
Excessive Innate Immunity Steers Pathogenic Adaptive Immunity in the Development of Theiler’s Virus-Induced Demyelinating Disease

Abstract
:1. Theiler’s Virus-Induced Demyelinating Disease as an Infectious Model of Multiple Sclerosis
2. Factors Affecting Permissiveness to TMEV Infection
2.1. Antigen-Presenting Cells
2.2. Role of Innate Immunity Associated with TMEV Infection
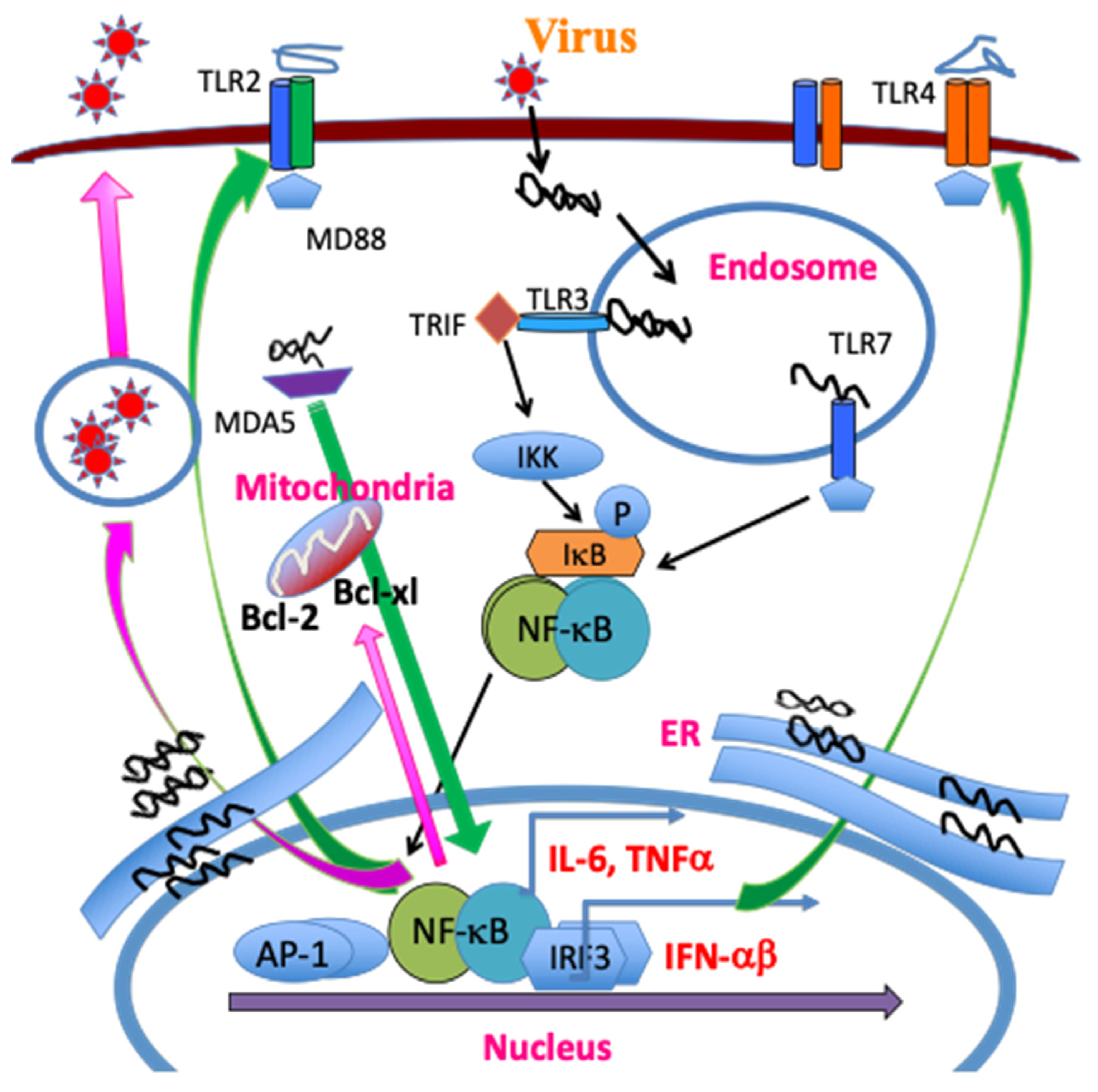
2.2.1. Critical Roles of Pattern Recognition Receptors (TLRs and MDA-5)
2.2.2. NLRP3 Inflammasome
2.2.3. Initial Chemokines and Cytokines
3. Role of Virus-Specific Adaptive Immunity in TMEV-Induced Demyelination
3.1. CD4+ T Cells
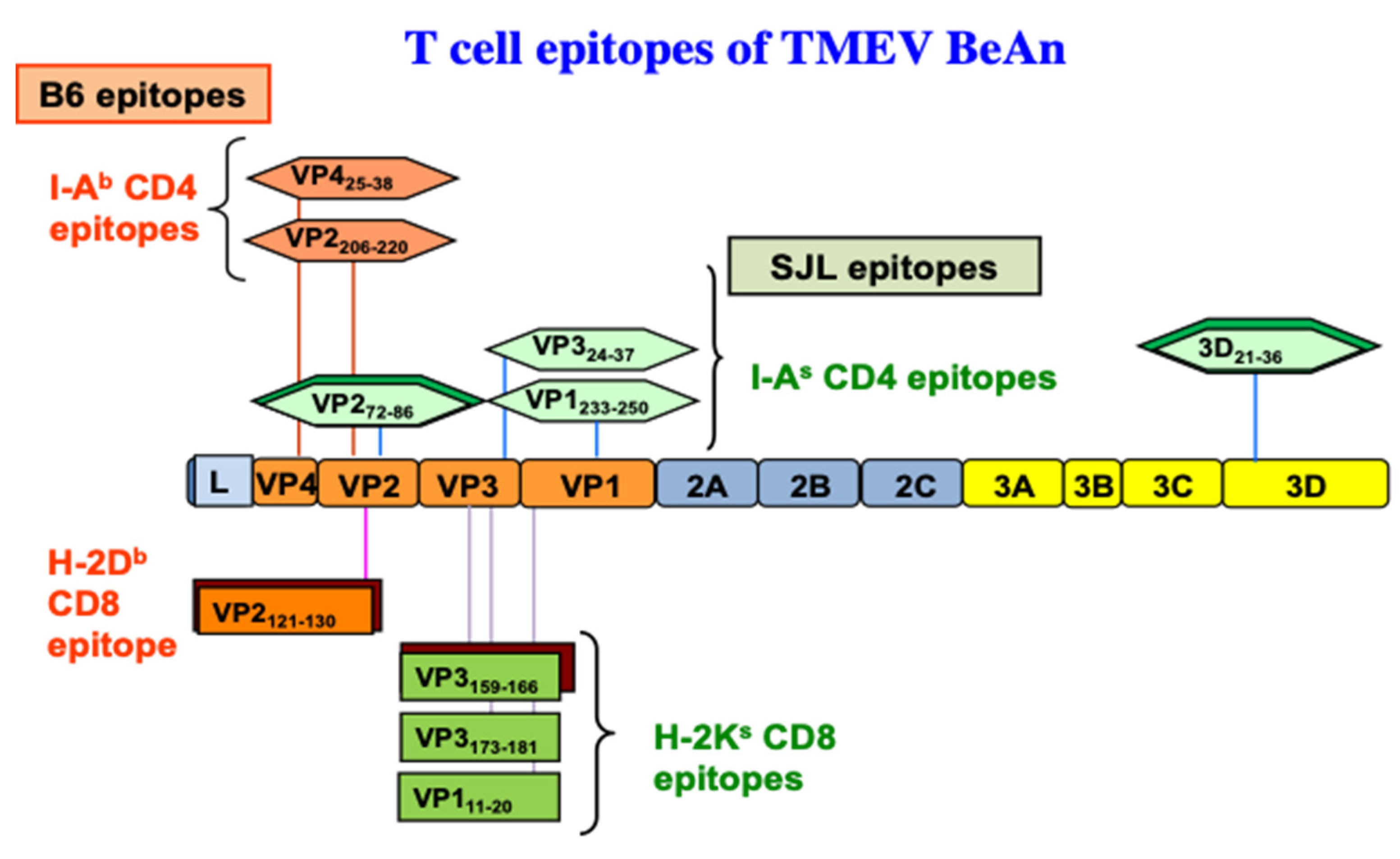
3.1.1. Early Studies of CD4+ T Responses
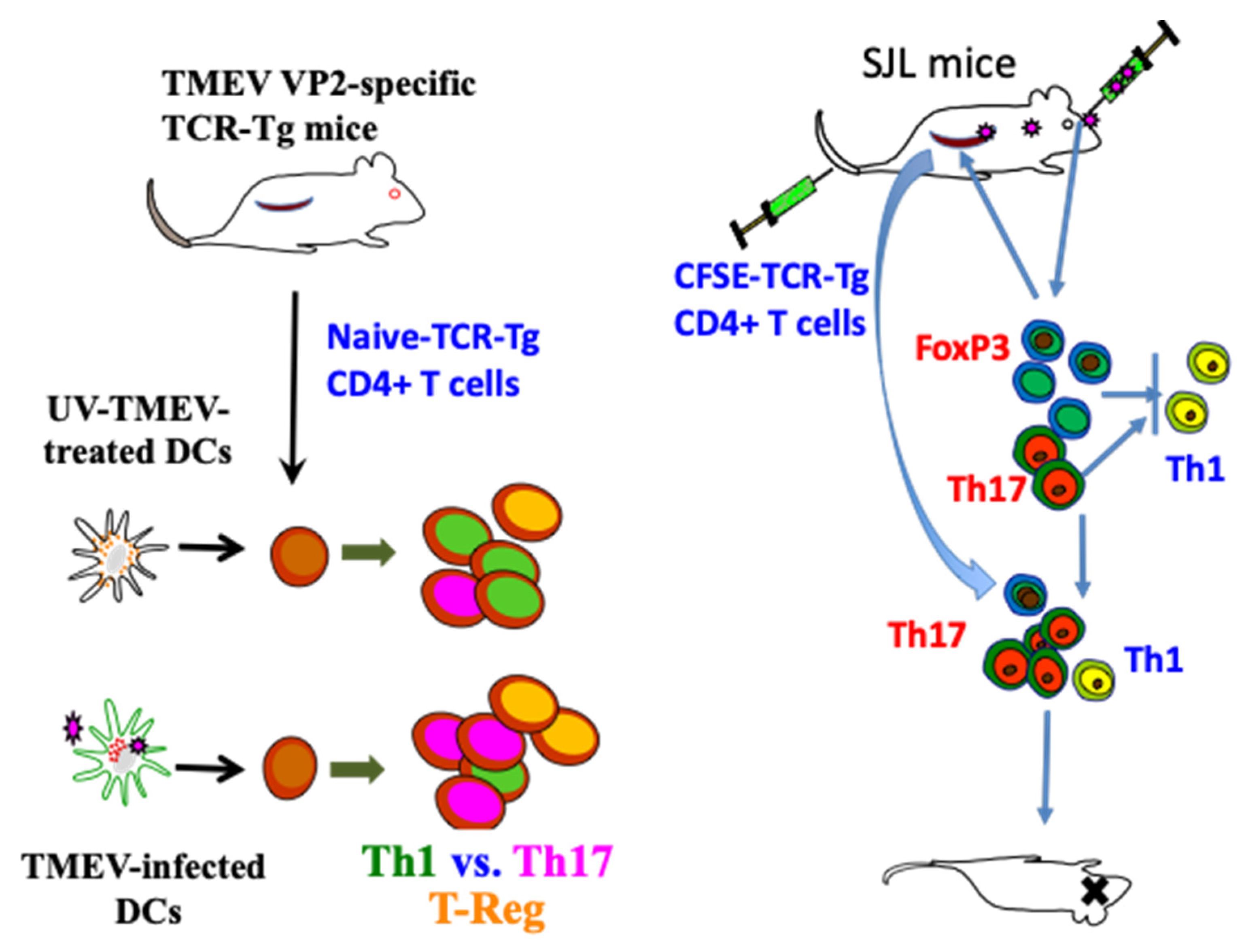
3.1.2. Utilization of Virus-Specific CD4+ T Cell Receptor Transgenic Mice
3.1.3. Involvement of Th1 Cells
3.1.4. Role of Th17 Cells
3.1.5. Participation of FoxP3+ Regulatory T Cells
3.2. Roles of CD8+ T Cells in the Pathogenesis of TMEV-IDD
3.2.1. Role of Tc1
3.2.2. Role of Tc17
3.3. Role of B Cells in the Development of TMEV-IDD
3.3.1. Anti-TMEV Antibody Responses
3.3.2. B Cells as a Viral Reservoir and Their Role in T Cell Activation
4. Development of Autoimmune Responses during TMEV-IDD
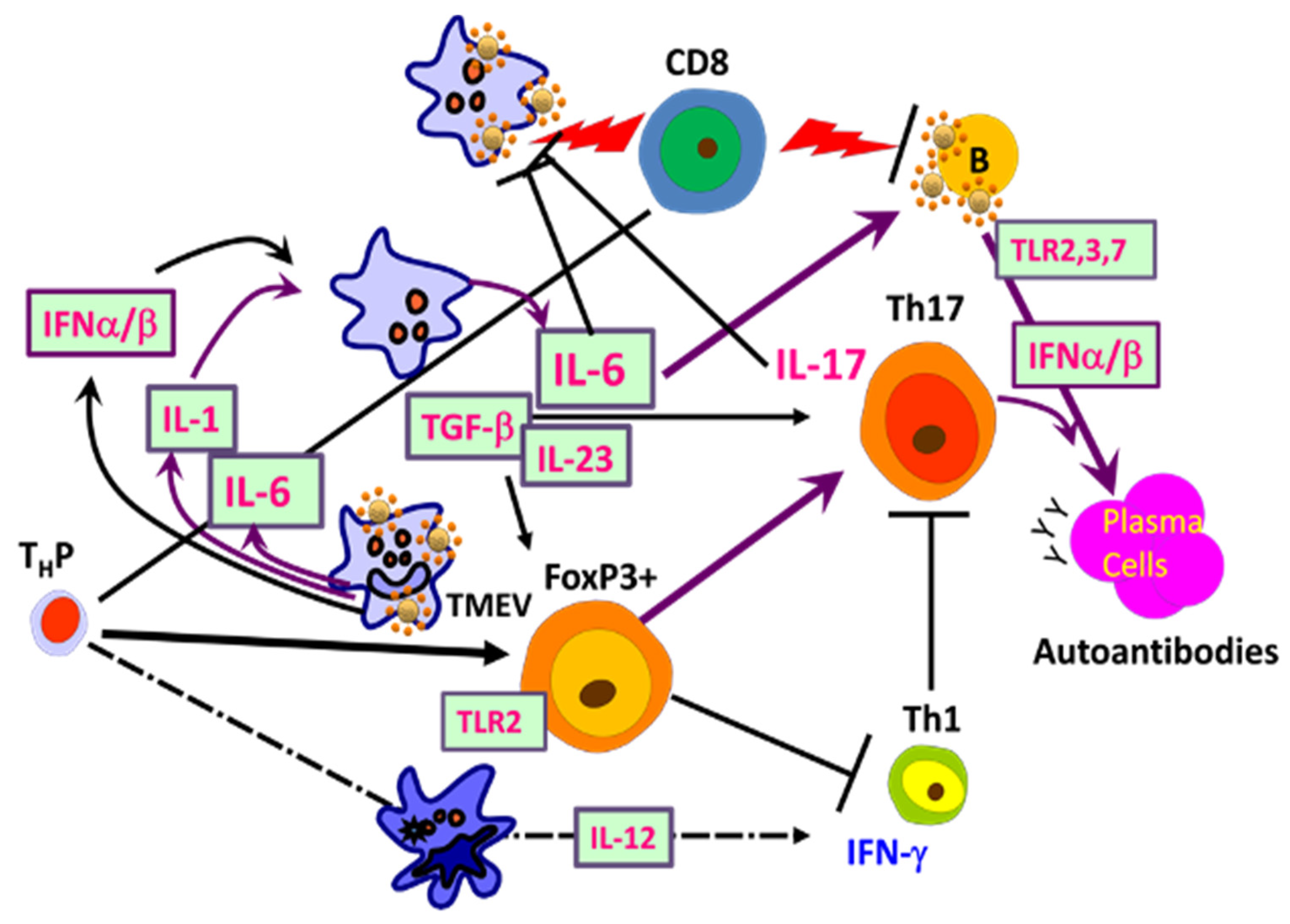
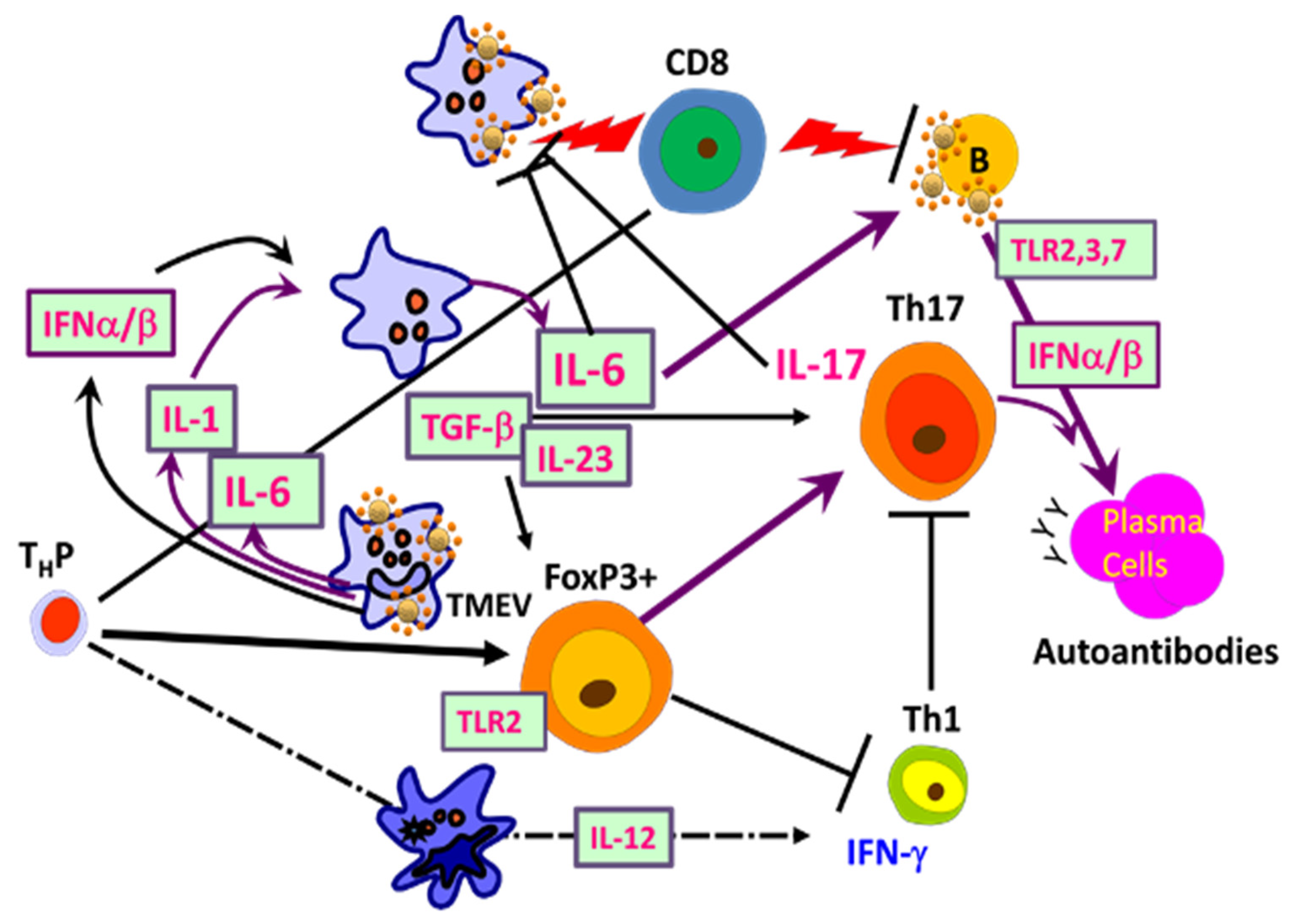
5. Shaping Adaptive Immune Responses by Innate Immunity after TMEV Infection
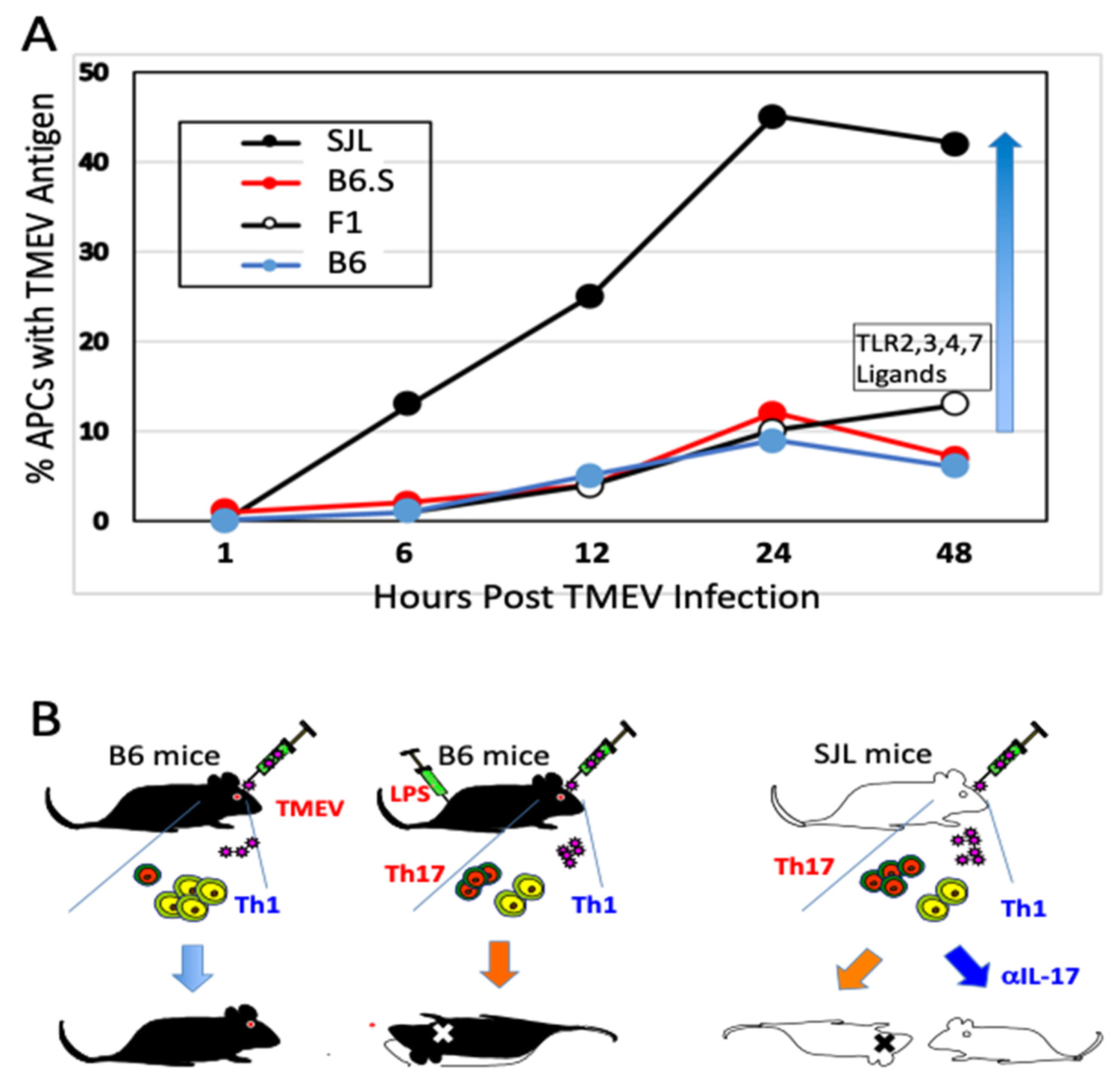
5.1. Excessive Innate Immunity Initiates the Pathogenesis of TMEV-IDD
5.2. Viral Load and Persistence
5.3. Relevance of TMEV-IDD in Understanding Other Chronic Viral Inflammatory Diseases
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MS | multiple sclerosis |
| CNS | central nervous system |
| TMEV | Theiler’s murine encephalomyelitis virus |
| TMEV-IDD | TMEV-induced demyelinating disease |
| PCR | polymerase chain reaction |
| PFU | plaque-forming unit |
| PI | post-infection |
| S mix | peptide mix derived from structural proteins |
| NS mix | peptide mix derived from nonstructural proteins |
| Tg | transgenic mice |
| TCR | T cell receptor |
| MHC | major histocompatibility complex |
References
- Adams, R.M. Principles of Neurology; McGraw Hill: New York, NY, USA, 1977; p. 1041. [Google Scholar]
- Johnson, R.T. The possible viral etiology of multiple sclerosis. Adv. Neurol. 1975, 13, 1–46. [Google Scholar] [PubMed]
- McFarlin, D.E.; McFarland, H.F. Multiple sclerosis (first of two parts). N. Engl. J. Med. 1982, 307, 1183–1188. [Google Scholar] [CrossRef]
- Soldan, S.S.; Berti, R.; Salem, N.; Secchiero, P.; Flamand, L.; Calabresi, P.A.; Brennan, M.B.; Maloni, H.W.; McFarland, H.F.; Lin, H.C.; et al. Association of human herpes virus 6 (HHV-6) with multiple sclerosis: Increased IgM response to HHV-6 early antigen and detection of serum HHV-6 DNA [see comments]. Nat. Med. 1997, 3, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Akhyani, N.; Berti, R.; Brennan, M.B.; Soldan, S.S.; Eaton, J.M.; McFarland, H.F.; Jacobson, S. Tissue distribution and variant characterization of human herpesvirus (HHV)-6: Increased prevalence of HHV-6A in patients with multiple sclerosis. J. Infect. Dis. 2000, 182, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lafuente, R.; Martin-Estefania, C.; de las Heras, V.; Castrillo, C.; Cour, I.; Picazo, J.J.; Varela De Seijas, E.; Arroyo, R. Prevalence of herpesvirus DNA in MS patients and healthy blood donors. Acta Neurol. Scand. 2002, 105, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Alvarez-Lafuente, R.; Mas, A.; Bartolome, M.; Garcia-Montojo, M.; de Las Heras, V.; de la Concha, E.G.; Arroyo, R.; Urcelay, E. Environment-gene interaction in multiple sclerosis: Human herpesvirus 6 and MHC2TA. Hum. Immunol. 2007, 68, 685–689. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; Lane, T.E. Viral-induced neurodegenerative disease. Curr. Opin. Microbiol. 1999, 2, 398–402. [Google Scholar] [CrossRef]
- Daniels, J.B.; Pappenheimer, A.M.; Richardson, S. Observations on encephalomyelitis of mice (DA strain). J. Exp. Med. 1952, 96, 517. [Google Scholar] [CrossRef] [Green Version]
- Lipton, H.L. Theiler’s virus infection in mice: An unusual biphasic disease process leading to demyelination. Infect. Immun. 1975, 11, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Wege, H.; Siddell, S.; ter Meulen, V. The biology and pathogenesis of coronaviruses. Curr. Top. Microbiol. Immunol. 1982, 99, 165–200. [Google Scholar]
- Alford, E.C. Experimental Allergic Encephalomyelitis: A Useful Model for Multiple Sclerosis; A.R. Liss: New York, NY, USA, 1984; p. 554. [Google Scholar]
- Dal Canto, M.C.; Kim, B.S.; Miller, S.D.; Melvold, R.W. Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelination: A model for human multiple clerosis. Methods 1996, 10, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.; Vanderlugt, C.L.; Begolka, W.S.; Pao, W.; Yauch, R.L.; Neville, K.L.; Katz-Levy, Y.; Carrizosa, A.; Kim, B.S. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997, 3, 1133–1136. [Google Scholar] [CrossRef]
- Liang, Z.; Kumar, A.S.; Jones, M.S.; Knowles, N.J.; Lipton, H.L. Phylogenetic Analysis of the Species Theilovirus: Emerging Murine and Human Pathogens. J. Virol. 2008, 82, 11545–11554. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.Y.; Greninger, A.L.; Kanada, K.; Kwok, T.; Fischer, K.F.; Runckel, C.; Louie, J.K.; Glaser, C.A.; Yagi, S.; Schnurr, D.P.; et al. Identification of cardioviruses related to Theiler’s murine encephalomyelitis virus in human infections. Proc. Natl. Acad. Sci. USA 2008, 105, 14124–14129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoll, J.; Erkens Hulshof, S.; Lanke, K.; Verduyn Lunel, F.; Melchers, W.J.; Schoondermark-van de Ven, E.; Roivainen, M.; Galama, J.M.; van Kuppeveld, F.J. Saffold virus, a human Theiler’s-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog 2009, 5, e1000416. [Google Scholar] [CrossRef]
- Theiler, M.; Gard, S. Encephalomyelitis of mice. J. Exp. Med. 1940, 72, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Friedmann, A. Purification of Theiler’s murine encephalomyelitis virus and analysis of the structural virion polypeptides: Correlation of the polypeptide profile with virulence. J.Virol. 1980, 33, 1165–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Canto, M.C.; Lipton, H.L. Primary demyelination in Theiler’s virus infection. An ultrastructural study. Lab. Investig. 1975, 33, 626–637. [Google Scholar]
- Lehrich, J.R.; Arnason, B.G.; Hochberg, F.H. Demyelinative myelopathy in mice induced by the DA virus. J. Neurol. Sci. 1976, 29, 149–160. [Google Scholar] [CrossRef]
- Kang, B.S.; Yahikozawa, H.; Koh, C.S.; Kim, B.S. Oral administration of live virus protects susceptible mice from developing Theiler’s virus-induced demyelinating disease. Virology 2007, 366, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Lipton, H.L.; Kim, B.S.; Yahikozawa, H.; Nadler, C.F. Serological evidence that Mus musculus is the natural host of Theiler’s murine encephalomyelitis virus. Virus Res. 2001, 76, 79–86. [Google Scholar] [CrossRef]
- DePaula-Silva, A.B.; Hanak, T.J.; Libbey, J.E.; Fujinami, R.S. Theiler’s murine encephalomyelitis virus infection of SJL/J and C57BL/6J mice: Models for multiple sclerosis and epilepsy. J. Neuroimmunol. 2017, 308, 30–42. [Google Scholar] [CrossRef]
- Gerhauser, I.; Alldinger, S.; Baumgartner, W. Ets-1 represents a pivotal transcription factor for viral clearance, inflammation, and demyelination in a mouse model of multiple sclerosis. J. Neuroimmunol. 2007, 188, 86–94. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Bloom, B.R. Primary demyelination as a nonspecific consequence of a cell-mediated immune reaction. J. Exp. Med. 1975, 141, 346–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clatch, R.J.; Lipton, H.L.; Miller, S.D. Characterization of Theiler’s murine encephalomyelitis virus (TMEV)-specific delayed-type hypersensitivity responses in TMEV-induced demyelinating disease: Correlation with clinical signs. J. Immunol. 1986, 136, 920–927. [Google Scholar]
- Rauch, H.C.; Montgomery, I.N.; Hinman, C.L.; Harb, W.; Benjamins, J.A. Chronic Theiler’s virus infection in mice: Appearance of myelin basic protein in the cerebrospinal fluid and serum antibody directed against MBP. J. Neuroimmunol. 1987, 14, 35–48. [Google Scholar] [CrossRef]
- Yamada, M.; Zurbriggen, A.; Fujinami, R.S. Monoclonal antibody to Theiler’s murine encephalomyelitis virus defines a determinant on myelin and oligodendrocytes, and augments demyelination in experimental allergic encephalomyelitis. J. Exp. Med. 1990, 171, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.H.; Kim, C.X.; Huang, J.; Kim, B.S. Infection and Activation of B Cells by Theiler’s Murine Encephalomyelitis Virus (TMEV) Leads to Autoantibody Production in an Infectious Model of Multiple Sclerosis. Cells 2020, 9, 1787. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Dal Canto, M.C. Susceptibility of inbred mice to chronic central nervous system infection by Theiler’s murine encephalomyelitis virus. Infect. Immun. 1979, 26, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.; David, C.S. Demyelination induced by Theiler’s virus: Influence of the H-2 haplotype. J. Immunol. 1985, 135, 2145–2148. [Google Scholar]
- Clatch, R.J.; Melvold, R.W.; Miller, S.D.; Lipton, H.L. Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease in mice is influenced by the H-2D region: Correlation with TEMV-specific delayed-type hypersensitivity. J. Immunol. 1985, 135, 1408–1414. [Google Scholar] [PubMed]
- Rodriguez, M.; Leibowitz, J.; David, C.S. Susceptibility to Theiler’s virus-induced demyelination. Mapping of the gene within the H-2D region. J. Exp. Med. 1986, 163, 620–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, B.S.; Lyman, M.A.; Kim, B.S. The majority of infiltrating CD8+ T cells in the central nervous system of susceptible SJL/J mice infected with Theiler’s virus are virus specific and fully functional. J. Virol. 2002, 76, 6577–6585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, H.L.; Melvold, R. Genetic analysis of susceptibility to Theiler’s virus-induced demyelinating disease in mice. J. Immunol. 1984, 132, 1821–1825. [Google Scholar] [PubMed]
- Jin, Y.H.; Kang, H.S.; Mohindru, M.; Kim, B.S. Preferential induction of protective T cell responses to Theiler’s virus in resistant (C57BL/6 x SJL)F1 mice. J. Virol. 2011, 85, 3033–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.H.; Kang, H.S.; Hou, W.; Meng, L.; Kim, B.S. The level of viral infection of antigen-presenting cells correlates with the level of development of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Virol. 2015, 89, 1867–1878. [Google Scholar] [CrossRef] [Green Version]
- Bureau, J.F.; Montagutelli, X.; Lefebvre, S.; Guenet, J.L.; Pla, M.; Brahic, M. The interaction of two groups of murine genes determines the persistence of Theiler’s virus in the central nervous system. J. Virol. 1992, 66, 4698–4704. [Google Scholar] [CrossRef] [Green Version]
- Bureau, J.F.; Montagutelli, X.; Bihl, F.; Lefebvre, S.; Guenet, J.L.; Brahic, M. Mapping loci influencing the persistence of Theiler’s virus in the murine central nervous system. Nat. Genet. 1993, 5, 87–91. [Google Scholar] [CrossRef]
- Bureau, J.F.; Drescher, K.M.; Pease, L.R.; Vikoren, T.; Delcroix, M.; Zoecklein, L.; Brahic, M.; Rodriguez, M. Chromosome 14 contains determinants that regulate susceptibility to Theiler’s virus-induced demyelination in the mouse. Genetics 1998, 148, 1941–1949. [Google Scholar] [CrossRef]
- Rodriguez, M.; Leibowitz, J.L.; Lampert, P.W. Persistent infection of oligodendrocytes in Theiler’s virus-induced encephalomyelitis. Ann. Neurol. 1983, 13, 426–433. [Google Scholar] [CrossRef]
- Lipton, H.L.; Kratochvil, J.; Sethi, P.; Dal Canto, M.C. Theiler’s virus antigen detected in mouse spinal cord 2 1/2 years after infection. Neurology 1984, 34, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Zurbriggen, A.; Fujinami, R.S. The relationship between viral RNA, myelin-specific mRNAs, and demyelination in central nervous system disease during Theiler’s virus infection. Am. J. Pathol. 1990, 137, 1467–1479. [Google Scholar] [PubMed]
- McAllister, A.; Tangy, F.; Aubert, C.; Brahic, M. Genetic mapping of the ability of Theiler’s virus to persist and demyelinate. J. Virol. 1990, 64, 4252–4257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangy, F.; McAllister, A.; Aubert, C.; Brahic, M. Determinants of persistence and demyelination of the DA strain of Theiler’s virus are found only in the VP1 gene. J. Virol. 1991, 65, 1616–1618. [Google Scholar] [CrossRef] [Green Version]
- Lipton, H.L.; Calenoff, M.; Bandyopadhyay, P.; Miller, S.D.; Dal Canto, M.C.; Gerety, S.; Jensen, K. The 5′ noncoding sequences from a less virulent Theiler’s virus dramatically attenuate GDVII neurovirulence. J. Virol. 1991, 65, 4370–4377. [Google Scholar] [CrossRef] [Green Version]
- Pullen, L.C.; Park, S.H.; Miller, S.D.; Dal Canto, M.C.; Kim, B.S. Treatment with bacterial LPS renders genetically resistant C57BL/6 mice susceptible to Theiler’s virus-induced demyelinating disease. J. Immunol. 1995, 155, 4497–4503. [Google Scholar]
- Myoung, J.; Il Bahk, Y.; Kang, H.S.; Dal Canto, M.C.; Kim, B.S. Anti-capsid immunity level, not viral persistence level, correlates with the progression of Theiler’s virus-induced demyelinating disease in viral P1-transgenic mice. J. Virol. 2008, 82, 5606–5617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.; Harting, E.; Frey, M.S.; Leibowitz, J.L.; Miranda, R.C. Theiler’s murine encephalomyelitis virus induces rapid necrosis and delayed apoptosis in myelinated mouse cerebellar explant cultures. Brain Res. 2000, 868, 259–267. [Google Scholar] [CrossRef]
- Palma, J.P.; Kim, B.S. Induction of selected chemokines in glial cells infected with Theiler’s virus. J. Neuroimmunol. 2001, 117, 166–170. [Google Scholar] [CrossRef]
- Zheng, L.; Calenoff, M.A.; Dal Canto, M.C. Astrocytes, not microglia, are the main cells responsible for viral persistence in Theiler’s murine encephalomyelitis virus infection leading to demyelination. J. Neuroimmunol. 2001, 118, 256–267. [Google Scholar] [CrossRef]
- Lipton, H.L.; Kumar, A.S.; Trottier, M. Theiler’s virus persistence in the central nervous system of mice is associated with continuous viral replication and a difference in outcome of infection of infiltrating macrophages versus oligodendrocytes. Virus Res. 2005, 111, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Katz-Levy, Y.; Neville, K.L.; Girvin, A.M.; Vanderlugt, C.L.; Pope, J.G.; Tan, L.J.; Miller, S.D. Endogenous presentation of self myelin epitopes by CNS-resident APCs in Theiler’s virus-infected mice. J. Clin. Investig. 1999, 104, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Palma, J.P.; Yauch, R.L.; Lang, S.; Kim, B.S. Potential role of CD4+ T cell-mediated apoptosis of activated astrocytes in Theiler’s virus-induced demyelination. J. Immunol. 1999, 162, 6543–6551. [Google Scholar]
- Olson, J.K.; Girvin, A.M.; Miller, S.D. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler’s virus. J. Virol. 2001, 75, 9780–9789. [Google Scholar] [CrossRef] [Green Version]
- Clatch, R.J.; Miller, S.D.; Metzner, R.; Dal Canto, M.C.; Lipton, H.L. Monocytes/macrophages isolated from the mouse central nervous system contain infectious Theiler’s murine encephalomyelitis virus (TMEV). Virology 1990, 176, 244–254. [Google Scholar] [CrossRef]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.H.; Mohindru, M.; Kang, M.H.; Fuller, A.C.; Kang, B.; Gallo, D.; Kim, B.S. Differential virus replication, cytokine production, and antigen-presenting function by microglia from susceptible and resistant mice infected with Theiler’s virus. J. Virol. 2007, 81, 11690–11702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, K.N.; Becker, R.P.; Kallio, P.; Lipton, H.L. Theiler’s virus-induced intrinsic apoptosis in M1-D macrophages is Bax mediated and restricts virus infectivity: A mechanism for persistence of a cytolytic virus. J. Virol. 2008, 82, 4502–4510. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.H.; So, E.Y.; Park, H.; Kim, B.S. Replication of Theiler’s virus requires NF-kappaB-activation: Higher viral replication and spreading in astrocytes from susceptible mice. Glia 2008, 56, 942–953. [Google Scholar] [CrossRef]
- Schneider, K.M.; Watson, N.B.; Minchenberg, S.B.; Massa, P.T. The influence of macrophage growth factors on Theiler’s Murine Encephalomyelitis Virus (TMEV) infection and activation of macrophages. Cytokine 2018, 102, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; So, E.Y.; Kim, B.S. Role of dendritic cells in differential susceptibility to viral demyelinating disease. PLoS Pathog 2007, 3, e124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, E.J.; Bailey, S.L.; Castenada, C.V.; Waldner, H.; Miller, S.D. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 2005, 11, 335–339. [Google Scholar] [CrossRef]
- Hou, W.; Kang, H.S.; Kim, B.S. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J. Exp. Med. 2009, 206, 313–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.H.; Kaneyama, T.; Kang, M.H.; Kang, H.S.; Koh, C.S.; Kim, B.S. TLR3 signaling is either protective or pathogenic for the development of Theiler’s virus-induced demyelinating disease depending on the time of viral infection. J. Neuroinflamm. 2011, 8, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.R.; Zaczynska, E.; Katsetos, C.D.; Platsoucas, C.D.; Oleszak, E.L. Differential expression of TGF-beta, IL-2, and other cytokines in the CNS of Theiler’s murine encephalomyelitis virus-infected susceptible and resistant strains of mice. Virology 2000, 278, 346–360. [Google Scholar] [CrossRef] [Green Version]
- Petro, T.M. Disparate expression of IL-12 by SJL/J and B10.S macrophages during Theiler’s virus infection is associated with activity of TLR7 and mitogen-activated protein kinases. Microbes Infect. 2005, 7, 224–232. [Google Scholar] [CrossRef]
- Moore, T.C.; Bush, K.L.; Cody, L.; Brown, D.M.; Petro, T.M. Control of early Theiler’s murine encephalomyelitis virus replication in macrophages by interleukin-6 occurs in conjunction with STAT1 activation and nitric oxide production. J. Virol. 2012, 86, 10841–10851. [Google Scholar] [CrossRef] [Green Version]
- So, E.Y.; Kang, M.H.; Kim, B.S. Induction of chemokine and cytokine genes in astrocytes following infection with Theiler’s murine encephalomyelitis virus is mediated by the Toll-like receptor 3. Glia 2006, 53, 858–867. [Google Scholar] [CrossRef]
- Turrin, N.P. Central nervous system Toll-like receptor expression in response to Theiler’s murine encephalomyelitis virus-induced demyelination disease in resistant and susceptible mouse strains. Virol. J. 2008, 5, 154. [Google Scholar] [CrossRef] [Green Version]
- So, E.Y.; Kim, B.S. Theiler’s virus infection induces TLR3-dependent upregulation of TLR2 critical for proinflammatory cytokine production. Glia 2009, 57, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Palma, J.P.; Kwon, D.; Clipstone, N.A.; Kim, B.S. Infection with Theiler’s murine encephalomyelitis virus directly induces proinflammatory cytokines in primary astrocytes via NF-kappaB activation: Potential role for the initiation of demyelinating disease. J. Virol. 2003, 77, 6322–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.H.; Kim, S.J.; So, E.Y.; Meng, L.; Colonna, M.; Kim, B.S. Melanoma differentiation-associated gene 5 is critical for protection against Theiler’s virus-induced demyelinating disease. J. Virol. 2012, 86, 1531–1543. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Jin, Y.H.; Kang, H.S.; Kim, B.S. Interleukin-6 (IL-6) and IL-17 Synergistically Promote Viral Persistence by Inhibiting Cellular Apoptosis and Cytotoxic T Cell Function. J. Virol. 2014, 88, 8479–8489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, P.A.; Williams, B.R.; Miller, S.D. Distinct roles of protein kinase R and toll-like receptor 3 in the activation of astrocytes by viral stimuli. Glia 2007, 55, 239–252. [Google Scholar] [CrossRef]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [Green Version]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaloye, J.; Roger, T.; Steiner-Tardivel, Q.G.; Le Roy, D.; Knaup Reymond, M.; Akira, S.; Petrilli, V.; Gomez, C.E.; Perdiguero, B.; Tschopp, J.; et al. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog 2009, 5, e1000480. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Jin, Y.H.; Kim, B.S. Prostaglandin E2 produced following infection with Theiler’s virus promotes the pathogenesis of demyelinating disease. PLoS ONE 2017, 12, e0176406. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.S.; Jin, Y.H.; Meng, L.; Hou, W.; Kang, H.S.; Park, H.S.; Koh, C.S. IL-1 signal affects both protection and pathogenesis of virus-induced chronic CNS demyelinating disease. J. Neuroinflamm. 2012, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidotti, L.G.; Chisari, F.V. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 2001, 19, 65–91. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Constam, D.; Frei, K.; Koedel, U.; Pfister, W.; Weller, M. Cytokines and defense against CNS infection. In Cytokines and the CNS; Ransohoff, R.M., Benveniste, E.N., Eds.; CRC Press: Boca Raton, FL, USA, 1996; pp. 188–219. [Google Scholar]
- Ludewig, B.; Junt, T.; Hengartner, H.; Zinkernagel, R.M. Dendritic cells in autoimmune diseases. Curr. Opin. Immunol. 2001, 13, 657–662. [Google Scholar] [CrossRef]
- Asensio, V.C.; Campbell, I.L. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J. Virol. 1997, 71, 7832–7840. [Google Scholar] [CrossRef] [Green Version]
- Lane, T.E.; Asensio, V.C.; Yu, N.; Paoletti, A.D.; Campbell, I.L.; Buchmeier, M.J. Dynamic regulation of alpha- and beta-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J. Immunol. 1998, 160, 970–978. [Google Scholar]
- Hoffman, L.M.; Fife, B.T.; Begolka, W.S.; Miller, S.D.; Karpus, W.J. Central nervous system chemokine expression during Theiler’s virus-induced demyelinating disease. J. Neurovirol. 1999, 5, 635–642. [Google Scholar] [CrossRef]
- Noe, K.H.; Cenciarelli, C.; Moyer, S.A.; Rota, P.A.; Shin, M.L. Requirements for measles virus induction of RANTES chemokine in human astrocytoma-derived U373 cells. J. Virol. 1999, 73, 3117–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, T.E.; Liu, M.T.; Chen, B.P.; Asensio, V.C.; Samawi, R.M.; Paoletti, A.D.; Campbell, I.L.; Kunkel, S.L.; Fox, H.S.; Buchmeier, M.J. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J. Virol. 2000, 74, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Theil, D.J.; Tsunoda, I.; Libbey, J.E.; Derfuss, T.J.; Fujinami, R.S. Alterations in cytokine but not chemokine mRNA expression during three distinct Theiler’s virus infections. J. Neuroimmunol. 2000, 104, 22–30. [Google Scholar] [CrossRef]
- Palma, J.P.; Kim, B.S. The scope and activation mechanisms of chemokine gene expression in primary astrocytes following infection with Theiler’s virus. J. Neuroimmunol. 2004, 149, 121–129. [Google Scholar] [CrossRef]
- Rubio, N.; Sanz-Rodriguez, F.; Lipton, H.L. Theiler’s virus induces the MIP-2 chemokine (CXCL2) in astrocytes from genetically susceptible but not from resistant mouse strains. Cell. Immunol. 2006, 239, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Rubio, N.; Sanz-Rodriguez, F. Induction of the CXCL1 (KC) chemokine in mouse astrocytes by infection with the murine encephalomyelitis virus of Theiler. Virology 2007, 358, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpus, W.J.; Kennedy, K.J.; Fife, B.T.; Bennett, J.L.; Dal Canto, M.C.; Kunkel, S.L.; Lukacs, N.W. Anti-CCL2 treatment inhibits Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neurovirol. 2006, 12, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.H.; Jin, Y.H.; Kim, B.S. Effects of Keratinocyte-Derived Cytokine (CXCL-1) on the Development of Theiler’s Virus-Induced Demyelinating Disease. Front. Cell. Infect. Microbiol. 2018, 8, 9. [Google Scholar] [CrossRef]
- Olson, J.K. Effect of the innate immune response on development of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neurovirol. 2014, 20, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Koh, C.S.; Yamazaki, M.; Ichikawa, M.; Isobe, M.; Ishihara, Y.; Yagita, H.; Kim, B.S. Anti-adhesion molecule therapy in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. Int. Immunol. 1997, 9, 1837–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozner, R.G.; Berria, M.I.; Negrotto, S.; Schattner, M.; Gomez, R.M. Differential astrocyte response to Theiler’s murine encephalomyelitis virus infection. Intervirology 2005, 48, 279–284. [Google Scholar] [CrossRef]
- Rubio, N.; Sanz-Rodriguez, F.; Arevalo, M.A. Up-regulation of the vascular cell adhesion molecule-1 (VCAM-1) induced by Theiler’s murine encephalomyelitis virus infection of murine brain astrocytes. Cell Commun. Adhes. 2010, 17, 57–68. [Google Scholar] [CrossRef]
- Jin, Y.H.; Kang, B.; Kang, H.S.; Koh, C.S.; Kim, B.S. Endothelin-1 contributes to the development of virus-induced demyelinating disease. J. Neuroinflamm. 2020, 17, 307. [Google Scholar] [CrossRef]
- Inoue, A.; Koh, C.S.; Yahikozawa, H.; Yanagisawa, N.; Yagita, H.; Ishihara, Y.; Kim, B.S. The level of tumor necrosis factor-alpha producing cells in the spinal cord correlates with the degree of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. Int. Immunol. 1996, 8, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.; Zoecklein, L.; Papke, L.; Gamez, J.; Denic, A.; Macura, S.; Howe, C. Tumor necrosis factor alpha is reparative via TNFR2 [corrected] in the hippocampus and via TNFR1 [corrected] in the striatum after virus-induced encephalitis. Brain Pathol. 2009, 19, 12–26. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Weinlich, R.; Bortoluci, K.R.; Chehab, C.F.; Serezani, C.H.; Ulbrich, A.G.; Peters-Golden, M.; Russo, M.; Amarante-Mendes, G.P. TLR4/MYD88-dependent, LPS-induced synthesis of PGE2 by macrophages or dendritic cells prevents anti-CD3-mediated CD95L upregulation in T cells. Cell Death Differ. 2008, 15, 1901–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.H.; Hou, W.; Kang, H.S.; Koh, C.S.; Kim, B.S. The role of interleukin-6 in the expression of PD-1 and PDL-1 on central nervous system cells following infection with Theiler’s murine encephalomyelitis virus. J. Virol. 2013, 87, 11538–11551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.C.; Li, C.W.; Xia, W.; Hsu, J.M.; Lee, H.H.; Cha, J.H.; Wang, H.L.; Yang, W.H.; Yen, E.Y.; Chang, W.C.; et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J. Clin. Investig. 2019, 129, 3324–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhaye, S.; Paul, S.; Blakqori, G.; Minet, M.; Weber, F.; Staeheli, P.; Michiels, T. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. USA 2006, 103, 7835–7840. [Google Scholar] [CrossRef] [Green Version]
- Rubio, N.; Palomo, M.; Alcami, A. Interferon-alpha/beta genes are up-regulated in murine brain astrocytes after infection with Theiler’s murine encephalomyelitis virus. J. Interferon Cytokine Res. 2010, 30, 253–262. [Google Scholar] [CrossRef]
- Njenga, M.K.; Pease, L.R.; Wettstein, P.; Mak, T.; Rodriguez, M. Interferon alpha/beta mediates early virus-induced expression of H-2D and H-2K in the central nervous system. Lab. Investig. 1997, 77, 71–84. [Google Scholar] [PubMed]
- Rodriguez, M.; Roos, R.P.; McGavern, D.; Zoecklein, L.; Pavelko, K.; Sang, H.; Lin, X. The CD4-mediated immune response is critical in determining the outcome of infection using Theiler’s viruses with VP1 capsid protein point mutations. Virology 2000, 275, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.H.; Hou, W.; Kim, S.J.; Fuller, A.C.; Kang, B.; Goings, G.; Miller, S.D.; Kim, B.S. Type I interferon signals control Theiler’s virus infection site, cellular infiltration and T cell stimulation in the CNS. J. Neuroimmunol. 2010, 226, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sasson, S.Z.; Hu-Li, J.; Quiel, J.; Cauchetaux, S.; Ratner, M.; Shapira, I.; Dinarello, C.A.; Paul, W.E. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 7119–7124. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.; Pavelko, K.D.; McKinney, C.W.; Leibowitz, J.L. Recombinant human IL-6 suppresses demyelination in a viral model of multiple sclerosis. J. Immunol. 1994, 153, 3811–3821. [Google Scholar] [PubMed]
- Lauder, S.N.; Jones, E.; Smart, K.; Bloom, A.; Williams, A.S.; Hindley, J.P.; Ondondo, B.; Taylor, P.R.; Clement, M.; Fielding, C.; et al. Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. Eur. J. Immunol. 2013, 43, 2613–2625. [Google Scholar] [CrossRef] [Green Version]
- Harker, J.A.; Lewis, G.M.; Mack, L.; Zuniga, E.I. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 2011, 334, 825–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, H.L.; Canto, C.D. Contrasting effects of immunosuppression on Theiler’s virus infection in mice. Infect. Immun. 1977, 15, 903–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.; Lafuse, W.P.; Leibowitz, J.; David, C.S. Partial suppression of Theiler’s virus-induced demyelination in vivo by administration of monoclonal antibodies to immune-response gene products (Ia antigens). Neurology 1986, 36, 964–970. [Google Scholar] [CrossRef]
- Welsh, C.J.; Tonks, P.; Nash, A.A.; Blakemore, W.F. The effect of L3T4 T cell depletion on the pathogenesis of Theiler’s murine encephalomyelitis virus infection in CBA mice. J.Gen.Virol. 1987, 68, 1659–1667. [Google Scholar] [CrossRef]
- Clatch, R.J.; Lipton, H.L.; Miller, S.D. Class II-restricted T cell responses in Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease. II. Survey of host immune responses and central nervous system virus titers in inbred mouse strains. Microb. Pathog. 1987, 3, 327–337. [Google Scholar] [CrossRef]
- Inoue, A.; Choe, Y.K.; Kim, B.S. Analysis of antibody responses to predominant linear epitopes of Theiler’s murine encephalomyelitis virus. J. Virol. 1994, 68, 3324–3333. [Google Scholar] [CrossRef] [Green Version]
- Melvold, R.W.; Jokinen, D.M.; Knobler, R.L.; Lipton, H.L. Variations in genetic control of susceptibility to Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease. I. Differences between susceptible SJL/J and resistant BALB/c strains map near the T cell beta-chain constant gene on chromosome 6. J. Immunol. 1987, 138, 1429–1433. [Google Scholar]
- Bahk, Y.Y.; Kappel, C.A.; Rasmussen, G.; Kim, B.S. Association between susceptibility to Theiler’s virus-induced demyelination and T-cell receptor J beta1-C beta1 polymorphism rather than V beta deletion. J. Virol. 1997, 71, 4181–4185. [Google Scholar] [CrossRef] [Green Version]
- Gerety, S.J.; Rundell, M.K.; Dal Canto, M.C.; Miller, S.D. Class II-restricted T cell responses in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. VI. Potentiation of demyelination with and characterization of an immunopathologic CD4+ T cell line specific for an immunodominant VP2 epitope. J. Immunol. 1994, 152, 919–929. [Google Scholar] [PubMed]
- Yauch, R.L.; Kim, B.S. A predominant viral epitope recognized by T cells from the periphery and demyelinating lesions of SJL/J mice infected with Theiler’s virus is located within VP1(233-244). J. Immunol. 1994, 153, 4508–4519. [Google Scholar]
- Yauch, R.L.; Palma, J.P.; Yahikozawa, H.; Koh, C.S.; Kim, B.S. Role of individual T-cell epitopes of Theiler’s virus in the pathogenesis of demyelination correlates with the ability to induce a Th1 response. J. Virol. 1998, 72, 6169–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, M.A.; Yauch, R.; Dal Canto, M.C.; Kim, B.S. Effect of immunization with Theiler’s virus on the course of demyelinating disease. J. Neuroimmunol. 1993, 45, 67–73. [Google Scholar] [CrossRef]
- Njenga, M.K.; Pavelko, K.D.; Baisch, J.; Lin, X.; David, C.; Leibowitz, J.; Rodriguez, M. Theiler’s virus persistence and demyelination in major histocompatibility complex class II-deficient mice. J. Virol. 1996, 70, 1729–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karls, K.A.; Denton, P.W.; Melvold, R.W. Susceptibility to Theiler’s murine encephalomyelitis virus-induced demyelinating disease in BALB/cAnNCr mice is related to absence of a CD4+ T-cell subset. Mult. Scler. 2002, 8, 469–474. [Google Scholar] [CrossRef]
- Gerety, S.J.; Karpus, W.J.; Cubbon, A.R.; Goswami, R.G.; Rundell, M.K.; Peterson, J.D.; Miller, S.D. Class II-restricted T cell responses in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. V. Mapping of a dominant immunopathologic VP2 T cell epitope in susceptible SJL/J mice. J. Immunol. 1994, 152, 908–918. [Google Scholar]
- Yauch, R.L.; Kerekes, K.; Saujani, K.; Kim, B.S. Identification of a major T-cell epitope within VP3 amino acid residues 24 to 37 of Theiler’s virus in demyelination-susceptible SJL/J mice. J. Virol. 1995, 69, 7315–7318. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.H.; Kang, B.; Kim, B.S. Theiler’s virus infection induces a predominant pathogenic CD4+ T cell response to RNA polymerase in susceptible SJL/J mice. J. Virol. 2009, 83, 10981–10992. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.; Kang, H.K.; Kim, B.S. Identification of capsid epitopes of Theiler’s virus recognized by CNS-infiltrating CD4(+) T cells from virus-infected C57BL/6 mice. Virus Res. 2005, 108, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Mendez-Fernandez, Y.; Moyer, A.M.; Sloma, C.R.; Pirko, I.; Block, M.S.; Rodriguez, M.; Pease, L.R. Antigen-specific CD8+ T cells mediate a peptide-induced fatal syndrome. J. Immunol. 2005, 174, 6854–6862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.S.; Hou, W.; Kim, B.S. Rapid expansion of virus-specific CD4+ T cell types in the CNS of susceptible mice infected with Theiler’s virus. Int. J. Mol. Sci. 2020, 21, 7719. [Google Scholar] [CrossRef]
- Palma, J.P.; Yauch, R.L.; Kang, H.K.; Lee, H.G.; Kim, B.S. Preferential induction of IL-10 in APC correlates with a switch from Th1 to Th2 response following infection with a low pathogenic variant of Theiler’s virus. J. Immunol. 2002, 168, 4221–4230. [Google Scholar] [CrossRef] [PubMed]
- Mohindru, M.; Kang, B.; Kim, B.S. Initial capsid-specific CD4(+) T cell responses protect against Theiler’s murine encephalomyelitisvirus-induced demyelinating disease. Eur. J. Immunol. 2006, 36, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Fiette, L.; Aubert, C.; Muller, U.; Huang, S.; Aguet, M.; Brahic, M.; Bureau, J.F. Theiler’s virus infection of 129Sv mice that lack the interferon alpha/beta or interferon gamma receptors. J. Exp. Med. 1995, 181, 2069–2076. [Google Scholar] [CrossRef]
- Rodriguez, M.; Zoecklein, L.J.; Howe, C.L.; Pavelko, K.D.; Gamez, J.D.; Nakane, S.; Papke, L.M. Gamma interferon is critical for neuronal viral clearance and protection in a susceptible mouse strain following early intracranial Theiler’s murine encephalomyelitis virus infection. J. Virol. 2003, 77, 12252–12265. [Google Scholar] [CrossRef] [Green Version]
- Pullen, L.C.; Miller, S.D.; Dal Canto, M.C.; Van der Meide, P.H.; Kim, B.S. Alteration in the level of interferon-gamma results in acceleration of Theiler’s virus-induced demyelinating disease. J. Neuroimmunol. 1994, 55, 143–152. [Google Scholar] [CrossRef]
- Rodriguez, M.; Pavelko, K.; Coffman, R.L. Gamma interferon is critical for resistance to Theiler’s virus-induced demyelination. J. Virol. 1995, 69, 7286–7290. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.D.; McGavern, D.B.; Pease, L.R.; Rodriguez, M. Cellular sources and targets of IFN-gamma-mediated protection against viral demyelination and neurological deficits. Eur. J. Immunol. 2002, 32, 606–615. [Google Scholar] [CrossRef]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat. Med. 2007, 13, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.E.; Sato, F.; Kawai, E.; Omura, S.; Takahashi, S.; Yoh, K.; Tsunoda, I. Th17-biased RORgammat transgenic mice become susceptible to a viral model for multiple sclerosis. Brain Behav. Immun. 2015, 43, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Richards, M.H.; Getts, M.T.; Podojil, J.R.; Jin, Y.H.; Kim, B.S.; Miller, S.D. Virus expanded regulatory T cells control disease severity in the Theiler’s virus mouse model of MS. J. Autoimmun. 2011, 36, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhao, J.; Perlman, S. Virus-specific regulatory T cells ameliorate encephalitis by repressing effector T cell functions from priming to effector stages. PLoS Pathog 2014, 10, e1004279. [Google Scholar] [CrossRef] [PubMed]
- Nawijn, M.C.; Motta, A.C.; Gras, R.; Shirinbak, S.; Maazi, H.; van Oosterhout, A.J. TLR-2 activation induces regulatory T cells and long-term suppression of asthma manifestations in mice. PLoS ONE 2013, 8, e55307. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, S.; Okada, K.; Maruyama, A.; Matsumoto, M.; Yagita, H.; Seya, T. TLR2-dependent induction of IL-10 and Foxp3+ CD25+ CD4+ regulatory T cells prevents effective anti-tumor immunity induced by Pam2 lipopeptides in vivo. PLoS ONE 2011, 6, e18833. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Beineke, A.; Iskandar, C.D.; Gudi, V.; Herder, V.; Gerhauser, I.; Haist, V.; Teich, R.; Huehn, J.; Baumgartner, W.; et al. Limited role of regulatory T cells during acute Theiler virus-induced encephalitis in resistant C57BL/6 mice. J. Neuroinflamm. 2014, 11, 180. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.R.; Kasprowicz, D.J.; Gersuk, V.H.; Benard, A.; Van Landeghen, M.; Buckner, J.H.; Ziegler, S.F. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J. Clin. Investig. 2003, 112, 1437–1443. [Google Scholar] [CrossRef] [Green Version]
- Zelenay, S.; Lopes-Carvalho, T.; Caramalho, I.; Moraes-Fontes, M.F.; Rebelo, M.; Demengeot, J. Foxp3+ CD25- CD4 T cells constitute a reservoir of committed regulatory cells that regain CD25 expression upon homeostatic expansion. Proc. Natl. Acad. Sci. USA 2005, 102, 4091–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoop, J.N.; Claassen, M.A.; Woltman, A.M.; Binda, R.S.; Kuipers, E.J.; Janssen, H.L.; van der Molen, R.G.; Boonstra, A. Intrahepatic regulatory T cells are phenotypically distinct from their peripheral counterparts in chronic HBV patients. Clin. Immunol. 2008, 129, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Savitskaya, A.; Steiner, C.W.; Rath, E.; Smolen, J.S.; Scheinecker, C. Phenotypic and functional analysis of CD4+ CD25- Foxp3+ T cells in patients with systemic lupus erythematosus. J. Immunol. 2009, 182, 1689–1695. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.; Cabrera, R.; Schenk, E.L.; Nava-Parada, P.; Bell, M.P.; Van Keulen, V.P.; Marler, R.J.; Felts, S.J.; Pease, L.R. Reprogrammed FoxP3+ T regulatory cells become IL-17+ antigen-specific autoimmune effectors in vitro and in vivo. J. Immunol. 2008, 181, 3137–3147. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Veiga-Parga, T.; Sehrawat, S.; Rouse, B.T. Role of regulatory T cells during virus infection. Immunol. Rev. 2013, 255, 182–196. [Google Scholar] [CrossRef]
- Suvas, S.; Azkur, A.K.; Kim, B.S.; Kumaraguru, U.; Rouse, B.T. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J. Immunol. 2004, 172, 4123–4132. [Google Scholar] [CrossRef] [Green Version]
- Trandem, K.; Anghelina, D.; Zhao, J.; Perlman, S. Regulatory T cells inhibit T cell proliferation and decrease demyelination in mice chronically infected with a coronavirus. J. Immunol. 2010, 184, 4391–4400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef]
- Martinez, N.E.; Karlsson, F.; Sato, F.; Kawai, E.; Omura, S.; Minagar, A.; Grisham, M.B.; Tsunoda, I. Protective and detrimental roles for regulatory T cells in a viral model for multiple sclerosis. Brain Pathol. 2014, 24, 436–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, S.; Iwakura, Y.; Suto, H.; Galli, S.J. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J. Leukoc. Biol. 2007, 81, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.D.; McGavern, D.B.; Lin, X.; Njenga, M.K.; Leibowitz, J.; Pease, L.R.; Rodriguez, M. Perforin-dependent neurologic injury in a viral model of multiple sclerosis. J. Neurosci. 1998, 18, 7306–7314. [Google Scholar] [CrossRef] [Green Version]
- Palma, J.P.; Lee, H.G.; Mohindru, M.; Kang, B.S.; Dal Canto, M.; Miller, S.D.; Kim, B.S. Enhanced susceptibility to Theiler’s virus-induced demyelinating disease in perforin-deficient mice. J. Neuroimmunol. 2001, 116, 125–135. [Google Scholar] [CrossRef]
- Pena Rossi, C.; McAllister, A.; Fiette, L.; Brahic, M. Theiler’s virus infection induces a specific cytotoxic T lymphocyte response. Cell. Immunol. 1991, 138, 341–348. [Google Scholar] [CrossRef]
- Lindsley, M.D.; Thiemann, R.; Rodriguez, M. Cytotoxic T cells isolated from the central nervous systems of mice infected with Theiler’s virus. J. Virol. 1991, 65, 6612–6620. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Quinones, C.; McGavern, D.; Schmelzer, J.D.; Hunter, S.F.; Low, P.A.; Rodriguez, M. Absence of neurological deficits following extensive demyelination in a class I-deficient murine model of multiple sclerosis. Nat. Med. 1998, 4, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Qin, Y.; Chluba, J.; Epplen, J.T.; Wekerle, H. Suppression of experimentally induced autoimmune encephalomyelitis by cytolytic T-T cell interactions. Nature 1988, 332, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Borrow, P.; Tonks, P.; Welsh, C.J.; Nash, A.A. The role of CD8+T cells in the acute and chronic phases of Theiler’s murine encephalomyelitis virus-induced disease in mice. J. Gen. Virol. 1992, 73, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Fiette, L.; Aubert, C.; Brahic, M.; Rossi, C.P. Theiler’s virus infection of beta 2-microglobulin-deficient mice. J. Virol. 1993, 67, 589–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, L.C.; Miller, S.D.; Dal Canto, M.C.; Kim, B.S. Class I-deficient resistant mice intracerebrally inoculated with Theiler’s virus show an increased T cell response to viral antigens and susceptibility to demyelination. Eur. J. Immunol. 1993, 23, 2287–2293. [Google Scholar] [CrossRef]
- Rodriguez, M.; Dunkel, A.J.; Thiemann, R.L.; Leibowitz, J.; Zijlstra, M.; Jaenisch, R. Abrogation of resistance to Theiler’s virus-induced demyelination in H-2b mice deficient in beta 2-microglobulin. J. Immunol. 1993, 151, 266–276. [Google Scholar]
- Begolka, W.S.; Haynes, L.M.; Olson, J.K.; Padilla, J.; Neville, K.L.; Dal Canto, M.; Palma, J.; Kim, B.S.; Miller, S.D. CD8-deficient SJL mice display enhanced susceptibility to Theiler’s virus infection and increased demyelinating pathology. J. Neurovirol. 2001, 7, 409–420. [Google Scholar]
- Dethlefs, S.; Brahic, M.; Larsson-Sciard, E.L. An early, abundant cytotoxic T-lymphocyte response against Theiler’s virus is critical for preventing viral persistence. J. Virol. 1997, 71, 8875–8878. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, S.M.; Dal Canto, M.C.; Miller, S.D.; Melvold, R.W. Adoptively transferred CD8+ T lymphocytes provide protection against TMEV-induced demyelinating disease in BALB/c mice. J. Immunol. 1996, 156, 1276–1283. [Google Scholar]
- Borson, N.D.; Paul, C.; Lin, X.; Nevala, W.K.; Strausbauch, M.A.; Rodriguez, M.; Wettstein, P.J. Brain-infiltrating cytolytic T lymphocytes specific for Theiler’s virus recognize H2Db molecules complexed with a viral VP2 peptide lacking a consensus anchor residue. J. Virol. 1997, 71, 5244–5250. [Google Scholar] [CrossRef] [Green Version]
- Dethlefs, S.; Escriou, N.; Brahic, M.; van der Werf, S.; Larsson-Sciard, E.L. Theiler’s virus and Mengo virus induce cross-reactive cytotoxic T lymphocytes restricted to the same immunodominant VP2 epitope in C57BL/6 mice. J. Virol. 1997, 71, 5361–5365. [Google Scholar] [CrossRef] [Green Version]
- Lyman, M.A.; Lee, H.G.; Kang, B.S.; Kang, H.K.; Kim, B.S. Capsid-specific cytotoxic T lymphocytes recognize three distinct H-2D(b)-restricted regions of the BeAn strain of Theiler’s virus and exhibit different cytokine profiles. J. Virol. 2002, 76, 3125–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, L.C.; Kim, B.S. Identification of a suitable target cell line for Theiler’s murine encephalomyelitis virus specific CTL. FASEB J. 1991, 5, A1094. [Google Scholar]
- Baenziger, J.; Hengartner, H.; Zinkernagel, R.; Cole, G. Induction or prevention of immunopathological disease by cloned cytotoxic T cell lines specific for lymphocytic choriomeningitis virus. Eur. J. Immunol. 1986, 16, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.; Allan, J.; Lynch, F.; Ceredig, R. Dissection of an inflammatory process induced by CD8+ T cells. Immulol. Today 1990, 11, 55–59. [Google Scholar] [CrossRef]
- Huber, S.A.; Lodge, P.A. Coxsackievirus B-3 myocarditis in Balb/c mice. Evidence for autoimmunity to myocyte antigens. Am. J. Pathol. 1984, 116, 21–29. [Google Scholar] [PubMed]
- Chang, Y.; Nadigel, J.; Boulais, N.; Bourbeau, J.; Maltais, F.; Eidelman, D.H.; Hamid, Q. CD8 positive T cells express IL-17 in patients with chronic obstructive pulmonary disease. Respir. Res. 2011, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.M.; Peng, C.; Zhao, Q.; Wu, Y.; Zhu, L.Y.; Wang, J.; Yin, X.Y.; Li, L.; Zheng, L. Tumor-activated monocytes promote expansion of IL-17-producing CD8+ T cells in hepatocellular carcinoma patients. J. Immunol. 2010, 185, 1544–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myoung, J.; Kang, H.S.; Hou, W.; Meng, L.; Dal Canto, M.C.; Kim, B.S. Epitope-specific CD8+ T cells play a differential pathogenic role in the development of a viral disease model for multiple sclerosis. J. Virol. 2012, 86, 13717–13728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, R.P.; Stein, S.; Routbort, M.; Senkowski, A.; Bodwell, T.; Wollmann, R. Theiler’s murine encephalomyelitis virus neutralization escape mutants have a change in disease phenotype. J.Virol. 1989, 63, 4469–4473. [Google Scholar] [CrossRef] [Green Version]
- Zurbriggen, A.; Fujinami, R.S. A neutralization-resistant Theiler’s virus variant produces an altered disease pattern in the mouse central nervous system. J.Virol. 1989, 63, 1505–1513. [Google Scholar] [CrossRef] [Green Version]
- Roos, R.P.; Nalefski, E.A.; Nitayaphan, S.; Variakojis, R.; Singh, K.K. An isoelectric focusing overlay study of the humoral immune response in Theiler’s virus demyelinating disease. J. Neuroimmunol. 1987, 13, 305–314. [Google Scholar] [CrossRef]
- Mattson, D.H.; Roos, R.P.; Arnason, B.G. Comparison of agar gel electrophoresis and isoelectric focusing in multiple sclerosis and subacute sclerosing panencephalitis. Ann. Neurol. 1981, 9, 34–41. [Google Scholar] [CrossRef]
- Cash, E.; Bandeira, A.; Chirinian, S.; Brahic, M. Characterization of B lymphocytes present in the demyelinating lesions induced by Theiler’s virus. J. Immunol. 1989, 143, 984–988. [Google Scholar] [PubMed]
- Pachner, A.R.; Li, L.; Lagunoff, D. Plasma cells in the central nervous system in the Theiler’s virus model of multiple sclerosis. J. Neuroimmunol. 2011, 232, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Crane, M.A.; Jue, C.; Mitchell, M.; Lipton, H.; Kim, B.S. Detection of restricted predominant epitopes of Theiler’s murine encephalomyelitis virus capsid proteins expressed in the lambda gt11 system: Differential patterns of antibody reactivity among different mouse strains. J. Neuroimmunol. 1990, 27, 173–186. [Google Scholar] [CrossRef]
- Kim, B.S.; Choe, Y.K.; Crane, M.A.; Jue, C.R. Identification and localization of a limited number of predominant conformation-independent antibody epitopes of Theiler’s murine encephalomyelitus virus. Immunol. Lett. 1992, 31, 199–205. [Google Scholar] [CrossRef]
- Cameron, K.; Zhang, X.; Seal, B.; Rodriguez, M.; Njenga, M.K. Antigens to viral capsid and non-capsid proteins are present in brain tissues and antibodies in sera of Theiler’s virus-infected mice. J. Virol. Methods 2001, 91, 11–19. [Google Scholar] [CrossRef]
- Rodriguez, M.; Kenny, J.J.; Thiemann, R.L.; Woloschak, G.E. Theiler’s virus-induced demyelination in mice immunosuppressed with anti-IgM and in mice expressing the xid gene. Microb. Pathog. 1990, 8, 23–35. [Google Scholar] [CrossRef]
- Kang, B.S.; Palma, J.P.; Lyman, M.A.; Dal Canto, M.; Kim, B.S. Antibody response is required for protection from Theiler’s virus-induced encephalitis in C57BL/6 mice in the absence of CD8(+) T cells. Virology 2005, 340, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilli, F.; Li, L.; Campbell, S.J.; Anthony, D.C.; Pachner, A.R. The effect of B-cell depletion in the Theiler’s model of multiple sclerosis. J. Neurol. Sci. 2015, 359, 40–47. [Google Scholar] [CrossRef]
- McCright, I.J.; Fujinami, R.S. Lack of correlation of Theiler’s virus binding to cells with infection. J. Neurovirol. 1997, 3 (Suppl. 1), S68–S70. [Google Scholar]
- Chesnut, R.G.; Grey, H.M. Antigen presentation by B cells and its significance in T-B interactions. Adv. Immunol. 1986, 39, 51–94. [Google Scholar] [PubMed]
- Santiago-Raber, M.L.; Baudino, L.; Izui, S. Emerging roles of TLR7 and TLR9 in murine SLE. J. Autoimmun. 2009, 33, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H. TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 2011, 11, 807–822. [Google Scholar] [CrossRef]
- Kiefer, K.; Oropallo, M.A.; Cancro, M.P.; Marshak-Rothstein, A. Role of type I interferons in the activation of autoreactive B cells. Immunol. Cell Biol. 2012, 90, 498–504. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, T. Interleukin-6: From basic science to medicine—40 years in immunology. Annu. Rev. Immunol. 2005, 23, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuno, K.; Matsushima, K. The IL-1 receptor signaling pathway. J. Leukoc. Biol. 1994, 56, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; Zurbriggen, A.; Powell, H.C. Monoclonal antibody defines determinant between Theiler’s virus and lipid -like structures. J. Neuroimmunol. 1988, 20, 25–32. [Google Scholar] [CrossRef]
- Tsunoda, I.; Kuang, L.Q.; Kobayashi-Warren, M.; Fujinami, R.S. Central nervous system pathology caused by autoreactive CD8+ T-cell clones following virus infection. J. Virol. 2005, 79, 14640–14646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinami, R.S.; Zurbriggen, A. Is Theiler’s murine encephalomyelitis virus infection of mice an autoimmune disease? APMIS 1989, 97, 1–8. [Google Scholar] [CrossRef]
- Miller, S.D.; Vanderlugt, C.L.; Begolka, W.S.; Pao, W.; Neville, K.L.; Yauch, R.L.; Kim, B.S. Epitope spreading leads to myelin-specific autoimmune responses in SJL mice chronically infected with Theiler’s virus. J. Neurovirol. 1997, 3 (Suppl. 1), S62–S65. [Google Scholar]
- Miller, D.J.; Njenga, M.K.; Murray, P.D.; Leibowitz, J.; Rodriguez, M. A monoclonal natural autoantibody that promotes remyelination suppresses central nervous system inflammation and increases virus expression after Theiler’s virus-induced demyelination. Int. Immunol. 1996, 8, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drescher, K.M.; Pease, L.R.; Rodriguez, M. Antiviral immune responses modulate the nature of central nervous system (CNS) disease in a murine model of multiple sclerosis. Immunol. Rev. 1997, 159, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Miller, D.J.; Pease, L.R.; Rodriguez, M. Targeting of IgMkappa antibodies to oligodendrocytes promotes CNS remyelination. J. Neurosci. 1998, 18, 7700–7708. [Google Scholar] [CrossRef] [PubMed]
- Palma, J.P.; Park, S.H.; Kim, B.S. Treatment with lipopolysaccharide enhances the pathogenicity of a low-pathogenic variant of Theiler’s murine encephalomyelitis virus. J. Neurosci. Res. 1996, 45, 776–785. [Google Scholar] [CrossRef]
- Fuller, A.; Yahikozawa, H.; So, E.Y.; Dal Canto, M.; Koh, C.S.; Welsh, C.J.; Kim, B.S. Castration of male C57L/J mice increases susceptibility and estrogen treatment restores resistance to Theiler’s virus-induced demyelinating disease. J. Neurosci. Res. 2007, 85, 871–881. [Google Scholar] [CrossRef]
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Subbarao, K. The Immunobiology of SARS*. Annu. Rev. Immunol. 2007, 25, 443–472. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Strains | Background Genes | MHC | Susceptibility | Th Epitopes | CTL Epitopes |
| SJL | SJL | H-2s | Yes | VP272−86, 3D21−36, VP324−37, VP1233−250 | VP3159−166, VP3173−181, VP111−20 |
| C57BL/6 (B6) | B6 | H-2b | No | VP2206−220, VP425−38, | VP2121−130 |
| B10.S | B10 | H-2s | No/weak | VP272−86, 3D21−36, VP324−37, VP1233−250 | VP3159−166, VP3173−181, VP111−20 |
| B6.S | B6 | H-2s | No/weak | VP272−86, 3D21−36, VP324−37, VP1233−250 | VP3159−166, VP3173−181, VP111−20 |
| (SJLxB6)F1 | SJL + B6 | H-2s/H-2b | No/weak | VP2206−220, VP425−38, VP272−86, 3D21−36 | VP2121−130, VP3159−166, VP3173−181, VP111−20 |
| Transgene | Background Genes | MHC | Susceptibility | Th Epitopes | CTL Epitopes |
| VP2-TCR-Tg | SJL | H-2s | >Yes | >>>VP272−86, | <<VP3159−166, VP3173−181, VP111−20 |
| TMEV P1-Tg | SJL | H-2s | No | 3D21−86 | |
| TMEV P2/P3-Tg | SJL | H-2s | Yes | VP272−86, VP324−37, VP1233−250 | VP3159−166, VP3173−181, VP111−20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.S. Excessive Innate Immunity Steers Pathogenic Adaptive Immunity in the Development of Theiler’s Virus-Induced Demyelinating Disease. Int. J. Mol. Sci. 2021, 22, 5254. https://doi.org/10.3390/ijms22105254
Kim BS. Excessive Innate Immunity Steers Pathogenic Adaptive Immunity in the Development of Theiler’s Virus-Induced Demyelinating Disease. International Journal of Molecular Sciences. 2021; 22(10):5254. https://doi.org/10.3390/ijms22105254
Chicago/Turabian StyleKim, Byung S. 2021. "Excessive Innate Immunity Steers Pathogenic Adaptive Immunity in the Development of Theiler’s Virus-Induced Demyelinating Disease" International Journal of Molecular Sciences 22, no. 10: 5254. https://doi.org/10.3390/ijms22105254
APA StyleKim, B. S. (2021). Excessive Innate Immunity Steers Pathogenic Adaptive Immunity in the Development of Theiler’s Virus-Induced Demyelinating Disease. International Journal of Molecular Sciences, 22(10), 5254. https://doi.org/10.3390/ijms22105254