
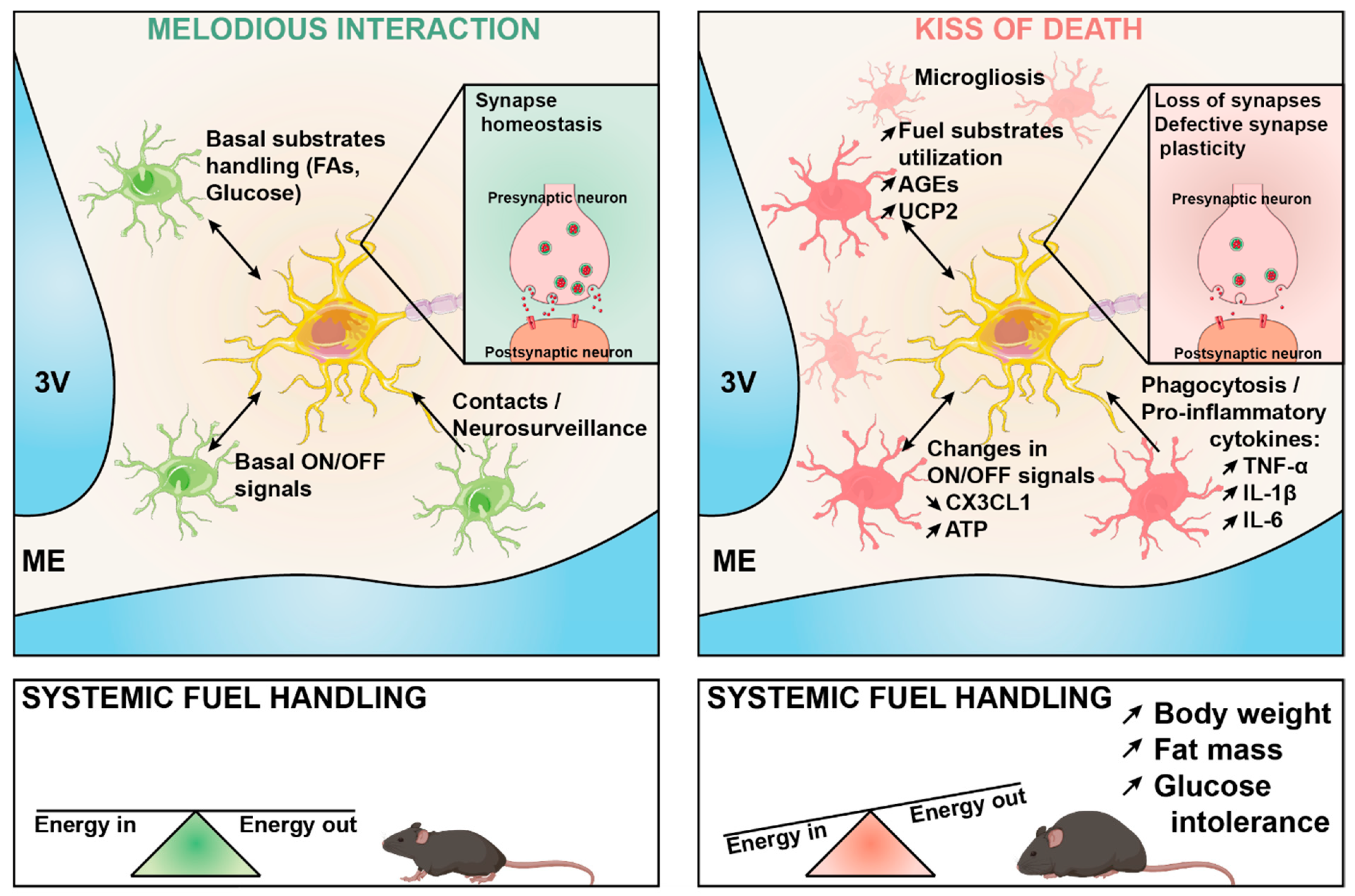
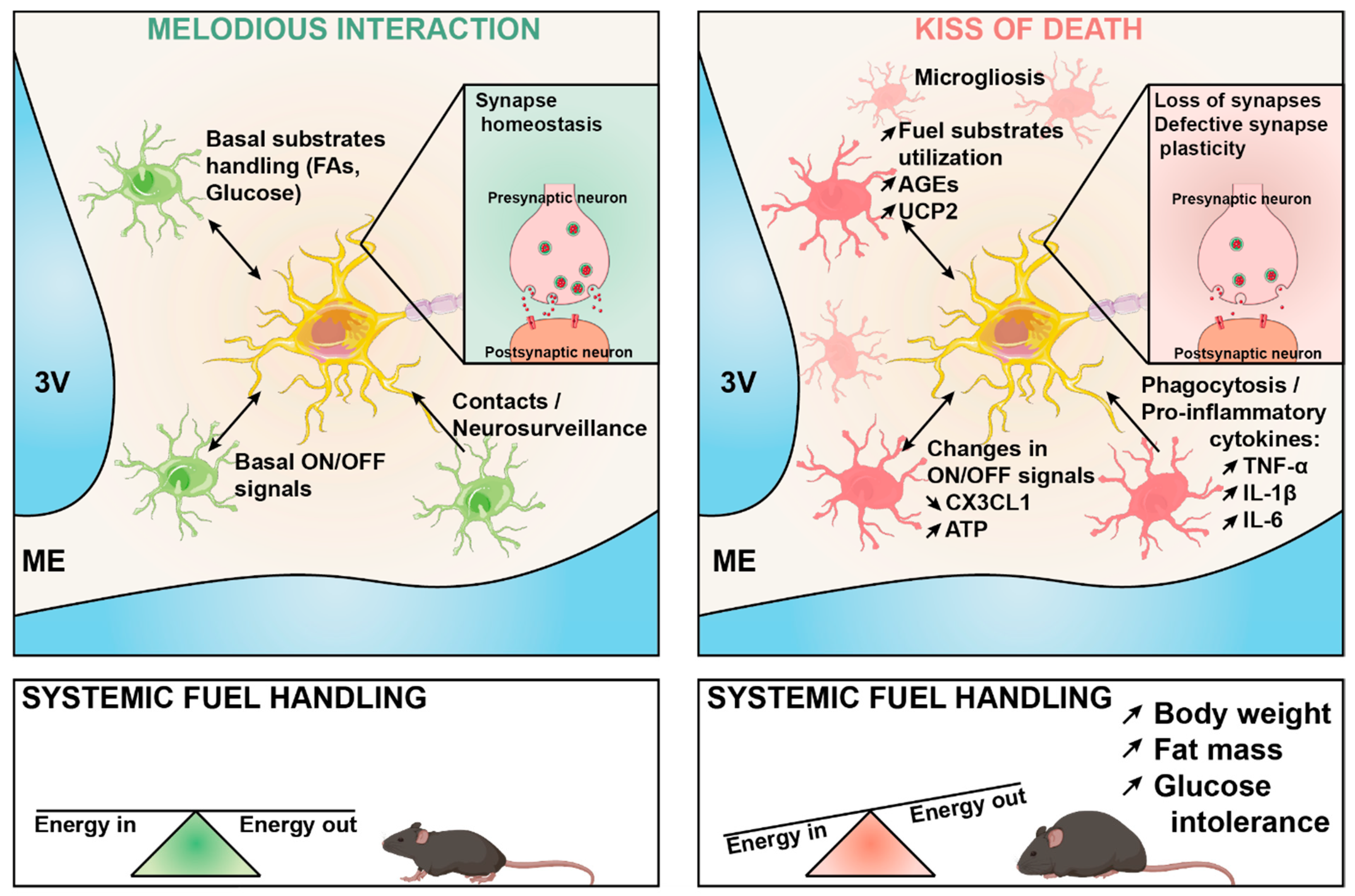
Microglia–Neuron Crosstalk in Obesity: Melodious Interaction or Kiss of Death?


{kind=link}
Abstract
:1. Introduction
2. The Emerging Neuroimmune Theory of Obesity
3. Role of Microglia in the Remodeling of Neuronal Networks in Obesity
4. Impact of Fuel Substrates on Microglia–Neuron Communication in Obesity
5. Role of Cytokines in Microglia–Neuron Communication in Obesity
6. “ON/OFF” Communication between Microglia and Neurons in Obesity
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedman, J. The Long Road to Leptin. J. Clin. Investig. 2016, 126, 4727–4734. [Google Scholar] [CrossRef] [Green Version]
- Elmquist, J.K.; Maratos-Flier, E.; Saper, C.B.; Flier, J.S. Unraveling the Central Nervous System Pathways Underlying Responses to Leptin. Nat. Neurosci. 1998, 1, 445–450. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic Studies of Body Mass Index Yield New Insights for Obesity Biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity Is Associated with Hypothalamic Injury in Rodents and Humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, T.L.; Sarman, B.; García-Cáceres, C.; Enriori, P.J.; Sotonyi, P.; Shanabrough, M.; Borok, E.; Argente, J.; Chowen, J.A.; Perez-Tilve, D.; et al. Synaptic Input Organization of the Melanocortin System Predicts Diet-Induced Hypothalamic Reactive Gliosis and Obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 14875–14880. [Google Scholar] [CrossRef] [Green Version]
- Paeger, L.; Pippow, A.; Hess, S.; Paehler, M.; Klein, A.C.; Husch, A.; Pouzat, C.; Brüning, J.C.; Kloppenburg, P. Energy Imbalance Alters Ca2+ Handling and Excitability of POMC Neurons. eLife 2017, 6, e25641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarta, C.; Fioramonti, X.; Cota, D. POMC Neurons Dysfunction in Diet-Induced Metabolic Disease: Hallmark or Mechanism of Disease? Neuroscience 2019. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Woods, S.C.; Porte, D.; Seeley, R.J.; Baskin, D.G. Central Nervous System Control of Food Intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef]
- Cota, D.; Proulx, K.; Seeley, R.J. The Role of CNS Fuel Sensing in Energy and Glucose Regulation. Gastroenterology 2007, 132, 2158–2168. [Google Scholar] [CrossRef]
- Koch, M.; Horvath, T.L. Molecular and Cellular Regulation of Hypothalamic Melanocortin Neurons Controlling Food Intake and Energy Metabolism. Mol. Psychiatry 2014, 19, 752–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, M.O.; Horvath, T.L. Hypothalamic Control of Energy Balance: Insights into the Role of Synaptic Plasticity. Trends Neurosci. 2013, 36, 65–73. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite Synapses: Astrocytes Process and Control Synaptic Information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Eroglu, C.; Barres, B.A. Regulation of Synaptic Connectivity by Glia. Nature 2010, 468, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [Green Version]
- André, C.; Guzman-Quevedo, O.; Rey, C.; Rémus-Borel, J.; Clark, S.; Castellanos-Jankiewicz, A.; Ladeveze, E.; Leste-Lasserre, T.; Nadjar, A.; Abrous, D.N.; et al. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 2017, 66, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Myers, M.G.; Koliwad, S.K. Hypothalamic Microglia as Potential Regulators of Metabolic Physiology. Nat. Metab. 2019, 1, 314–320. [Google Scholar] [CrossRef]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia-Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef]
- Dantzer, R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef] [PubMed]
- Pohl, J.; Sheppard, M.; Luheshi, G.N.; Woodside, B. Diet-Induced Weight Gain Produces a Graded Increase in Behavioral Responses to an Acute Immune Challenge. Brain Behav. Immun. 2014, 35, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Pohl, J.; Woodside, B.; Luheshi, G.N. Leptin Modulates the Late Fever Response to LPS in Diet-Induced Obese Animals. Brain Behav. Immun. 2014, 42, 41–47. [Google Scholar] [CrossRef]
- Leyrolle, Q.; Layé, S.; Nadjar, A. Direct and Indirect Effects of Lipids on Microglia Function. Neurosci. Lett. 2019, 708, 134348. [Google Scholar] [CrossRef]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia Dictate the Impact of Saturated Fat Consumption on Hypothalamic Inflammation and Neuronal Function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Bielohuby, M.; Fleming, T.; Grabner, G.F.; Foppen, E.; Bernhard, W.; Guzmán-Ruiz, M.; Layritz, C.; Legutko, B.; Zinser, E.; et al. Dietary Sugars, Not Lipids, Drive Hypothalamic Inflammation. Mol. Metab. 2017, 6, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Vidal-Itriago, A.; Milanova, I.; Korpel, N.L.; Kalsbeek, M.J.; Tom, R.Z.; Kalsbeek, A.; Hofmann, S.M.; Yi, C.-X. Deficiency of Leptin Receptor in Myeloid Cells Disrupts Hypothalamic Metabolic Circuits and Causes Body Weight Increase. Mol. Metab. 2018, 7, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Velloso, L.A.; Schwartz, M.W. Altered Hypothalamic Function in Diet-Induced Obesity. Int. J. Obes. 2011, 35, 1455–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, W.L.; Bikman, B.T.; Wang, L.-P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-Induced Insulin Resistance Mediated by the Proinflammatory Receptor TLR4 Requires Saturated Fatty Acid-Induced Ceramide Biosynthesis in Mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Kleinridders, A.; Schenten, D.; Könner, A.C.; Belgardt, B.F.; Mauer, J.; Okamura, T.; Wunderlich, F.T.; Medzhitov, R.; Brüning, J.C. MyD88 Signaling in the CNS Is Required for Development of Fatty Acid-Induced Leptin Resistance and Diet-Induced Obesity. Cell Metab. 2009, 10, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.L.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G.; Ozcan, U. Endoplasmic Reticulum Stress Plays a Central Role in Development of Leptin Resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-KappaB and ER Stress Link Overnutrition to Energy Imbalance and Obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.A.; Velloso, L.A. Consumption of a Fat-Rich Diet Activates a Proinflammatory Response and Induces Insulin Resistance in the Hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [Green Version]
- Kreutzer, C.; Peters, S.; Schulte, D.M.; Fangmann, D.; Türk, K.; Wolff, S.; van Eimeren, T.; Ahrens, M.; Beckmann, J.; Schafmayer, C.; et al. Hypothalamic Inflammation in Human Obesity Is Mediated by Environmental and Genetic Factors. Diabetes 2017, 66, 2407–2415. [Google Scholar] [CrossRef] [Green Version]
- Schur, E.A.; Melhorn, S.J.; Oh, S.-K.; Lacy, J.M.; Berkseth, K.E.; Guyenet, S.J.; Sonnen, J.A.; Tyagi, V.; Rosalynn, M.; De Leon, B.; et al. Radiologic Evidence That Hypothalamic Gliosis Is Associated with Obesity and Insulin Resistance in Humans. Obesity 2015, 23, 2142–2148. [Google Scholar] [CrossRef]
- Baufeld, C.; Osterloh, A.; Prokop, S.; Miller, K.R.; Heppner, F.L. High-Fat Diet-Induced Brain Region-Specific Phenotypic Spectrum of CNS Resident Microglia. Acta Neuropathol. 2016, 132, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Kalsbeek, M.J.; Wolff, S.E.; Korpel, N.L.; la Fleur, S.E.; Romijn, J.A.; Fliers, E.; Kalsbeek, A.; Swaab, D.F.; Huitinga, I.; Hol, E.M.; et al. The Impact of Antidiabetic Treatment on Human Hypothalamic Infundibular Neurons and Microglia. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197.e3. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Xu, A.W.; Koliwad, S.K. Hypothalamic Inflammation in the Control of Metabolic Function. Annu. Rev. Physiol. 2015, 77, 131–160. [Google Scholar] [CrossRef]
- Quarta, C.; Clemmensen, C.; Zhu, Z.; Yang, B.; Joseph, S.S.; Lutter, D.; Yi, C.-X.; Graf, E.; García-Cáceres, C.; Legutko, B.; et al. Molecular Integration of Incretin and Glucocorticoid Action Reverses Immunometabolic Dysfunction and Obesity. Cell Metab. 2017, 26, 620–632.e6. [Google Scholar] [CrossRef] [Green Version]
- Clasadonte, J.; Prevot, V. The Special Relationship: Glia-Neuron Interactions in the Neuroendocrine Hypothalamus. Nat. Rev. Endocrinol. 2018, 14, 25–44. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Thaler, J.P.; Guyenet, S.J.; Dorfman, M.D.; Wisse, B.E.; Schwartz, M.W. Hypothalamic Inflammation: Marker or Mechanism of Obesity Pathogenesis? Diabetes 2013, 62, 2629–2634. [Google Scholar] [CrossRef] [Green Version]
- Cansell, C.; Stobbe, K.; Sanchez, C.; Le Thuc, O.; Mosser, C.-A.; Ben-Fradj, S.; Leredde, J.; Lebeaupin, C.; Debayle, D.; Fleuriot, L.; et al. Dietary Fat Exacerbates Postprandial Hypothalamic Inflammation Involving Glial Fibrillary Acidic Protein-Positive Cells and Microglia in Male Mice. Glia 2021, 69, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Münzberg, H.; Flier, J.S.; Bjørbæk, C. Region-Specific Leptin Resistance within the Hypothalamus of Diet-Induced Obese Mice. Endocrinology 2004, 145, 4880–4889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olofsson, L.E.; Unger, E.K.; Cheung, C.C.; Xu, A.W. Modulation of AgRP-Neuronal Function by SOCS3 as an Initiating Event in Diet-Induced Hypothalamic Leptin Resistance. Proc. Natl. Acad. Sci. USA 2013, 110, E697–E706. [Google Scholar] [CrossRef] [Green Version]
- Cavadas, C.; Aveleira, C.A.; Souza, G.F.P.; Velloso, L.A. The Pathophysiology of Defective Proteostasis in the Hypothalamus—From Obesity to Ageing. Nat. Rev. Endocrinol. 2016, 12, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, S.; Claret, M. Hypothalamic ER Stress: A Bridge between Leptin Resistance and Obesity. FEBS Lett. 2015, 589, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.C.; Coope, A.; Morari, J.; Cintra, D.E.; Roman, E.A.; Pauli, J.R.; Romanatto, T.; Carvalheira, J.B.; Oliveira, A.L.R.; Saad, M.J.; et al. High-Fat Diet Induces Apoptosis of Hypothalamic Neurons. PLoS ONE 2009, 4, e5045. [Google Scholar] [CrossRef]
- Nyamugenda, E.; Trentzsch, M.; Russell, S.; Miles, T.; Boysen, G.; Phelan, K.D.; Baldini, G. Injury to Hypothalamic Sim1 Neurons Is a Common Feature of Obesity by Exposure to High-Fat Diet in Male and Female Mice. J. Neurochem. 2019, 149, 73–97. [Google Scholar] [CrossRef]
- Lemus, M.B.; Bayliss, J.A.; Lockie, S.H.; Santos, V.V.; Reichenbach, A.; Stark, R.; Andrews, Z.B. A Stereological Analysis of NPY, POMC, Orexin, GFAP Astrocyte, and Iba1 Microglia Cell Number and Volume in Diet-Induced Obese Male Mice. Endocrinology 2015, 156, 1701–1713. [Google Scholar] [CrossRef]
- Bessis, A.; Béchade, C.; Bernard, D.; Roumier, A. Microglial Control of Neuronal Death and Synaptic Properties. Glia 2007, 55, 233–238. [Google Scholar] [CrossRef]
- Reis, W.L.; Yi, C.-X.; Gao, Y.; Tschöp, M.H.; Stern, J.E. Brain Innate Immunity Regulates Hypothalamic Arcuate Neuronal Activity and Feeding Behavior. Endocrinology 2015, 156, 1303–1315. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; Dey, A.; Yu, X.; Stranahan, A.M. Dietary Obesity Reversibly Induces Synaptic Stripping by Microglia and Impairs Hippocampal Plasticity. Brain Behav. Immun. 2016, 51, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Cope, E.C.; LaMarca, E.A.; Monari, P.K.; Olson, L.B.; Martinez, S.; Zych, A.D.; Katchur, N.J.; Gould, E. Microglia Play an Active Role in Obesity-Associated Cognitive Decline. J. Neurosci. 2018, 38, 8889–8904. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, T.M.; Kleopoulos, S.P.; Bergen, H.T.; Roberts, J.L.; Priest, C.A.; Mobbs, C.V. Hypothalamic Pro-Opiomelanocortin MRNA Is Reduced by Fasting and [Corrected] in Ob/Ob and Db/Db Mice, but Is Stimulated by Leptin. Diabetes 1998, 47, 294–297. [Google Scholar] [CrossRef] [Green Version]
- Horvath, T.L. Synaptic Plasticity in Energy Balance Regulation. Obesity 2006, 14 (Suppl. S5), 228S–233S. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Ottaway, N.; Schriever, S.C.; Legutko, B.; García-Cáceres, C.; de la Fuente, E.; Mergen, C.; Bour, S.; Thaler, J.P.; Seeley, R.J.; et al. Hormones and Diet, but Not Body Weight, Control Hypothalamic Microglial Activity. Glia 2014, 62, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Lafrance, V.; Inoue, W.; Kan, B.; Luheshi, G.N. Leptin Modulates Cell Morphology and Cytokine Release in Microglia. Brain Behav. Immun. 2010, 24, 358–365. [Google Scholar] [CrossRef]
- Bernier, L.-P.; York, E.M.; Kamyabi, A.; Choi, H.B.; Weilinger, N.L.; MacVicar, B.A. Microglial Metabolic Flexibility Supports Immune Surveillance of the Brain Parenchyma. Nat. Commun. 2020, 11, 1559. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced Glycoxidation and Lipoxidation End Products (AGEs and ALEs): An Overview of Their Mechanisms of Formation. Free Radic. Res. 2013, 47 (Suppl. S1), 3–27. [Google Scholar] [CrossRef] [Green Version]
- Horvath, T.L.; Andrews, Z.B.; Diano, S. Fuel Utilization by Hypothalamic Neurons: Roles for ROS. Trends Endocrinol. Metab. 2009, 20, 78–87. [Google Scholar] [CrossRef]
- Parton, L.E.; Ye, C.P.; Coppari, R.; Enriori, P.J.; Choi, B.; Zhang, C.-Y.; Xu, C.; Vianna, C.R.; Balthasar, N.; Lee, C.E.; et al. Glucose Sensing by POMC Neurons Regulates Glucose Homeostasis and Is Impaired in Obesity. Nature 2007, 449, 228–232. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Liu, Z.-W.; Walllingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschöp, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.I.; et al. UCP2 Mediates Ghrelin’s Action on NPY/AgRP Neurons by Lowering Free Radicals. Nature 2008, 454, 846–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.D.; Yoon, N.A.; Jin, S.; Diano, S. Microglial UCP2 Mediates Inflammation and Obesity Induced by High-Fat Feeding. Cell Metab. 2019, 30, 952–962.e5. [Google Scholar] [CrossRef]
- Gao, Y.; Vidal-Itriago, A.; Kalsbeek, M.J.; Layritz, C.; García-Cáceres, C.; Tom, R.Z.; Eichmann, T.O.; Vaz, F.M.; Houtkooper, R.H.; van der Wel, N.; et al. Lipoprotein Lipase Maintains Microglial Innate Immunity in Obesity. Cell Rep. 2017, 20, 3034–3042. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, M.; Niidome, T.; Matsuda, S.; Akaike, A.; Kihara, T.; Sugimoto, H. Microglia-Derived Interleukin-6 and Leukaemia Inhibitory Factor Promote Astrocytic Differentiation of Neural Stem/Progenitor Cells. Eur. J. Neurosci. 2007, 25, 649–658. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia Protect Neurons against Ischemia by Synthesis of Tumor Necrosis Factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef]
- Ropelle, E.R.; Flores, M.B.; Cintra, D.E.; Rocha, G.Z.; Pauli, J.R.; Morari, J.; de Souza, C.T.; Moraes, J.C.; Prada, P.O.; Guadagnini, D.; et al. IL-6 and IL-10 Anti-Inflammatory Activity Links Exercise to Hypothalamic Insulin and Leptin Sensitivity through IKKbeta and ER Stress Inhibition. PLoS Biol. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.X.; Walter, M.; Gao, Y.; Pitra, S.; Legutko, B.; Kälin, S.; Layritz, C.; García-Cáceres, C.; Bielohuby, M.; Bidlingmaier, M.; et al. TNFα Drives Mitochondrial Stress in POMC Neurons in Obesity. Nat. Commun. 2017, 8, 15143. [Google Scholar] [CrossRef] [Green Version]
- Jais, A.; Brüning, J.C. Hypothalamic Inflammation in Obesity and Metabolic Disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef]
- Bobbo, V.C.D.; Jara, C.P.; Mendes, N.F.; Morari, J.; Velloso, L.A.; Araújo, E.P. Interleukin-6 Expression by Hypothalamic Microglia in Multiple Inflammatory Contexts: A Systematic Review. Available online: https://www.hindawi.com/journals/bmri/2019/1365210/ (accessed on 23 February 2021).
- Schöbitz, B.; Pezeshki, G.; Pohl, T.; Hemmann, U.; Heinrich, P.C.; Holsboer, F.; Reul, J.M. Soluble Interleukin-6 (IL-6) Receptor Augments Central Effects of IL-6 in Vivo. FASEB J. 1995, 9, 659–664. [Google Scholar] [CrossRef]
- Wallenius, K.; Wallenius, V.; Sunter, D.; Dickson, S.L.; Jansson, J.-O. Intracerebroventricular Interleukin-6 Treatment Decreases Body Fat in Rats. Biochem. Biophys. Res. Commun. 2002, 293, 560–565. [Google Scholar] [CrossRef]
- Wallenius, V.; Wallenius, K.; Ahrén, B.; Rudling, M.; Carlsten, H.; Dickson, S.L.; Ohlsson, C.; Jansson, J.-O. Interleukin-6-Deficient Mice Develop Mature-Onset Obesity. Nat. Med. 2002, 8, 75–79. [Google Scholar] [CrossRef]
- Dascombe, M.J.; Rothwell, N.J.; Sagay, B.O.; Stock, M.J. Pyrogenic and Thermogenic Effects of Interleukin 1 Beta in the Rat. Am. J. Physiol. 1989, 256, E7–E11. [Google Scholar] [CrossRef] [PubMed]
- Plata-Salamán, C.R. Cytokine-Induced Anorexia. Behavioral, Cellular, and Molecular Mechanisms. Ann. N. Y. Acad. Sci. 1998, 856, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Benrick, A.; Schéle, E.; Pinnock, S.B.; Wernstedt-Asterholm, I.; Dickson, S.L.; Karlsson-Lindahl, L.; Jansson, J.-O. Interleukin-6 Gene Knockout Influences Energy Balance Regulating Peptides in the Hypothalamic Paraventricular and Supraoptic Nuclei. J. Neuroendocrinol. 2009, 21, 620–628. [Google Scholar] [CrossRef]
- Schéle, E.; Benrick, A.; Grahnemo, L.; Egecioglu, E.; Anesten, F.; Pálsdóttir, V.; Jansson, J.-O. Inter-Relation between Interleukin (IL)-1, IL-6 and Body Fat Regulating Circuits of the Hypothalamic Arcuate Nucleus. J. Neuroendocrinol. 2013, 25, 580–589. [Google Scholar] [CrossRef]
- Timper, K.; Denson, J.L.; Steculorum, S.M.; Heilinger, C.; Engström-Ruud, L.; Wunderlich, C.M.; Rose-John, S.; Wunderlich, F.T.; Brüning, J.C. IL-6 Improves Energy and Glucose Homeostasis in Obesity via Enhanced Central IL-6 Trans-Signaling. Cell Rep. 2017, 19, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Le Thuc, O.; Stobbe, K.; Cansell, C.; Nahon, J.-L.; Blondeau, N.; Rovère, C. Hypothalamic Inflammation and Energy Balance Disruptions: Spotlight on Chemokines. Front. Endocrinol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Plata-Salamán, C.R.; Borkoski, J.P. Chemokines/Intercrines and Central Regulation of Feeding. Am. J. Physiol. 1994, 266, R1711–R1715. [Google Scholar] [CrossRef]
- Le Thuc, O.; Cansell, C.; Bourourou, M.; Denis, R.G.; Stobbe, K.; Devaux, N.; Guyon, A.; Cazareth, J.; Heurteaux, C.; Rostène, W.; et al. Central CCL2 Signaling onto MCH Neurons Mediates Metabolic and Behavioral Adaptation to Inflammation. EMBO Rep. 2016, 17, 1738–1752. [Google Scholar] [CrossRef] [Green Version]
- Morari, J.; Anhe, G.F.; Nascimento, L.F.; de Moura, R.F.; Razolli, D.; Solon, C.; Guadagnini, D.; Souza, G.; Mattos, A.H.; Tobar, N.; et al. Fractalkine (CX3CL1) Is Involved in the Early Activation of Hypothalamic Inflammation in Experimental Obesity. Diabetes 2014, 63, 3770–3784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, K.; Barson, J.R.; Ho, H.T.; Leibowitz, S.F. Relationship of the Chemokine, CXCL12, to Effects of Dietary Fat on Feeding-Related Behaviors and Hypothalamic Neuropeptide Systems. Front. Behav. Neurosci. 2016, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.-H.; Kim, J.; Kim, C.-S.; Tu, T.H.; Kim, M.-S.; Suk, K.; Kim, D.H.; Lee, B.J.; Choi, H.-S.; Park, T.; et al. Hypothalamic Lipid-Laden Astrocytes Induce Microglia Migration and Activation. FEBS Lett. 2017, 591, 1742–1751. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Tsirka, S.E. Monocyte Chemoattractant Protein-1 and the Blood-Brain Barrier. Cell. Mol. Life Sci. 2014, 71, 683–697. [Google Scholar] [CrossRef] [Green Version]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W.G.M. Neuronal “On” and “Off” Signals Control Microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D. The P2Y12 Receptor Regulates Microglial Activation by Extracellular Nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia Monitor and Protect Neuronal Function through Specialized Somatic Purinergic Junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative Feedback Control of Neuronal Activity by Microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Galea, I.; Bechmann, I.; Perry, V.H. What Is Immune Privilege (Not)? Trends Immunol. 2007, 28, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.-H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, H.; Misgeld, T.; Matsumuro, K.; Wekerle, H. Neurotrophins Inhibit Major Histocompatibility Class II Inducibility of Microglia: Involvement of the P75 Neurotrophin Receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 5779–5784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Loh, K.H.; Wu, M.; Morgan, D.A.; Schneeberger, M.; Yu, X.; Chi, J.; Kosse, C.; Kim, D.; Rahmouni, K.; et al. A Leptin-BDNF Pathway Regulating Sympathetic Innervation of Adipose Tissue. Nature 2020, 583, 839–844. [Google Scholar] [CrossRef]
- Xu, B.; Xie, X. Neurotrophic Factor Control of Satiety and Body Weight. Nat. Rev. Neurosci. 2016, 17, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trang, T.; Beggs, S.; Wan, X.; Salter, M.W. P2X4-Receptor-Mediated Synthesis and Release of Brain-Derived Neurotrophic Factor in Microglia Is Dependent on Calcium and P38-Mitogen-Activated Protein Kinase Activation. J. Neurosci. 2009, 29, 3518–3528. [Google Scholar] [CrossRef]
- Urabe, H.; Kojima, H.; Chan, L.; Terashima, T.; Ogawa, N.; Katagi, M.; Fujino, K.; Kumagai, A.; Kawai, H.; Asakawa, A.; et al. Haematopoietic Cells Produce BDNF and Regulate Appetite upon Migration to the Hypothalamus. Nat. Commun. 2013, 4, 1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pocock, J.M.; Kettenmann, H. Neurotransmitter Receptors on Microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Fontainhas, A.M.; Wang, M.; Liang, K.J.; Chen, S.; Mettu, P.; Damani, M.; Fariss, R.N.; Li, W.; Wong, W.T. Microglial Morphology and Dynamic Behavior Is Regulated by Ionotropic Glutamatergic and GABAergic Neurotransmission. PLoS ONE 2011, 6, e15973. [Google Scholar] [CrossRef]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic Modulation of Microglial Activation by Alpha 7 Nicotinic Receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Nakanishi, H.; Nabekura, J.; Akaike, N. AMPA-Kainate Subtypes of Glutamate Receptor in Rat Cerebral Microglia. J. Neurosci. 2000, 20, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.G.; O’Driscoll, C.M.; Bressler, J.; Kaufmann, W.; Rojas, C.J.; Slusher, B.S. Small Molecule Glutaminase Inhibitors Block Glutamate Release from Stimulated Microglia. Biochem. Biophys. Res. Commun. 2014, 443, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Fuente-Martín, E.; García-Cáceres, C.; Granado, M.; de Ceballos, M.L.; Sánchez-Garrido, M.Á.; Sarman, B.; Liu, Z.-W.; Dietrich, M.O.; Tena-Sempere, M.; Argente-Arizón, P.; et al. Leptin Regulates Glutamate and Glucose Transporters in Hypothalamic Astrocytes. J. Clin. Investig. 2012, 122, 3900–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, K.; Müller, T.D.; DiMarchi, R.D.; Pfluger, P.T.; Tschöp, M.H. CNS-Targeting Pharmacological Interventions for the Metabolic Syndrome. J. Clin. Investig. 2019, 129, 4058–4071. [Google Scholar] [CrossRef] [PubMed]
- Delpech, J.-C.; Herron, S.; Botros, M.B.; Ikezu, T. Neuroimmune Crosstalk through Extracellular Vesicles in Health and Disease. Trends Neurosci. 2019, 42, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E.; et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef]
- Thion, M.S.; Low, D.; Silvin, A.; Chen, J.; Grisel, P.; Schulte-Schrepping, J.; Blecher, R.; Ulas, T.; Squarzoni, P.; Hoeffel, G.; et al. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell 2018, 172, 500–516.e16. [Google Scholar] [CrossRef] [Green Version]
- Guneykaya, D.; Ivanov, A.; Hernandez, D.P.; Haage, V.; Wojtas, B.; Meyer, N.; Maricos, M.; Jordan, P.; Buonfiglioli, A.; Gielniewski, B.; et al. Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep. 2018, 24, 2773–2783.e6. [Google Scholar] [CrossRef] [Green Version]
- Dorfman, M.D.; Krull, J.E.; Douglass, J.D.; Fasnacht, R.; Lara-Lince, F.; Meek, T.H.; Shi, X.; Damian, V.; Nguyen, H.T.; Matsen, M.E.; et al. Sex Differences in Microglial CX3CR1 Signalling Determine Obesity Susceptibility in Mice. Nat. Commun. 2017, 8, 14556. [Google Scholar] [CrossRef] [Green Version]
- Argente-Arizón, P.; Díaz, F.; Ros, P.; Barrios, V.; Tena-Sempere, M.; García-Segura, L.M.; Argente, J.; Chowen, J.A. The Hypothalamic Inflammatory/Gliosis Response to Neonatal Overnutrition Is Sex and Age Dependent. Endocrinology 2018, 159, 368–387. [Google Scholar] [CrossRef] [Green Version]
- García-Cáceres, C.; Balland, E.; Prevot, V.; Luquet, S.; Woods, S.C.; Koch, M.; Horvath, T.L.; Yi, C.-X.; Chowen, J.A.; Verkhratsky, A.; et al. Role of Astrocytes, Microglia, and Tanycytes in Brain Control of Systemic Metabolism. Nat. Neurosci. 2019, 22, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Cha, D.; Kim, S.; Jiang, L.; Cooke, P.; Adebesin, M.; Wolfe, A.; Riddle, R.; Aja, S.; Blackshaw, S. Tanycyte Ablation in the Arcuate Nucleus and Median Eminence Increases Obesity Susceptibility by Increasing Body Fat Content in Male Mice. Glia 2020, 68, 1987–2000. [Google Scholar] [CrossRef]
- Balland, E.; Dam, J.; Langlet, F.; Caron, E.; Steculorum, S.; Messina, A.; Rasika, S.; Falluel-Morel, A.; Anouar, Y.; Dehouck, B.; et al. Hypothalamic Tanycytes Are an ERK-Gated Conduit for Leptin into the Brain. Cell Metab. 2014, 19, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graupera, M.; Claret, M. Endothelial Cells: New Players in Obesity and Related Metabolic Disorders. Trends Endocrinol. Metab. 2018, 29, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Campbell, J.N.; Macosko, E.Z.; Fenselau, H.; Pers, T.H.; Lyubetskaya, A.; Tenen, D.; Goldman, M.; Verstegen, A.M.J.; Resch, J.M.; McCarroll, S.A.; et al. A Molecular Census of Arcuate Hypothalamus and Median Eminence Cell Types. Nat. Neurosci. 2017, 20, 484–496. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Wu, X.; Jiang, L.; Zhang, Y. Single-Cell RNA-Seq Reveals Hypothalamic Cell Diversity. Cell Rep. 2017, 18, 3227–3241. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.; Claret, M.; Zeltser, L.M.; Williams, K.W.; Yeo, G.S.H.; Tschöp, M.H.; Diano, S.; Brüning, J.C.; Cota, D. POMC Neuronal Heterogeneity in Energy Balance and beyond: An Integrated View. Nat. Metab. 2021, 3, 299–308. [Google Scholar] [CrossRef]
- Mendes, N.F.; Jara, C.P.; Zanesco, A.M.; de Araújo, E.P. Hypothalamic Microglial Heterogeneity and Signature under High Fat Diet–Induced Inflammation. Int. J. Mol. Sci. 2021, 22, 2256. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Léon, S.; Nadjar, A.; Quarta, C. Microglia–Neuron Crosstalk in Obesity: Melodious Interaction or Kiss of Death? Int. J. Mol. Sci. 2021, 22, 5243. https://doi.org/10.3390/ijms22105243
Léon S, Nadjar A, Quarta C. Microglia–Neuron Crosstalk in Obesity: Melodious Interaction or Kiss of Death? International Journal of Molecular Sciences. 2021; 22(10):5243. https://doi.org/10.3390/ijms22105243
Chicago/Turabian StyleLéon, Stéphane, Agnès Nadjar, and Carmelo Quarta. 2021. "Microglia–Neuron Crosstalk in Obesity: Melodious Interaction or Kiss of Death?" International Journal of Molecular Sciences 22, no. 10: 5243. https://doi.org/10.3390/ijms22105243
APA StyleLéon, S., Nadjar, A., & Quarta, C. (2021). Microglia–Neuron Crosstalk in Obesity: Melodious Interaction or Kiss of Death? International Journal of Molecular Sciences, 22(10), 5243. https://doi.org/10.3390/ijms22105243