGF/CCN2 in human skeletal muscle. J. Appl. Physiol. 2007, 103, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Klein, J.D.; Hassounah, F.; Zhang, J.; Zhang, C.; Wang, X.H. Aging increases CCN1 expression leading to muscle senescence. Am. J. Physiol. Cell Physiol. 2014, 306, C28–C36. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.S.; Mills, S.T.; Jones, K.A.; Ho, S.N.; Pavlath, G.K. A combinatorial role for NFAT5 in both myoblast migration and differentiation during skeletal muscle myogenesis. J. Cell Sci. 2006, 120, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, D.; Andermarcher, E.; Schofield, P.N.; Boulter, C.A. Mouse Nov gene is expressed in hypaxial musculature and cranial structures derived from neural crest cells and placodes. Dev. Dyn. 2000, 219, 417–425. [Google Scholar] [CrossRef]
- Heath, E.; Tahri, D.; Andermarcher, E.; Schofield, P.; Fleming, S.; Boulter, C.A. Abnormal skeletal and cardiac development, cardiomyopathy, muscle atrophy and cataracts in mice with a targeted disruption of the Nov (Ccn3) gene. BMC Dev. Biol. 2008, 8, 18. [Google Scholar] [CrossRef]
- Lafont, J.; Thibout, H.; Dubois, C.; Laurent, M.; Martinerie, C. NOV/CCN3 induces adhesion of muscle skeletal cells and cooperates with FGF2 and IGF-1 to promote proliferation and survival. Cell Commun. Adhes. 2005, 12, 41–57. [Google Scholar] [CrossRef][Green Version]
- Calhabeu, F.; Lafont, J.; Le Dreau, G.; Laurent, M.; Kazazian, C.; Schaeffer, L.; Martinerie, C.; Dubois, C. NOV/CCN3 impairs muscle cell commitment and differentiation. Exp. Cell Res. 2006, 312, 1876–1889. [Google Scholar] [CrossRef]
- Cabello-Verrugio, C.; Córdova, G.; Vial, C.; Zúñiga, L.M.; Brandan, E. Connective tissue growth factor induction by lysophosphatidic acid requires transactivation of transforming growth factor type β receptors and the JNK pathway. Cell. Signal. 2011, 23, 449–457. [Google Scholar] [CrossRef]
- Vial, C.; Gutiérrez, J.; Santander, C.; Cabrera, D.; Brandan, E. Decorin interacts with connective tissue growth factor (CTGF)/CCN2 by LRR12 inhibiting its biological activity. J. Biol. Chem. 2011, 286, 24242–24252. [Google Scholar] [CrossRef]
- Vial, C.; Zuniga, L.M.; Cabello-Verrugio, C.; Canon, P.; Fadic, R.; Brandan, E. Skeletal muscle cells express the profibrotic cytokine connective tissue growth factor (CTGF/CCN2), which induces their dedifferentiation. J. Cell. Physiol. 2008, 215, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Kubota, S.; Aoyama, E.; Janune, D.; Lyons, K.M.; Takigawa, M. CCN family protein 2 (CCN2) promotes the early differentiation, but inhibits the terminal differentiation of skeletal myoblasts. J. Biochem. 2015, 157, 91–100. [Google Scholar] [CrossRef]
- Ivkovic, S.; Yoon, B.S.; Popoff, S.N.; Safadi, F.F.; Libuda, D.E.; Stephenson, R.C.; Daluiski, A.; Lyons, K.M. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 2003, 130, 2779–2791. [Google Scholar] [CrossRef]
- Morales, M.G.; Gutierrez, J.; Cabello-Verrugio, C.; Cabrera, D.; Lipson, K.E.; Goldschmeding, R.; Brandan, E. Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum. Mol. Genet. 2013, 22, 4938–4951. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Y.; Zhang, W.; Tong, H.; Li, S.; Yan, Y. WISP1 promotes bovine MDSC differentiation via recruitment of ANXA1 for the regulation of the TGF-β signalling pathway. Mol. Cell. Biochem. 2020, 470, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Biferali, B.; Proietti, D.; Mozzetta, C.; Madaro, L. Fibro–adipogenic progenitors cross-talk in skeletal muscle: The social network. Front. Physiol. 2019, 10, 1074. [Google Scholar] [CrossRef] [PubMed]
- Lukjanenko, L.; Karaz, S.; Stuelsatz, P.; Gurriaran-Rodriguez, U.; Michaud, J.; Dammone, G.; Sizzano, F.; Mashinchian, O.; Ancel, S.; Migliavacca, E.; et al. Aging disrupts muscle stem cell function by impairing matricellular WISP1 secretion from fibro-adipogenic progenitors. Cell Stem Cell 2019, 24, 433–446.e7. [Google Scholar] [CrossRef]
- Kivelä, R.; Silvennoinen, M.; Lehti, M.; Jalava, S.; Vihko, V.; Kainulainen, H. Exercise-induced expression of angiogenic growth factors in skeletal muscle and in capillaries of healthy and diabetic mice. Cardiovasc. Diabetol. 2008, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, S.A.; Byrne, K.A.; Reverter, A.; Nattrass, G.S.; Greenwood, P.L.; Wang, Y.H.; Hudson, N.J.; Harper, G.S. Gene expression profiling of bovine skeletal muscle in response to and during recovery from chronic and severe undernutrition1. J. Anim. Sci. 2006, 84, 3239–3250. [Google Scholar] [CrossRef]
- Rabinovich, R.A.; Drost, E.; Manning, J.R.; Dunbar, D.R.; Díaz-Ramos, M.; Lakhdar, R.; Bastos, R.; MacNee, W. Genome-wide mRNA expression profiling in vastus lateralis of COPD patients with low and normal fat free mass index and healthy controls. Respir. Res. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Rebolledo, D.L.; Correa, L.M.; Court, F.A.; Cerpa, W.; Lipson, K.E.; van Zundert, B.; Brandan, E. The inhibition of CTGF/CCN2 activity improves muscle and locomotor function in a murine ALS model. Hum. Mol. Genet. 2018, 27, 2913–2926. [Google Scholar] [CrossRef]
- Hu, F.; Lin, Y.; Zuo, Y.; Chen, R.; Luo, S.; Su, Z. CCN1 induces adipogenic differentiation of fibro/adipogenic progenitors in a chronic kidney disease model. Biochem. Biophys. Res. Commun. 2019, 520, 385–391. [Google Scholar] [CrossRef]
- Contreras, O.; Rebolledo, D.L.; Oyarzún, J.E.; Olguín, H.C.; Brandan, E. Connective tissue cells expressing fibro/adipogenic progenitor markers increase under chronic damage: Relevance in fibroblast-myofibroblast differentiation and skeletal muscle fibrosis. Cell Tissue Res. 2016, 364, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; McCoy, S.C.; Ross, H.H.; Bernardo, J.A.; Beharry, A.W.; Senf, S.M.; Judge, A.R.; Beck, D.T.; Conover, C.F.; Cannady, D.F.; et al. Transcriptional regulation of myotrophic actions by testosterone and trenbolone on androgen-responsive muscle. Steroids 2014, 87, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Valle-Tenney, R.; Rebolledo, D.; Acuña, M.J.; Brandan, E. HIF-hypoxia signaling in skeletal muscle physiology and fibrosis. J. Cell Commun. Signal. 2020, 14, 147–158. [Google Scholar] [CrossRef]
- Valle-Tenney, R.; Rebolledo, D.L.; Lipson, K.E.; Brandan, E. Role of hypoxia in skeletal muscle fibrosis: Synergism between hypoxia and TGF-β signaling upregulates CCN2/CTGF expression specifically in muscle fibers. Matrix Biol. 2020, 87, 48–65. [Google Scholar] [CrossRef]
- Martin, N.R.W.; Aguilar-Agon, K.; Robinson, G.P.; Player, D.J.; Turner, M.C.; Myers, S.D.; Lewis, M.P. Hypoxia impairs muscle function and reduces myotube size in tissue engineered skeletal muscle. J. Cell. Biochem. 2017, 118, 2599–2605. [Google Scholar] [CrossRef]
- Fataccioli, V.; Abergel, V.; Wingertsmann, L.; Neuville, P.; Spitz, E.; Adnot, S.; Calenda, V.; Teiger, E. Stimulation of angiogenesis by Cyr61 gene: A new therapeutic candidate. Hum. Gene Ther. 2002, 13, 1461–1470. [Google Scholar] [CrossRef]
- Frey, S.P.; Yorumazel, B.; Hölscher-Doht, S.; Eden, L.; Schütze, N.; Meffert, R.H.; Jansen, H. CYR61 improves muscle force recreation in a rabbit trauma model. Technol. Health Care 2019, 1–12, pre-press. [Google Scholar] [CrossRef]
- Kivelä, R.; Silvennoinen, M.; Touvra, A.-M.; Maarit Lehti, T.; Kainulainen, H.; Vihko, V.; Kivelä, R.; Silvennoinen, M.; Touvra, A.-M.; Lehti, T.M.; et al. Effects of experimental type 1 diabetes and exercise training on angiogenic gene expression and capillarization in skeletal muscle. FASEB J. 2006, 20, 1570–1572. [Google Scholar] [CrossRef]
- Inoki, I.; Shiomi, T.; Hashimoto, G.; Enomoto, H.; Nakamura, H.; Makino, K.; Ikeda, E.; Takata, S.; Kobayashi, K.; Okada, Y. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002, 16, 219–221. [Google Scholar] [CrossRef]
- Chaqour, B. Caught between a “Rho” and a hard place: Are CCN1/CYR61 and CCN2/CTGF the arbiters of microvascular stiffness? J. Cell Commun. Signal. 2020, 14, 21–29. [Google Scholar] [CrossRef]
- Brandan, E.; Gutierrez, J. Role of proteoglycans in the regulation of the skeletal muscle fibrotic response. FEBS J. 2013, 280, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—a common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Muñoz-Cánoves, P.; Serrano, A.L. Macrophages decide between regeneration and fibrosis in muscle. Trends Endocrinol. Metab. 2015, 26, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Wells, R.G. Tissue mechanics and fibrosis. Biochim. Biophys. Acta 2013, 1832, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Goetsch, K.P.; Niesler, C.U. The extracellular matrix regulates the effect of decorin and transforming growth factor beta-2 (TGF-β2) on myoblast migration. Biochem. Biophys. Res. Commun. 2016, 479, 351–357. [Google Scholar] [CrossRef]
- Droguett, R.; Cabello-Verrugio, C.; Riquelme, C.; Brandan, E. Extracellular proteoglycans modify TGF-beta bio-availability attenuating its signaling during skeletal muscle differentiation. Matrix Biol. 2006, 25, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Gargioli, C.; Coletta, M.; De Grandis, F.; Cannata, S.M.; Cossu, G. PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat. Med. 2008, 14, 973–978. [Google Scholar] [CrossRef]
- Cabrera, D.; Gutiérrez, J.; Cabello-Verrugio, C.; Morales, M.G.; Mezzano, S.; Fadic, R.; Casar, J.C.; Hancke, J.L.; Brandan, E. Andrographolide attenuates skeletal muscle dystrophy in mdx mice and increases efficiency of cell therapy by reducing fibrosis. Skelet. Muscle 2014, 4, 6. [Google Scholar] [CrossRef]
- Györfi, A.H.; Matei, A.E.; Distler, J.H.W. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Accornero, F.; Kanisicak, O.; Tjondrokoesoemo, A.; Attia, A.C.; McNally, E.M.; Molkentin, J.D. Myofiber-specific inhibition of TGFβ signaling protects skeletal muscle from injury and dystrophic disease in mice. Hum. Mol. Genet. 2014, 23, 6903–6915. [Google Scholar] [CrossRef] [PubMed]
- Acuña, M.J.; Brandan, E. Analysis of pathological activities of CCN2/CTGF in muscle dystrophy. Methods Mol. Biol. 2017, 1489, 513–521. [Google Scholar]
- Petrosino, J.M.; Leask, A.; Accornero, F. Genetic manipulation of CCN2/CTGF unveils cell-specific ECM-remodeling effects in injured skeletal muscle. FASEB J. 2019, 33, 2047–2057. [Google Scholar] [CrossRef]
- Morales, M.G.; Acuña, M.J.; Cabrera, D.; Goldschmeding, R.; Brandan, E. The pro-fibrotic connective tissue growth factor (CTGF/CCN2) correlates with the number of necrotic-regenerative foci in dystrophic muscle. J. Cell Commun. Signal. 2018, 12, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.G.; Cabello-Verrugio, C.; Santander, C.; Cabrera, D.; Goldschmeding, R.; Brandan, E. CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J. Pathol. 2011, 225, 490–501. [Google Scholar] [CrossRef]
- Au, C.G.; Butler, T.L.; Sherwood, M.C.; Egan, J.R.; North, K.N.; Winlaw, D.S. Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. Int. J. Exp. Pathol. 2011, 92, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Haginoya, K.; Wu, Y.; Chiba, Y.; Nakanishi, T.; Onuma, A.; Sato, Y.; Takigawa, M.; Iinuma, K.; Tsuchiya, S. Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J. Neurol. Sci. 2008, 267, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Putker, K.; Van De Vijver, D.; Tanganyika-De Winter, C.L.; Pasteuning-Vuhman, S.; Plomp, J.J.; Aartsma-Rus, A.M.; Van Putten, M. Cross-sectional study into age-related pathology of mouse models for limb girdle muscular dystrophy types 2D and 2F. PLoS ONE 2019, 14, e0220665. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Contreras, O.; Rebolledo, D.L.; Espinoza, J.P.; van Zundert, B.; Brandan, E. ALS skeletal muscle shows enhanced TGF-β signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS ONE 2017, 12, e0177649. [Google Scholar] [CrossRef]
- Spliet, W.G.; Aronica, E.; Ramkema, M.; Aten, J.; Troost, D. Increased expression of connective tissue growth factor in amyotrophic lateral sclerosis human spinal cord. Acta Neuropathol. 2003, 106, 449–457. [Google Scholar] [CrossRef]
- Liu, F.; Tang, W.; Chen, D.; Li, M.; Gao, Y.; Zheng, H.; Chen, S. Expression of TGF-β1 and CTGF is associated with fibrosis of denervated sternocleidomastoid muscles in mice. Tohoku J. Exp. Med. 2016, 238, 49–56. [Google Scholar] [CrossRef]
- Magnusson, C.; Svensson, A.; Christerson, U.; Tågerud, S. Denervation-induced alterations in gene expression in mouse skeletal muscle. Eur. J. Neurosci. 2005, 21, 577–580. [Google Scholar] [CrossRef]
- Rebolledo, D.L.; Gonzalez, D.; Faundez-Contreras, J.; Contreras, O.; Vio, C.P.; Murphy-Ullrich, J.E.; Lipson, K.E.; Brandan, E. Denervation-induced skeletal muscle fibrosis is mediated by CTGF/CCN2 independently of TGF-beta. Matrix Biol. 2019, 82, 20–37. [Google Scholar] [CrossRef]
- Barbe, M.F.; Hilliard, B.A.; Amin, M.; Harris, M.Y.; Hobson, L.J.; Cruz, G.E.; Popoff, S.N. Blocking CTGF/CCN2 reduces established skeletal muscle fibrosis in a rat model of overuse injury. FASEB J. 2020, 34, 6554–6569. [Google Scholar] [CrossRef]
- Song, Y.; Yao, S.; Liu, Y.; Long, L.; Yang, H.; Li, Q.; Liang, J.; Li, X.; Lu, Y.; Zhu, H.; et al. Expression levels of TGF-β1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med. 2017, 13, 1209–1214. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal muscle in ALS: An unappreciated therapeutic opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef] [PubMed]
- Barbe, M.F.; Hilliard, B.A.; Amin, M.; Harris, M.Y.; Hobson, L.J.; Cruz, G.E.; Dorotan, J.T.; Paul, R.W.; Klyne, D.M.; Popoff, S.N. Blocking CTGF/CCN2 reverses neural fibrosis and sensorimotor declines in a rat model of overuse-induced median mononeuropathy. J. Orthop. Res. 2020, 38, 2396–2408. [Google Scholar] [CrossRef]
- Lipson, K.; Elmankabadi, B.; Kouchakjii, E. An open-label, single-arm Phase 2 trial evaluating pamrevlumab, a monoclonal antibody to connective tissue growth factor (CTGF) in non-ambulatory subjects with duchenne muscular dystrophy (NCT02606136). In Proceedings of the Parent Project Muscular Dystrophy Annual Meeting, Orlando, FL, USA, 26–30 June 2019. [Google Scholar]
- Riser, B.L.; Najmabadi, F.; Garchow, K.; Barnes, J.L.; Peterson, D.R.; Sukowski, E.J. Treatment with the matricellular protein CCN3 blocks and/or reverses fibrosis development in obesity with diabetic nephropathy. Am. J. Pathol. 2014, 184, 2908–2921. [Google Scholar] [CrossRef]
- Riser, B.L.; Najmabadi, F.; Perbal, B.; Peterson, D.R.; Rambow, J.A.; Riser, M.L.; Sukowski, E.; Yeger, H.; Riser, S.C. CCN3 (NOV) is a negative regulator of CCN2 (CTGF) and a novel endogenous inhibitor of the fibrotic pathway in an in vitro model of renal disease. Am. J. Pathol. 2009, 174, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Najmabadi, F.; Perbal, B.; Rambow, J.A.; Riser, M.L.; Sukowski, E.; Yeger, H.; Riser, S.C.; Peterson, D.R. CCN3/CCN2 regulation and the fibrosis of diabetic renal disease. J. Cell Commun. Signal. 2010, 4, 39–50. [Google Scholar] [CrossRef]
- Ren, Z.; Hou, Y.; Ma, S.; Tao, Y.; Li, J.; Cao, H.; Ji, L. Effects of CCN3 on fibroblast proliferation, apoptosis and extracellular matrix production. Int. J. Mol. Med. 2014, 33, 1607–1612. [Google Scholar] [CrossRef][Green Version]
- Jones, J.A.; Gray, M.R.; Oliveira, B.E.; Koch, M.; Castellot, J.J. CCN5 expression in mammals. J. Cell Commun. Signal. 2007, 1, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Li, H.; Zeng, C.; Chen, W.; Wei, L.; Liu, Y.; Qi, X. Endogenous CCN5 participates in angiotensin II/TGF-β(1) networking of cardiac fibrosis in high angiotensin II-Induced hypertensive heart failure. Front. Pharmacol. 2020, 11, 1235. [Google Scholar] [CrossRef] [PubMed]
- Yoon, P.O.; Lee, M.A.; Cha, H.; Jeong, M.H.; Kim, J.; Jang, S.P.; Choi, B.Y.; Jeong, D.; Yang, D.K.; Hajjar, R.J.; et al. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J. Mol. Cell. Cardiol. 2010, 49, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Joo, S.; Eom, G.H.; Lee, S.H.; Lee, M.A.; Lee, M.; Kim, K.W.; Kim, D.H.; Kook, H.; Kwak, T.H.; et al. CCN5 knockout mice exhibit lipotoxic cardiomyopathy with mild obesity and diabetes. PLoS ONE 2018, 13, e0207228. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Lee, M.A.; Li, Y.; Yang, D.K.; Kho, C.; Oh, J.G.; Hong, G.; Lee, A.; Song, M.H.; LaRocca, T.J.; et al. Matricellular protein CCN5 reverses established cardiac fibrosis. J. Am. Coll. Cardiol. 2016, 67, 1556–1568. [Google Scholar] [CrossRef]
- Vainio, L.E.; Szabó, Z.; Lin, R.; Ulvila, J.; Yrjölä, R.; Alakoski, T.; Piuhola, J.; Koch, W.J.; Ruskoaho, H.; Fouse, S.D.; et al. Connective tissue growth factor inhibition enhances cardiac repair and limits fibrosis after myocardial infarction. JACC Basic Transl. Sci. 2019, 4, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Accornero, F.; van Berlo, J.H.; Correll, R.N.; Elrod, J.W.; Sargent, M.A.; York, A.; Rabinowitz, J.E.; Leask, A.; Molkentin, J.D. Genetic analysis of connective tissue growth factor as an effector of transforming growth factor β signaling and cardiac remodeling. Mol. Cell. Biol. 2015, 35, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Dorn, L.E.; Petrosino, J.M.; Wright, P.; Accornero, F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J. Mol. Cell. Cardiol. 2018, 121, 205–211. [Google Scholar] [CrossRef]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef]
- Gallardo, F.S.; Córdova-Casanova, A.; Brandan, E. The linkage between inflammation and fibrosis in muscular dystrophies: The axis autotaxin–lysophosphatidic acid as a new therapeutic target? J. Cell Commun. Signal. 2021. in Press. [Google Scholar] [CrossRef]
- Riquelme-Guzmán, C.; Contreras, O.; Brandan, E. Expression of CTGF/CCN2 in response to LPA is stimulated by fibrotic extracellular matrix via the integrin/FAK axis. Am. J. Physiol. Cell Physiol. 2018, 314, C415–C427. [Google Scholar] [CrossRef]
- Felley-Bosco, E.; Stahel, R. Hippo/YAP pathway for targeted therapy. Transl. Lung Cancer Res. 2014, 3, 75–83. [Google Scholar] [PubMed]
- Watt, K.I.; Turner, B.J.; Hagg, A.; Zhang, X.; Davey, J.R.; Qian, H.; Beyer, C.; Winbanks, C.E.; Harvey, K.F.; Gregorevic, P. The Hippo pathway effector YAP is a critical regulator of skeletal muscle fibre size. Nat. Commun. 2015, 6, 6048. [Google Scholar] [CrossRef]
- Fischer, M.; Rikeit, P.; Knaus, P.; Coirault, C. YAP-mediated mechanotransduction in skeletal muscle. Front. Physiol. 2016, 7, 41. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef]
- Tao, J.; Calvisi, D.F.; Ranganathan, S.; Cigliano, A.; Zhou, L.; Singh, S.; Jiang, L.; Fan, B.; Terracciano, L.; Armeanu-Ebinger, S.; et al. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014, 147, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Et tu, CCN1…. J. Cell Commun. Signal. 2020, 14, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Lee, S.; Caesar, J.A.; Pruchenko, S.; Leask, A.; Knowles, J.A.; Sinon, J.; Chaqour, B. A CTGF-YAP regulatory pathway is essential for angiogenesis and barriergenesis in the retina. iScience 2020, 23, 101184. [Google Scholar] [CrossRef]
- Judson, R.N.; Gray, S.R.; Walker, C.; Carroll, A.M.; Itzstein, C.; Lionikas, A.; Zammit, P.S.; De Bari, C.; Wackerhage, H. Constitutive expression of yes-associated protein (Yap) in adult skeletal muscle fibres induces muscle atrophy and myopathy. PLoS ONE 2013, 8, e59622. [Google Scholar] [CrossRef]
- Jin, J.; Wang, T.; Park, W.; Li, W.; Kim, W.; Park, S.K.; Kang, K.P. Inhibition of yes-associated protein by verteporfin ameliorates unilateral ureteral obstruction-induced renal tubulointerstitial inflammation and fibrosis. Int. J. Mol. Sci. 2020, 21, 8184. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Gobeil, F. Kinins and peptide receptors. Biol. Chem. 2016, 397, 297–304. [Google Scholar] [CrossRef]
- Su, J.B. Different cross-talk sites between the renin-angiotensin and the kallikrein-kinin systems. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 319–328. [Google Scholar] [CrossRef]
- Cozzoli, A.; Nico, B.; Sblendorio, V.T.; Capogrosso, R.F.; Dinardo, M.M.; Longo, V.; Gagliardi, S.; Montagnani, M.; De Luca, A. Enalapril treatment discloses an early role of angiotensin II in inflammation- and oxidative stress-related muscle damage in dystrophic mdx mice. Pharmacol. Res. 2011, 64, 482–492. [Google Scholar] [CrossRef]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef]
- Bedair, H.S.; Karthikeyan, T.; Quintero, A.; Li, Y.; Huard, J. Angiotensin II receptor blockade administered after injury improves muscle regeneration and decreases fibrosis in normal skeletal muscle. Am. J. Sports Med. 2008, 36, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Tian, L.; Wang, B.; Ji, X.; Bai, Z.; Wang, F. Experimental study on losartan decreasing denervated skeletal muscle atrophy through reducing cell apoptosis. Chin. J. Reparative Reconstr. Surg. 2008, 22, 602–605. [Google Scholar]
- Burks, T.N.; Andres-Mateos, E.; Marx, R.; Mejias, R.; Van Erp, C.; Simmers, J.L.; Walston, J.D.; Ward, C.W.; Cohn, R.D. Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci. Transl. Med. 2011, 3, 82ra37. [Google Scholar] [CrossRef] [PubMed]
- Meinen, S.; Lin, S.; Ruegg, M.A. Angiotensin II type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-α2-deficient congenital muscular dystrophy (MDC1A). Skelet. Muscle 2012, 2, 18. [Google Scholar] [CrossRef]
- Lin, C.H.; Yang, H.; Xue, Q.L.; Chuang, Y.F.; Roy, C.N.; Abadir, P.; Walston, J.D. Losartan improves measures of activity, inflammation, and oxidative stress in older mice. Exp. Gerontol. 2014, 58, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Hall, S.E.; Sollanek, K.J.; Morton, A.B.; Talbert, E.E.; Toklu, H.Z.; Tumer, N.; Powers, S.K. AT1 receptor blocker losartan protects against mechanical ventilation-induced diaphragmatic dysfunction. J. Appl. Physiol. 2015, 119, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.M.; Kim, D.Y.; Kim, A.Y.; Lee, E.J.; Kim, S.H.; Lee, M.M.; Sung, S.E.; Park, J.K.; Jeong, K.S. Chronic effects of losartan on the muscles and the serologic profiles of mdx mice. Life Sci. 2015, 143, 35–42. [Google Scholar] [CrossRef]
- Hwang, O.K.; Park, J.K.; Lee, E.J.; Lee, E.M.; Kim, A.Y.; Jeong, K.S. Therapeutic effect of losartan, an angiotensin II Type 1 receptor antagonist, on CCl4-induced skeletal muscle injury. Int. J. Mol. Sci. 2016, 17, 227. [Google Scholar] [CrossRef]
- Han, S.O.; Haynes, A.C.; Li, S.; Abraham, D.M.; Kishnani, P.S.; Steet, R.; Koeberl, D.D. Evaluation of antihypertensive drugs in combination with enzyme replacement therapy in mice with Pompe disease. Mol. Genet. Metab. 2020, 129, 73–79. [Google Scholar] [CrossRef]
- Tawfik, V.L.; Quarta, M.; Paine, P.; Forman, T.E.; Pajarinen, J.; Takemura, Y.; Goodman, S.B.; Rando, T.A.; Clark, J.D. Angiotensin receptor blockade mimics the effect of exercise on recovery after orthopaedic trauma by decreasing pain and improving muscle regeneration. J. Physiol. 2020, 598, 317–329. [Google Scholar] [CrossRef]
- Elbaz, M.; Yanay, N.; Aga-Mizrachi, S.; Brunschwig, Z.; Kassis, I.; Ettinger, K.; Barak, V.; Nevo, Y. Losartan, a therapeutic candidate in congenital muscular dystrophy: Studies in the dy(2J) /dy(2J) mouse. Ann. Neurol. 2012, 71, 699–708. [Google Scholar] [CrossRef]
- Hord, J.M.; Garcia, M.M.; Farris, K.R.; Guzzoni, V.; Lee, Y.; Lawler, M.S.; Lawler, J.M. Nox2 signaling and muscle fiber remodeling are attenuated by losartan administration during skeletal muscle unloading. Physiol. Rep. 2021, 9, e14606. [Google Scholar] [CrossRef] [PubMed]
- Acuña, M.J.; Pessina, P.; Olguin, H.; Cabrera, D.; Vio, C.P.; Bader, M.; Muñoz-Canoves, P.; Santos, R.A.; Cabello-Verrugio, C.; Brandan, E. Restoration of muscle strength in dystrophic muscle by angiotensin-1-7 through inhibition of TGF-β signalling. Hum. Mol. Genet. 2014, 23, 1237–1249. [Google Scholar] [CrossRef]
- Acuña, M.J.; Salas, D.; Córdova-Casanova, A.; Cruz-Soca, M.; Céspedes, C.; Vio, C.P.; Brandan, E. Blockade of Bradykinin receptors worsens the dystrophic phenotype of mdx mice: Differential effects for B1 and B2 receptors. J. Cell Commun. Signal. 2018, 12, 589–601. [Google Scholar] [CrossRef]
- Cabello-Verrugio, C.; Morales, M.G.; Cabrera, D.; Vio, C.P.; Brandan, E. Angiotensin II receptor type 1 blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J. Cell. Mol. Med. 2012, 16, 752–764. [Google Scholar] [CrossRef]
- Morales, M.G.; Cabrera, D.; Céspedes, C.; Vio, C.P.; Vazquez, Y.; Brandan, E.; Cabello-Verrugio, C. Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res. 2013, 353, 173–187. [Google Scholar] [CrossRef]
- Cabello-Verrugio, C.; Rivera, J.C.; Garcia, D. Skeletal muscle wasting: New role of nonclassical renin-angiotensin system. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Igić, R.; Škrbić, R. The renin-angiotensin system and its blockers. Srp. Arh. Celok. Lek. 2014, 142, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Gobeil, F. Kallikrein–kinin system as the dominant mechanism to counteract hyperactive renin–angiotensin system. Can. J. Physiol. Pharmmacol. 2017, 95, 1117–1124. [Google Scholar] [CrossRef]
- Parreiras, E.S.L.T.; Reis, R.I.; Santos, G.A.; Pires-Oliveira, M.; Pesquero, J.B.; Gomes, M.D.; Godinho, R.O.; Costa-Neto, C.M. The kinin B1 receptor regulates muscle-specific E3 ligases expression and is involved in skeletal muscle mass control. Clin. Sci. 2014, 127, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Dabiré, H.; Barthélémy, I.; Blanchard-Gutton, N.; Sambin, L.; Sampedrano, C.C.; Gouni, V.; Unterfinger, Y.; Aguilar, P.; Thibaud, J.-L.; Ghaleh, B.; et al. Vascular endothelial dysfunction in Duchenne muscular dystrophy is restored by bradykinin through upregulation of eNOS and nNOS. Basic Res. Cardiol. 2011, 107, 240. [Google Scholar] [CrossRef][Green Version]
- Powers, S.K.; Morton, A.B.; Hyatt, H.; Hinkley, M.J. The renin-angiotensin system and skeletal muscle. Exerc. Sport Sci. Rev. 2018, 46, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhang, X.; Wang, J.; Wang, S.; Xiao, X.; Wang, R.; Li, P.; Wang, Y. Angiotensin II induces connective tissue growth factor expression in human hepatic stellate cells by a transforming growth factor β-independent mechanism. Sci. Rep. 2017, 7, 7841. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Øie, E.; Vinge, L.E.; Yndestad, A.; Øystein Andersen, G.; Andersson, Y.; Attramadal, T.; Attramadal, H. Connective tissue growth factor—A novel mediator of angiotensin II-stimulated cardiac fibroblast activation in heart failure in rats. J. Mol. Cell. Cardiol. 2004, 36, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chung, A.C.; Huang, X.R.; Lan, H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: The role of Smad3. Hypertension 2009, 54, 877–884. [Google Scholar] [CrossRef]
- Rupérez, M.; Ruiz-Ortega, M.; Esteban, V.; Lorenzo, O.; Mezzano, S.; Plaza, J.J.; Egido, J. Angiotensin II increases connective tissue growth factor in the kidney. Am. J. Pathol. 2003, 163, 1937–1947. [Google Scholar] [CrossRef][Green Version]
- Rodríguez-Vita, J.; Sánchez-López, E.; Esteban, V.; Rupérez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef]
- Cabello-Verrugio, C.; Acuna, M.J.; Morales, M.G.; Becerra, A.; Simon, F.; Brandan, E. Fibrotic response induced by angiotensin-II requires NAD(P)H oxidase-induced reactive oxygen species (ROS) in skeletal muscle cells. Biochem. Biophys. Res. Commun. 2011, 410, 665–670. [Google Scholar] [CrossRef]
- Morales, M.G.; Vazquez, Y.; Acuña, M.J.; Rivera, J.C.; Simon, F.; Salas, J.D.; Alvarez Ruf, J.; Brandan, E.; Cabello-Verrugio, C. Angiotensin II-induced pro-fibrotic effects require p38MAPK activity and transforming growth factor beta 1 expression in skeletal muscle cells. Int. J. Biochem. Cell Biol. 2012, 44, 1993–2002. [Google Scholar] [CrossRef]
- Passos-Silva, D.G.; Brandan, E.; Santos, R.A. Angiotensins as therapeutic targets beyond heart disease. Trends Pharmacol. Sci. 2015, 36, 310–320. [Google Scholar] [CrossRef]
- Sabharwal, R.; Chapleau, M.W. Autonomic, locomotor and cardiac abnormalities in a mouse model of muscular dystrophy: Targeting the renin-angiotensin system. Exp. Physiol. 2014, 99, 627–631. [Google Scholar] [CrossRef]
- Willey, J.S.; Bracey, D.N.; Gallagher, P.E.; Tallant, E.A.; Wiggins, W.F.; Callahan, M.F.; Smith, T.L.; Emory, C.L. Angiotensin-(1-7) attenuates skeletal muscle fibrosis and stiffening in a mouse model of extremity sarcoma radiation therapy. J. Bone Jt. Surg. Am. 2016, 98, 48–55. [Google Scholar] [CrossRef]
- Desposito, D.; Potier, L.; Chollet, C.; Gobeil, F., Jr.; Roussel, R.; Alhenc-Gelas, F.; Bouby, N.; Waeckel, L. Kinin receptor agonism restores hindlimb postischemic neovascularization capacity in diabetic mice. J. Pharmacol. Exp. Ther. 2015, 352, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Graiani, G.; Salis, M.B.; Gadau, S.; Desortes, E.; Madeddu, P. Prophylactic gene therapy with human tissue kallikrein ameliorates limb ischemia recovery in Type 1 diabetic mice. Diabetes 2004, 53, 1096–1103. [Google Scholar] [CrossRef]
- Hamid, S.; Rhaleb, I.A.; Kassem, K.M.; Rhaleb, N.E. Role of kinins in hypertension and heart failure. Pharmaceuticals 2020, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Gobeil, F., Jr. Critical insights into the beneficial and protective actions of the kallikrein-kinin system. Vasc. Pharmacol. 2015, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Sander, M.; Lau, K.S.; Huang, P.L.; Stull, J.T.; Victor, R.G. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. USA 1998, 95, 15090–15095. [Google Scholar] [CrossRef]
- Thomas, G.D.; Shaul, P.W.; Yuhanna, I.S.; Froehner, S.C.; Adams, M.E. Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal Nitric oxide synthase. Circ. Res. 2003, 92, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo, D.L.; Kim, M.J.; Whitehead, N.P.; Adams, M.E.; Froehner, S.C. Sarcolemmal targeting of nNOSμ improves contractile function of mdx muscle. Hum. Mol. Genet. 2016, 25, 158–166. [Google Scholar] [CrossRef]
- Alves, J.M.; Martins, A.H.; Lameu, C.; Glaser, T.; Boukli, N.M.; Bassaneze, V.; Dariolli, R.; Nascimento, I.C.; Martins, P.C.M.; de Souza, H.D.N.; et al. Kinin-B2 receptor activity in skeletal muscle regeneration and myoblast differentiation. Stem Cell Rev. Rep. 2019, 15, 48–58. [Google Scholar] [CrossRef]
- Bonda, T.A.; Kamiński, K.A.; Dziemidowicz, M.; Litvinovich, S.; Kożuch, M.; Hirnle, T.; Dmitruk, I.; Chyczewski, L.; Winnicka, M.M. Atrial expression of the CCN1 and CCN2 proteins in chronic heart failure. Folia Histochem. Cytobiol. 2012, 50, 99–103. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bonda, T.A.; Kożuch, M.; Litvinovich, S.; Bialuk, I.; Taranta, A.; Lipiec, P.; Szymczyk, E.; Musiał, W.J.; Winnicka, M.M.; Kamiński, K.A. Transcriptional and post-transcriptional regulation of CCN genes in failing heart. Pharmacol. Rep. 2015, 67, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guleria, R.S.; Thakur, S.; Zhang, C.L.; Pan, J.; Baker, K.M.; Gupta, S. Thymosin β4 prevents angiotensin II-induced cardiomyocyte growth by regulating Wnt/WISP signaling. J. Cell. Physiol. 2016, 231, 1737–1744. [Google Scholar] [CrossRef]
- Shanmugam, P.; Valente, A.J.; Prabhu, S.D.; Venkatesan, B.; Yoshida, T.; Delafontaine, P.; Chandrasekar, B. Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2011, 50, 928–938. [Google Scholar] [CrossRef] [PubMed][Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rebolledo, D.L.; Acuña, M.J.; Brandan, E. Role of Matricellular CCN Proteins in Skeletal Muscle: Focus on CCN2/CTGF and Its Regulation by Vasoactive Peptides. Int. J. Mol. Sci. 2021, 22, 5234. https://doi.org/10.3390/ijms22105234
Rebolledo DL, Acuña MJ, Brandan E. Role of Matricellular CCN Proteins in Skeletal Muscle: Focus on CCN2/CTGF and Its Regulation by Vasoactive Peptides. International Journal of Molecular Sciences. 2021; 22(10):5234. https://doi.org/10.3390/ijms22105234
Chicago/Turabian StyleRebolledo, Daniela L., María José Acuña, and Enrique Brandan. 2021. "Role of Matricellular CCN Proteins in Skeletal Muscle: Focus on CCN2/CTGF and Its Regulation by Vasoactive Peptides" International Journal of Molecular Sciences 22, no. 10: 5234. https://doi.org/10.3390/ijms22105234
APA StyleRebolledo, D. L., Acuña, M. J., & Brandan, E. (2021). Role of Matricellular CCN Proteins in Skeletal Muscle: Focus on CCN2/CTGF and Its Regulation by Vasoactive Peptides. International Journal of Molecular Sciences, 22(10), 5234. https://doi.org/10.3390/ijms22105234