
Reviewing Evidence for the Relationship of EEG Abnormalities and RTT Phenotype Paralleled by Insights from Animal Studies


Abstract
:1. Introduction
2. Results
2.1. Epilepsy/Seizures
2.1.1. Patient Studies
Seizure Semiology
Centro-Temporal Spikes as a Frequent Epileptiform Abnormality in RTT
Development of Seizures and Severity of RTT Symptoms
2.1.2. Animal Studies
2.2. Abnormalities in Resting EEG Spectra and Their Neurophysiological Underpinnings
2.2.1. Patient Studies
General Slowing of Background EEG Activity
Spectral Slope
Cortical Gamma Oscillations (30–100 Hz)
EEG Abnormalities in Relation to the Developmental Stage
2.2.2. Animal Studies
2.3. EEG Abnormalities Associated with Sleep Disturbances, and Their Neurophysiological Underpinnings
2.3.1. Patient Studies
2.3.2. Animal Studies
2.4. Behavioral and EEG Abnormalities in Relation to RTT Genotype
2.4.1. Patient Studies
2.4.2. Animal Studies
2.5. EEG as a Biomarker of Treatment Efficacy
3. Method
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hagberg, B.; Goutières, F.; Hanefeld, F.; Rett, A.; Wilson, J. Rett Syndrome: Criteria for Inclusion and Exclusion. Brain Dev. 1985, 7, 372–373. [Google Scholar] [CrossRef]
- Kozinetz, C.A.; Skender, M.L.; MacNaughton, N.; Almes, M.J.; Schultz, R.J.; Percy, A.K.; Glaze, D.G. Epidemiology of Rett Syndrome: A Population-Based Registry. Pediatrics 1993, 91, 445–450. [Google Scholar]
- Laurvick, C.L.; de Klerk, N.; Bower, C.; Christodoulou, J.; Ravine, D.; Ellaway, C.; Williamson, S.; Leonard, H. Rett Syndrome in Australia: A Review of the Epidemiology. J. Pediatr. 2006, 148, 347–352. [Google Scholar] [CrossRef]
- Wong, V.C.N.; Li, S.Y.H. Rett Syndrome: Prevalence Among Chinese and a Comparison of MECP2 Mutations of Classic Rett Syndrome With Other Neurodevelopmental Disorders. J. Child Neurol. 2007, 22, 1397–1400. [Google Scholar] [CrossRef]
- Hagberg, B.; Hanefeld, F.; Percy, A.; Skjeldal, O. An Update on Clinically Applicable Diagnostic Criteria in Rett Syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2002, 6, 293–297. [Google Scholar] [CrossRef]
- Young, D.; Bebbington, A.; Anderson, A.; Ravine, D.; Ellaway, C.; Kulkarni, A.; Klerk, N.; Kaufmann, W.; Leonard, H. The Diagnosis of Autism in a Female: Could It Be Rett Syndrome? Eur. J. Pediatr. 2008, 167, 661–669. [Google Scholar] [CrossRef]
- Weese-Mayer, D.E.; Lieske, S.P.; Boothby, C.M.; Kenny, A.S.; Bennett, H.L.; Ramirez, J.-M. Autonomic Dysregulation in Young Girls with Rett Syndrome during Nighttime In-Home Recordings. Pediatr. Pulmonol. 2008, 43, 1045–1060. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett Syndrome: Revised Diagnostic Criteria and Nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Merrick, J.; Lotan, M.; Morad, M.; Kandel, I. Rett Syndrome and Aging. Int. J. Disabil. Hum. Dev. 2006, 5. [Google Scholar] [CrossRef]
- Svedberg, L.; Herngren, B.; Michno, P. How Reconstructive Surgery Combined with Physiotherapy for a Painful Nontraumatic Patellar Dislocation Enabled a Woman with Rett Syndrome to Become Pain Free and Remain Physically Active: A Case Report. Clin. Case Rep. 2019, 7, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl-CpG-Binding Protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Bienvenu, T.; Carrié, A.; de Roux, N.; Vinet, M.C.; Jonveaux, P.; Couvert, P.; Villard, L.; Arzimanoglou, A.; Beldjord, C.; Fontes, M.; et al. MECP2 Mutations Account for Most Cases of Typical Forms of Rett Syndrome. Hum. Mol. Genet. 2000, 9, 1377–1384. [Google Scholar] [CrossRef]
- Cheadle, J.P.; Gill, H.; Fleming, N.; Maynard, J.; Kerr, A.; Leonard, H.; Krawczak, M.; Cooper, D.N.; Lynch, S.; Thomas, N.; et al. Long-Read Sequence Analysis of the MECP2 Gene in Rett Syndrome Patients: Correlation of Disease Severity with Mutation Type and Location. Hum. Mol. Genet. 2000, 9, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific Mutations in Methyl-CpG-Binding Protein 2 Confer Different Severity in Rett Syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A Mouse Mecp2-Null Mutation Causes Neurological Symptoms That Mimic Rett Syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of Methyl-CpG Binding Protein-2 in CNS Neurons Results in a Rett-like Phenotype in Mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef]
- Schaevitz, L.R.; Gómez, N.B.; Zhen, D.P.; Berger-Sweeney, J.E. MeCP2 R168X Male and Female Mutant Mice Exhibit Rett-like Behavioral Deficits. Genes Brain Behav. 2013, 12, 732–740. [Google Scholar] [CrossRef]
- Zhou, Z.; Goffin, D. Modeling Rett Syndrome with MeCP2 T158A Knockin Mice. In Comprehensive Guide to Autism; Patel, V.B., Preedy, V.R., Martin, C.R., Eds.; Springer: New York, NY, USA, 2014; pp. 2723–2739. ISBN 978-1-4614-4787-0. [Google Scholar]
- Pitcher, M.R.; Herrera, J.A.; Buffington, S.A.; Kochukov, M.Y.; Merritt, J.K.; Fisher, A.R.; Schanen, N.C.; Costa-Mattioli, M.; Neul, J.L. Rett Syndrome like Phenotypes in the R255X Mecp2 Mutant Mouse Are Rescued by MECP2 Transgene. Hum. Mol. Genet. 2015, 24, 2662–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangasamy, S.; Olfers, S.; Gerald, B.; Hilbert, A.; Svejda, S.; Narayanan, V. Reduced Neuronal Size and MTOR Pathway Activity in the Mecp2 A140V Rett Syndrome Mouse Model. F1000Research 2016, 5, 2269. [Google Scholar] [CrossRef] [PubMed]
- Vonhoff, F.; Williams, A.; Ryglewski, S.; Duch, C. Drosophila as a Model for MECP2 Gain of Function in Neurons. PLoS ONE 2012, 7, e31835. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, K.; Lin, Y.; Kubodera, R.; Shimizu, Y.; Tanaka, H.; Ohshima, T. Zebrafish Mecp2 Is Required for Proper Axonal Elongation of Motor Neurons and Synapse Formation. Dev. Neurobiol. 2017, 77, 1101–1113. [Google Scholar] [CrossRef]
- Pietri, T.; Roman, A.-C.; Guyon, N.; Romano, S.A.; Washbourne, P.; Moens, C.B.; de Polavieja, G.G.; Sumbre, G. The First Mecp2-Null Zebrafish Model Shows Altered Motor Behaviors. Front. Neural Circuits 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Veeraragavan, S.; Wan, Y.-W.; Connolly, D.R.; Hamilton, S.M.; Ward, C.S.; Soriano, S.; Pitcher, M.R.; McGraw, C.M.; Huang, S.G.; Green, J.R.; et al. Loss of MeCP2 in the Rat Models Regression, Impaired Sociability and Transcriptional Deficits of Rett Syndrome. Hum. Mol. Genet. 2016, 25, 3284–3302. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhong, W.; Cui, N.; Johnson, C.M.; Xing, H.; Zhang, S.; Jiang, C. Characterization of Rett Syndrome-like Phenotypes in Mecp2-Knockout Rats. J. Neurodev. Disord. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Alessio, N.; Riccitiello, F.; Squillaro, T.; Capasso, S.; Del Gaudio, S.; Di Bernardo, G.; Cipollaro, M.; Melone, M.A.B.; Peluso, G.; Galderisi, U. Neural Stem Cells from a Mouse Model of Rett Syndrome Are Prone to Senescence, Show Reduced Capacity to Cope with Genotoxic Stress, and Are Impaired in the Differentiation Process. Exp. Mol. Med. 2018, 50. [Google Scholar] [CrossRef] [Green Version]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.R.; Fernandes, T.G.; Cabral, J.M.S.; Diogo, M.M. Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 3751. [Google Scholar] [CrossRef]
- Goffin, D.; Allen, M.; Zhang, L.; Amorim, M.; Wang, I.-T.J.; Reyes, A.-R.S.; Mercado-Berton, A.; Ong, C.; Cohen, S.; Hu, L.; et al. Rett Syndrome Mutation MeCP2 T158A Disrupts DNA Binding, Protein Stability and ERP Responses. Nat. Neurosci. 2012, 15, 274–283. [Google Scholar] [CrossRef]
- Brust, V.; Schindler, P.M.; Lewejohann, L. Lifetime Development of Behavioural Phenotype in the House Mouse (Mus Musculus). Front. Zool. 2015, 12, S17. [Google Scholar] [CrossRef] [Green Version]
- Tudor, M.; Akbarian, S.; Chen, R.Z.; Jaenisch, R. Transcriptional Profiling of a Mouse Model for Rett Syndrome Reveals Subtle Transcriptional Changes in the Brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15536–15541. [Google Scholar] [CrossRef] [Green Version]
- Ehrhart, F.; Coort, S.L.M.; Cirillo, E.; Smeets, E.; Evelo, C.T.; Curfs, L.M.G. Rett Syndrome—Biological Pathways Leading from MECP2 to Disorder Phenotypes. Orphanet J. Rare Dis. 2016, 11. [Google Scholar] [CrossRef] [Green Version]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 Is Responsible for the Congenital Variant of Rett Syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Kadam, S.D.; Sullivan, B.J.; Goyal, A.; Blue, M.E.; Smith-Hicks, C. Rett Syndrome and CDKL5 Deficiency Disorder: From Bench to Clinic. Int. J. Mol. Sci. 2019, 20, 5098. [Google Scholar] [CrossRef] [Green Version]
- Dastidar, S.G.; Bardai, F.H.; Ma, C.; Price, V.; Rawat, V.; Verma, P.; Narayanan, V.; D’Mello, S.R. Isoform-Specific Toxicity of Mecp2 in Postmitotic Neurons: Suppression of Neurotoxicity by FoxG1. J. Neurosci. 2012, 32, 2846–2855. [Google Scholar] [CrossRef]
- Ward, L.M. Synchronous Neural Oscillations and Cognitive Processes. Trends Cogn. Sci. 2003, 7, 553–559. [Google Scholar] [CrossRef]
- Lopes da Silva, F. EEG and MEG: Relevance to Neuroscience. Neuron 2013, 80, 1112–1128. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A Progressive Syndrome of Autism, Dementia, Ataxia, and Loss of Purposeful Hand Use in Girls: Rett’s Syndrome: Report of 35 Cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef]
- Garofalo, E.A.; Drury, I.; Goldstein, G.W. EEG Abnormalities Aid Diagnosis of Rett Syndrome. Pediatr. Neurol. 1988, 4, 350–353. [Google Scholar] [CrossRef] [Green Version]
- Sysoeva, O.V.; Smirnov, K.; Stroganova, T.A. Sensory Evoked Potentials in Patients with Rett Syndrome through the Lens of Animal Studies: Systematic Review. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 213–224. [Google Scholar] [CrossRef]
- Krajnc, N. Management of Epilepsy in Patients with Rett Syndrome: Perspectives and Considerations. Ther. Clin. Risk Manag. 2015, 925. [Google Scholar] [CrossRef] [Green Version]
- Cutri-French, C.; Armstrong, D.; Saby, J.; Gorman, C.; Lane, J.; Fu, C.; Peters, S.U.; Percy, A.; Neul, J.L.; Marsh, E.D. Comparison of Core Features in Four Developmental Encephalopathies in the Rett Natural History Study. Ann. Neurol. 2020, 88, 396–406. [Google Scholar] [CrossRef]
- Tarquinio, D.C.; Hou, W.; Berg, A.; Kaufmann, W.E.; Lane, J.B.; Skinner, S.A.; Motil, K.J.; Neul, J.L.; Percy, A.K.; Glaze, D.G. Longitudinal Course of Epilepsy in Rett Syndrome and Related Disorders. Brain J. Neurol. 2017, 140, 306–318. [Google Scholar] [CrossRef]
- Pintaudi, M.; Calevo, M.G.; Vignoli, A.; Parodi, E.; Aiello, F.; Baglietto, M.G.; Hayek, Y.; Buoni, S.; Renieri, A.; Russo, S.; et al. Epilepsy in Rett Syndrome: Clinical and Genetic Features. Epilepsy Behav. 2010, 19, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Buoni, S.; Zannolli, R.; Felice, C.D.; Saponari, S.; Strambi, M.; Dotti, M.T.; Castrucci, E.; Corbini, L.; Orsi, A.; Hayek, J. Drug-Resistant Epilepsy and Epileptic Phenotype-EEG Association in MECP2 Mutated Rett Syndrome. Clin. Neurophysiol. 2008, 119, 2455–2458. [Google Scholar] [CrossRef]
- Operto, F.F.; Mazza, R.; Pastorino, G.M.G.; Verrotti, A.; Coppola, G. Epilepsy and Genetic in Rett Syndrome: A Review. Brain Behav. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Leung, H.T.T.; Ring, H. Epilepsy in Four Genetically Determined Syndromes of Intellectual Disability. J. Intellect. Disabil. Res. JIDR 2013, 57, 3–20. [Google Scholar] [CrossRef]
- Glaze, D.G.; Schultz, R.J.; Frost, J.D. Rett Syndrome: Characterization of Seizures versus Non-Seizures. Electroencephalogr. Clin. Neurophysiol. 1998, 106, 79–83. [Google Scholar] [CrossRef]
- Vignoli, A.; Fabio, R.A.; La Briola, F.; Giannatiempo, S.; Antonietti, A.; Maggiolini, S.; Canevini, M.P. Correlations between Neurophysiological, Behavioral, and Cognitive Function in Rett Syndrome. Epilepsy Behav. 2010, 17, 489–496. [Google Scholar] [CrossRef]
- Buoni, S.; Zannolli, R.; De Felice, C.; De Nicola, A.; Guerri, V.; Guerra, B.; Casali, S.; Pucci, B.; Corbini, L.; Mari, F.; et al. EEG Features and Epilepsy in MECP2-Mutated Patients with the Zappella Variant of Rett Syndrome. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2010, 121, 652–657. [Google Scholar] [CrossRef]
- Glaze, D.G.; Percy, A.K.; Skinner, S.; Motil, K.J.; Neul, J.L.; Barrish, J.O.; Lane, J.B.; Geerts, S.P.; Annese, F.; Graham, J.; et al. Epilepsy and the Natural History of Rett Syndrome. Neurology 2010, 74, 909–912. [Google Scholar] [CrossRef] [Green Version]
- Pintaudi, M.; Calevo, M.G.; Vignoli, A.; Baglietto, M.G.; Hayek, Y.; Traverso, M.; Giacomini, T.; Giordano, L.; Renieri, A.; Russo, S.; et al. Antiepileptic Drugs in Rett Syndrome. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2015, 19, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Downs, J.; Wong, K.; Williams, S.; Leonard, H. Using a Large International Sample to Investigate Epilepsy in Rett Syndrome. Dev. Med. Child Neurol. 2013, 55, 553–558. [Google Scholar] [CrossRef]
- Dolce, A.; Ben-Zeev, B.; Naidu, S.; Kossoff, E.H. Rett Syndrome and Epilepsy: An Update for Child Neurologists. Pediatr. Neurol. 2013, 48, 337–345. [Google Scholar] [CrossRef] [PubMed]
- d’Orsi, G.; Demaio, V.; Minervini, M.G. Myoclonic Status Misdiagnosed as Movement Disorders in Rett Syndrome: A Video-Polygraphic Study. Epilepsy Behav. EB 2009, 15, 260–262. [Google Scholar] [CrossRef]
- d’Orsi, G.; Trivisano, M.; Luisi, C.; Demaio, V.; Di Claudio, M.T.; Pascarella, M.G.; Sciruicchio, V.; Galeone, D.; La Neve, A.; Scarpelli, F.; et al. Epileptic Seizures, Movement Disorders, and Breathing Disturbances in Rett Syndrome: Diagnostic Relevance of Video-Polygraphy. Epilepsy Behav. 2012, 25, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Hagebeuk, E.E.O.; Koelman, J.H.T.M.; Duran, M.; Abeling, N.G.; Vyth, A.; Poll-The, B.-T. Clinical and Electroencephalographic Effects of Folinic Acid Treatment in Rett Syndrome Patients. J. Child Neurol. 2011, 26, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Parrini, E. Epilepsy in Rett Syndrome, and CDKL5- and FOXG1-Gene-Related Encephalopathies. Epilepsia 2012, 53, 2067–2078. [Google Scholar] [CrossRef]
- Gulati, P.; Jain, P.; Borlot, F.; Munn, R.; Ochi, A. Teaching Video NeuroImages: Needle-like Central Spikes Evoked by Hand Tapping in Rett Syndrome. Neurology 2019, 93, e422–e423. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Li, C.; Wang, X.; Yu, X.; Jiang, J. Tapping-Lips Aggravated Interictal Bilateral Discharges in EEG in the Patients with Rett Syndrome: A Case Report. BMC Neurol. 2019, 19, 77. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.R.B.; Ostendorf, A. Teaching NeuroImages: A Central Theta EEG Rhythm in Rett Syndrome Can Masquerade as Seizure. Neurology 2016, 87, e29–e30. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Liu, C.; Shi, M.; Cui, L. Clapping-Surpressed Focal Spikes in EEG May Be Unique for the Patients with Rett Syndrome: A Case Report. BMC Neurol. 2016, 16, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, E.; Raviglione, F.; Franceschetti, S.; D’Arrigo, S.; Milani, D.; Selicorni, A.; Riva, D.; Zuffardi, O.; Pantaleoni, C.; Binelli, S. Seizures and EEG Features in 74 Patients with Genetic-Dysmorphic Syndromes. Am. J. Med. Genet. A 2014, 164A, 3154–3161. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Ming, X.; Dekermenjian, R.; Chokroverty, S. Continuous Spike and Wave in Slow-Wave Sleep in a Patient with Rett Syndrome and in a Patient with Lhermitte-Duclos Syndrome and Neurofibromatosis 1. J. Child Neurol. 2014, 29, NP176–NP180. [Google Scholar] [CrossRef] [PubMed]
- Nissenkorn, A.; Ben-Zeev, B. Unilateral Rhythmic Hand Tapping in Rett Syndrome: Is This Stereotypy? J. Child Neurol. 2013, 28, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Roche Martínez, A.; Alonso Colmenero, M.I.; Gomes Pereira, A.; Sanmartí Vilaplana, F.X.; Armstrong Morón, J.; Pineda Marfa, M. Reflex Seizures in Rett Syndrome. Epileptic Disord. Int. Epilepsy J. Videotape 2011, 13, 389–393. [Google Scholar] [CrossRef]
- Pelc, K.; Dan, B. Postural Cortical Myoclonus during Gait in Rett Syndrome. Epilepsy Behav. 2009, 16, 188. [Google Scholar] [CrossRef]
- Moser, S.J.; Weber, P.; Lütschg, J. Rett Syndrome: Clinical and Electrophysiologic Aspects. Pediatr. Neurol. 2007, 36, 95–100. [Google Scholar] [CrossRef]
- Buoni, S.; Zannolli, R.; Colamaria, V.; Macucci, F.; di Bartolo, R.M.; Corbini, L.; Orsi, A.; Zappella, M.; Hayek, J. Myoclonic Encephalopathy in the CDKL5 Gene Mutation. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2006, 117, 223–227. [Google Scholar] [CrossRef]
- Valente, K.D. Another Rett Patient with a Typical Angelman EEG. Epilepsia 2003, 44, 873–874, author reply 874. [Google Scholar] [CrossRef] [Green Version]
- Bashina, V.M.; Simashkova, N.V.; Grachev, V.V.; Gorbachevskaya, N.L. Speech and Motor Disturbances in Rett Syndrome. Neurosci. Behav. Physiol. 2002, 32, 323–327. [Google Scholar] [CrossRef]
- Laan, L.A.E.M.; Vein, A.A. A Rett Patient with a Typical Angelman EEG. Epilepsia 2002, 43, 1590–1592. [Google Scholar] [CrossRef]
- Laan, L.A.E.M.; Brouwer, O.F.; Begeer, C.H.; Zwinderman, A.H.; Gert van Dijk, J. The Diagnostic Value of the EEG in Angelman and Rett Syndrome at a Young Age. Electroencephalogr. Clin. Neurophysiol. 1998, 106, 404–408. [Google Scholar] [CrossRef]
- Cooper, R.A.; Kerr, A.M.; Amos, P.M. Rett Syndrome: Critical Examination of Clinical Features, Serial EEG and Video-Monitoring in Understanding and Management. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 1998, 2, 127–135. [Google Scholar] [CrossRef]
- Lappalainen, R.; Liewendahl, K.; Sainio, K.; Nikkinen, P.; Riikonen, R.S. Brain Perfusion SPECT and EEG Findings in Rett Syndrome. Acta Neurol. Scand. 1997, 95, 44–50. [Google Scholar] [CrossRef]
- Niedermeyer, E.; Naidu, S.B.; Plate, C. Unusual EEG Theta Rhythms over Central Region in Rett Syndrome: Considerations of the Underlying Dysfunction. Clin. EEG Electroencephalogr. 1997, 28, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Elian, M.; Rudolf, N.D. EEG and Respiration in Rett Syndrome. Acta Neurol. Scand. 1991, 83, 123–128. [Google Scholar] [CrossRef]
- Kerr, A.; Southall, D.; Amos, P.; Cooper, R.; Samuels, M.; Mitchell, J.; Stephenson, J. Correlation of Electroencephalogram, Respiration and Movement in the Rett Syndrome. Brain Dev. 1990, 12, 61–68. [Google Scholar] [CrossRef]
- Aldrich, M.S.; Garofalo, E.A.; Drury, I. Epileptiform Abnormalities during Sleep in Rett Syndrome. Electroencephalogr. Clin. Neurophysiol. 1990, 75, 365–370. [Google Scholar] [CrossRef]
- Niedermeyer, E.; Naidu, S. Further EEG Observations in Children with the Rett Syndrome. Brain Dev. 1990, 12, 53–54. [Google Scholar] [CrossRef]
- Hagne, I.; Witt-Engerström, I.; Hagberg, B. EEG Development in Rett Syndrome. A Study of 30 Cases. Electroencephalogr. Clin. Neurophysiol. 1989, 72, 1–6. [Google Scholar] [CrossRef]
- Espinar-Sierra, J.; Toledano, M.A.; Franco, C.; Campos-Castello, J.; González-Hidalgo, M.; Oliete, F.; García-Nart, M. Rett’s Syndrome: A Neurophysiological Study. Neurophysiol. Clin. Clin. Neurophysiol. 1990, 20, 35–42. [Google Scholar] [CrossRef]
- Ishizaki, A.; Inoue, Y.; Sasaki, H.; Fukuyama, Y. Longitudinal Observation of Electroencephalograms in the Rett Syndrome. Brain Dev. 1989, 11, 407–412. [Google Scholar] [CrossRef]
- Robb, S.A.; Harden, A.; Boyd, S.G. Rett Syndrome: An EEG Study in 52 Girls. Neuropediatrics 1989, 20, 192–195. [Google Scholar] [CrossRef]
- Robertson, R.; Langill, L.; Wong, P.K.; Ho, H.H. Rett Syndrome: EEG Presentation. Electroencephalogr. Clin. Neurophysiol. 1988, 70, 388–395. [Google Scholar] [CrossRef]
- Percy, A.K.; Zoghbi, H.Y.; Glaze, D.G. Rett Syndrome: Discrimination of Typical and Variant Forms. Brain Dev. 1987, 9, 458–461. [Google Scholar] [CrossRef]
- Trauner, D.A.; Haas, R.H. Electroencephalographic Abnormalities in Rett Syndrome. Pediatr. Neurol. 1987, 3, 331–334. [Google Scholar] [CrossRef]
- Verma, N.P.; Chheda, R.L.; Nigro, M.A.; Hart, Z.H. Electroencephalographic Findings in Rett Syndrome. Electroencephalogr. Clin. Neurophysiol. 1986, 64, 394–401. [Google Scholar] [CrossRef]
- Rolando, S. Rett Syndrome: Report of Eight Cases. Brain Dev. 1985, 7, 290–296. [Google Scholar] [CrossRef]
- Brunel, R.; Gilly, R. A Case of the Rett Syndrome. Brain Dev. 1985, 7, 313–315. [Google Scholar] [CrossRef]
- Völkl-Kernstock, S.; Bauch-Prater, S.; Ponocny-Seliger, E.; Feucht, M. Speech and School Performance in Children with Benign Partial Epilepsy with Centro-Temporal Spikes (BCECTS). Seizure 2009, 18, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Vannest, J.; Tenney, J.R.; Gelineau-Morel, R.; Maloney, T.; Glauser, T.A. Cognitive and Behavioral Outcomes in Benign Childhood Epilepsy with Centrotemporal Spikes. Epilepsy Behav. EB 2015, 45, 85–91. [Google Scholar] [CrossRef]
- Niedermeyer, E.; Rocca, U. The Diagnostic Significance of Sleep Electroencephalograms in Temporal Lobe Epilepsy. A Comparison of Scalp and Depth Tracings. Eur. Neurol. 1972, 7, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Niedermeyer, E. Niedermeyer Frontal Lobe Disinhibition, Rett Syndrome and Attention Deficit Hyperactivity Disorder. Clin. Electroencephalogr. 2001, 32, 20–23. Available online: https://www.ncbi.nlm.nih.gov/pubmed/11202137 (accessed on 14 March 2017). [CrossRef]
- Jiang, Y.; Song, L.; Li, X.; Zhang, Y.; Chen, Y.; Jiang, S.; Hou, C.; Yao, D.; Wang, X.; Luo, C. Dysfunctional White-Matter Networks in Medicated and Unmedicated Benign Epilepsy with Centrotemporal Spikes. Hum. Brain Mapp. 2019, 40, 3113–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Yan, X.; Gao, Z.; Xu, K.; Zhou, X.; Chen, Q. Common and Distinctive Patterns of Cognitive Dysfunction in Children With Benign Epilepsy Syndromes. Pediatr. Neurol. 2017, 72, 36–41.e1. [Google Scholar] [CrossRef]
- Sceniak, M.P.; Lang, M.; Enomoto, A.C.; James Howell, C.; Hermes, D.J.; Katz, D.M. Mechanisms of Functional Hypoconnectivity in the Medial Prefrontal Cortex of Mecp2 Null Mice. Cereb. Cortex 2016, 26, 1938–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Peterson, M.; Beyer, B.; Frankel, W.N.; Zhang, Z.-W. Loss of MeCP2 From Forebrain Excitatory Neurons Leads to Cortical Hyperexcitation and Seizures. J. Neurosci. 2014, 34, 2754–2763. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, R.; Bonanni, P.; Parmeggiani, L.; Santucci, M.; Parmeggiani, A.; Sartucci, F. Cortical Reflex Myoclonus in Rett Syndrome. Ann. Neurol. 1998, 43, 472–479. [Google Scholar] [CrossRef]
- Yamanouchi, H.; Kaga, M.; Arima, M. Abnormal Cortical Excitability in Rett Syndrome. Pediatr. Neurol. 1993, 9, 202–206. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Kaga, M.; Suzuki, H.; Sakuragawa, N.; Arima, M. Giant Somatosensory Evoked Potentials in the Rett Syndrome. Brain Dev. 1991, 13, 36–39. [Google Scholar] [CrossRef]
- Steffenburg, U.; Hagberg, G.; Hagberg, B. Epilepsy in a Representative Series of Rett Syndrome. Acta Paediatr. 2001, 90, 34–39. [Google Scholar] [CrossRef]
- Nissenkorn, A.; Gak, E.; Vecsler, M.; Reznik, H.; Menascu, S.; Zeev, B.B. Epilepsy in Rett Syndrome—The Experience of a National Rett Center. Epilepsia 2010, 51, 1252–1258. [Google Scholar] [CrossRef]
- Jian, L.; Nagarajan, L.; de Klerk, N.; Ravine, D.; Christodoulou, J.; Leonard, H. Seizures in Rett Syndrome: An Overview from a One-Year Calendar Study. Eur. J. Paediatr. Neurol. 2007, 11, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, M.W.; Breck, H.; von Tetzchner, S.; Paus, B.; Skjeldal, O.H.; Brodtkorb, E. Epilepsy in Classic Rett Syndrome: Course and Characteristics in Adult Age. Epilepsy Res. 2018, 145, 134–139. [Google Scholar] [CrossRef] [Green Version]
- Shahbazian, M.D.; Young, J.I.; Yuva-Paylor, L.A.; Spencer, C.M.; Antalffy, B.A.; Noebels, J.L.; Armstrong, D.L.; Paylor, R.; Zoghbi, H.Y. Mice with Truncated MeCP2 Recapitulate Many Rett Syndrome Features and Display Hyperacetylation of Histone H3. Neuron 2002, 35, 243–254. [Google Scholar] [CrossRef] [Green Version]
- D’Cruz, J.A.; Wu, C.; Zahid, T.; El-Hayek, Y.; Zhang, L.; Eubanks, J.H. Alterations of Cortical and Hippocampal EEG Activity in MeCP2-Deficient Mice. Neurobiol. Dis. 2010, 38, 8–16. [Google Scholar] [CrossRef]
- Zhang, L.; Wither, R.G.; Lang, M.; Wu, C.; Sidorova-Darmos, E.; Netchev, H.; Matolcsy, C.B.; Snead, O.C.; Eubanks, J.H. A Role for Diminished GABA Transporter Activity in the Cortical Discharge Phenotype of MeCP2-Deficient Mice. Neuropsychopharmacology 2016, 41, 1467–1476. [Google Scholar] [CrossRef] [Green Version]
- Goffin, D.; Brodkin, E.S.; Blendy, J.A.; Siegel, S.J.; Zhou, Z. Cellular Origins of Auditory Event-Related Potential Deficits in Rett Syndrome. Nat. Neurosci. 2014, 17, 804–806. [Google Scholar] [CrossRef] [Green Version]
- Ito-Ishida, A.; Ure, K.; Chen, H.; Swann, J.W.; Zoghbi, H.Y. Loss of MeCP2 in Parvalbumin-and Somatostatin-Expressing Neurons in Mice Leads to Distinct Rett Syndrome-like Phenotypes. Neuron 2015, 88, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Lang, M.; Wither, R.G.; Colic, S.; Wu, C.; Monnier, P.P.; Bardakjian, B.L.; Zhang, L.; Eubanks, J.H. Rescue of Behavioral and EEG Deficits in Male and Female Mecp2-Deficient Mice by Delayed Mecp2 Gene Reactivation. Hum. Mol. Genet. 2014, 23, 303–318. [Google Scholar] [CrossRef] [Green Version]
- Wither, R.G.; Colic, S.; Wu, C.; Bardakjian, B.L.; Zhang, L.; Eubanks, J.H. Daily Rhythmic Behaviors and Thermoregulatory Patterns Are Disrupted in Adult Female MeCP2-Deficient Mice. PLoS ONE 2012, 7, e35396. [Google Scholar] [CrossRef]
- Wither, R.G.; Colic, S.; Bardakjian, B.L.; Snead, O.C.; Zhang, L.; Eubanks, J.H. Electrographic and Pharmacological Characterization of a Progressive Epilepsy Phenotype in Female MeCP2-Deficient Mice. Epilepsy Res. 2018, 140, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Letts, V.A.; Beyer, B.J.; Frankel, W.N. Hidden in Plain Sight: Spike-Wave Discharges in Mouse Inbred Strains. Genes Brain Behav. 2014, 13, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Sitnikova, E. Neonatal Sensory Deprivation Promotes Development of Absence Seizures in Adult Rats with Genetic Predisposition to Epilepsy. Brain Res. 2011, 1377, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wang, W.; Lu, H.; He, L.; Chen, W.; Chao, E.S.; Fiorotto, M.L.; Tang, B.; Herrera, J.A.; Seymour, M.L.; et al. Manipulations of MeCP2 in Glutamatergic Neurons Highlight Their Contributions to Rett and Other Neurological Disorders. eLife 2016, 5, e14199. [Google Scholar] [CrossRef]
- Johnston, M.V.; Ammanuel, S.; O’Driscoll, C.; Wozniak, A.; Naidu, S.; Kadam, S.D. Twenty-Four Hour Quantitative-EEG and in-Vivo Glutamate Biosensor Detects Activity and Circadian Rhythm Dependent Biomarkers of Pathogenesis in Mecp2 Null Mice. Front. Syst. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Lang, M.; Wither, R.G.; Brotchie, J.M.; Wu, C.; Zhang, L.; Eubanks, J.H. Selective Preservation of MeCP2 in Catecholaminergic Cells Is Sufficient to Improve the Behavioral Phenotype of Male and Female Mecp2-Deficient Mice. Hum. Mol. Genet. 2013, 22, 358–371. [Google Scholar] [CrossRef] [Green Version]
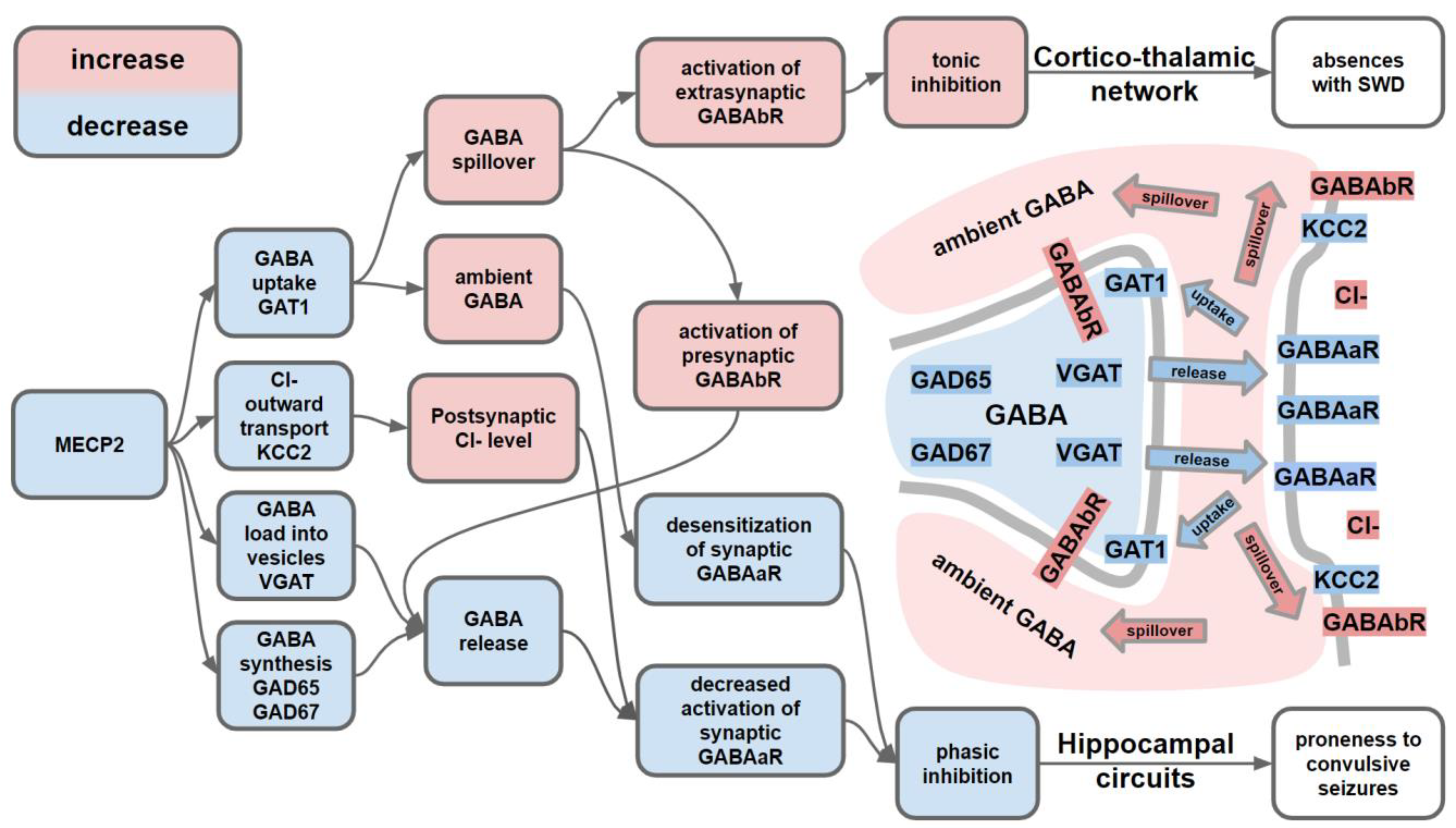
- Chao, H.-T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.-C.; Heintz, N.; et al. Dysfunction in GABA Signalling Mediates Autism-like Stereotypies and Rett Syndrome Phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef]
- Ure, K.; Lu, H.; Wang, W.; Ito-Ishida, A.; Wu, Z.; He, L.; Sztainberg, Y.; Chen, W.; Tang, J.; Zoghbi, H.Y. Restoration of Mecp2 Expression in GABAergic Neurons Is Sufficient to Rescue Multiple Disease Features in a Mouse Model of Rett Syndrome. eLife 2016, 5, e14198. [Google Scholar] [CrossRef]
- McLeod, F.; Ganley, R.; Williams, L.; Selfridge, J.; Bird, A.; Cobb, S.R. Reduced Seizure Threshold and Altered Network Oscillatory Properties in a Mouse Model of Rett Syndrome. Neuroscience 2013, 231, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Cui, N.; Zhong, W.; Jin, X.-T.; Jiang, C. GABAergic Synaptic Inputs of Locus Coeruleus Neurons in Wild-Type and Mecp2 -Null Mice. Am. J. Physiol.-Cell Physiol. 2013, 304, C844–C857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Vergnes, M.; Depaulis, A.; Marescaux, C. Involvement of Intrathalamic GABAb Neurotransmission in the Control of Absence Seizures in the Rat. Neuroscience 1992, 48, 87–93. [Google Scholar] [CrossRef]
- Crunelli, V.; Lőrincz, M.L.; McCafferty, C.; Lambert, R.C.; Leresche, N.; Di Giovanni, G.; David, F. Clinical and Experimental Insight into Pathophysiology, Comorbidity and Therapy of Absence Seizures. Brain 2020, 143, 2341–2368. [Google Scholar] [CrossRef]
- Destexhe, A. Spike-and-Wave Oscillations Based on the Properties of GABAB Receptors. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 9099–9111. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, N.; Miya, F.; Tsunoda, T.; Kato, M.; Saitoh, S.; Yamasaki, M.; Shimizu, A.; Torii, C.; Kanemura, Y.; Kosaki, K. Targeted Next-Generation Sequencing in the Diagnosis of Neurodevelopmental Disorders. Clin. Genet. 2015, 88, 288–292. [Google Scholar] [CrossRef]
- Dong, Q.; Kim, J.; Nguyen, L.; Bu, Q.; Chang, Q. An Astrocytic Influence on Impaired Tonic Inhibition in Hippocampal CA1 Pyramidal Neurons in a Mouse Model of Rett Syndrome. J. Neurosci. 2020, 40, 6250–6261. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl- Co-Transporter KCC2 Renders GABA Hyperpolarizing during Neuronal Maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Rikhye, R.V.; Breton-Provencher, V.; Tang, X.; Li, C.; Li, K.; Runyan, C.A.; Fu, Z.; Jaenisch, R.; Sur, M. Jointly Reduced Inhibition and Excitation Underlies Circuit-Wide Changes in Cortical Processing in Rett Syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E7287–E7296. [Google Scholar] [CrossRef] [Green Version]
- Duarte, S.T.; Armstrong, J.; Roche, A.; Ortez, C.; Pérez, A.; Maria del Mar, O.C.; Pereira, A.; Sanmartí, F.; Ormazábal, A.; Artuch, R.; et al. Abnormal Expression of Cerebrospinal Fluid Cation Chloride Cotransporters in Patients with Rett Syndrome. PLoS ONE 2013, 8, e68851. [Google Scholar] [CrossRef] [PubMed]
- Hinz, L.; Torrella Barrufet, J.; Heine, V.M. KCC2 Expression Levels Are Reduced in Post Mortem Brain Tissue of Rett Syndrome Patients. Acta Neuropathol. Commun. 2019, 7, 196. [Google Scholar] [CrossRef]
- Wright, R.; Newey, S.E.; Ilie, A.; Wefelmeyer, W.; Raimondo, J.V.; Ginham, R.; Mcllhinney, R.A.J.; Akerman, C.J. Neuronal Chloride Regulation via KCC2 Is Modulated through a GABAB Receptor Protein Complex. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 5447–5462. [Google Scholar] [CrossRef] [Green Version]
- Lozovaya, N.; Nardou, R.; Tyzio, R.; Chiesa, M.; Pons-Bennaceur, A.; Eftekhari, S.; Bui, T.-T.; Billon-Grand, M.; Rasero, J.; Bonifazi, P.; et al. Early Alterations in a Mouse Model of Rett Syndrome: The GABA Developmental Shift Is Abolished at Birth. Sci. Rep. 2019, 9, 9276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Drotar, J.; Li, K.; Clairmont, C.D.; Brumm, A.S.; Sullins, A.J.; Wu, H.; Liu, X.S.; Wang, J.; Gray, N.S.; et al. Pharmacological Enhancement of KCC2 Gene Expression Exerts Therapeutic Effects on Human Rett Syndrome Neurons and Mecp2 Mutant Mice. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, S.-X.; Sun, P.; He, H.-Y.; Yang, C.-H.; Chen, X.-J.; Shen, C.-J.; Wang, X.-D.; Chen, Z.; Berg, D.K.; et al. Loss of MeCP2 in Cholinergic Neurons Causes Part of RTT-like Phenotypes via A7 Receptor in Hippocampus. Cell Res. 2016, 26, 728–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaze, D.G. Neurophysiology of Rett Syndrome. J. Child Neurol. 2005, 20, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Gorbachevskaya, N.; Bashina, V.; Gratchev, V.; Iznak, A. Cerebrolysin Therapy in Rett Syndrome: Clinical and EEG Mapping Study. Brain Dev. 2001, 23, S90–S93. [Google Scholar] [CrossRef]
- Gratchev, V.V.; Bashina, V.M.; Klushnik, T.P.; Ulas, V.U.; Gorbachevskaya, N.L.; Vorsanova, S.G. Clinical, Neurophysiological and Immunological Correlations in Classical Rett Syndrome. Brain Dev. 2001, 23, S108–S112. [Google Scholar] [CrossRef]
- Roche, K.J.; LeBlanc, J.J.; Levin, A.R.; O’Leary, H.M.; Baczewski, L.M.; Nelson, C.A. Electroencephalographic Spectral Power as a Marker of Cortical Function and Disease Severity in Girls with Rett Syndrome. J. Neurodev. Disord. 2019, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Keogh, C.; Pini, G.; Dyer, A.H.; Bigoni, S.; DiMarco, P.; Gemo, I.; Reilly, R.; Tropea, D. Clinical and Genetic Rett Syndrome Variants Are Defined by Stable Electrophysiological Profiles. Bmc Pediatr. 2018, 18, 333. [Google Scholar] [CrossRef]
- Ammanuel, S.; Chan, W.C.; Adler, D.A.; Lakshamanan, B.M.; Gupta, S.S.; Ewen, J.B.; Johnston, M.V.; Marcus, C.L.; Naidu, S.; Kadam, S.D. Heightened Delta Power during Slow-Wave-Sleep in Patients with Rett Syndrome Associated with Poor Sleep Efficiency. PLoS ONE 2015, 10, e0138113. [Google Scholar] [CrossRef] [Green Version]
- Faienza, C.; Capone, C.; Sani, E.; Villani, D.; Prati, G. EEG Mapping in a Child with Rett Syndrome. Psychiatry Res. 1989, 29, 425–426. [Google Scholar] [CrossRef]
- Bader, G.G.; Witt-Engerström, I.; Hagberg, B. Neurophysiological Findings in the Rett Syndrome, II: Visual and Auditory Brainstem, Middle and Late Evoked Responses. Brain Dev. 1989, 11, 110–114. [Google Scholar] [CrossRef]
- Akshoomoff, N. Use of the Mullen Scales of Early Learning for the Assessment of Young Children with Autism Spectrum Disorders. Child Neuropsychol. J. Norm. Abnorm. Dev. Child. Adolesc. 2006, 12, 269–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, A.R.; Barry, R.J.; Johnstone, S.J.; McCarthy, R.; Selikowitz, M. EEG Development in Attention Deficit Hyperactivity Disorder: From Child to Adult. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2019, 130, 1256–1262. [Google Scholar] [CrossRef]
- Perera, M.P.N.; Bailey, N.W.; Herring, S.E.; Fitzgerald, P.B. Electrophysiology of Obsessive Compulsive Disorder: A Systematic Review of the Electroencephalographic Literature. J. Anxiety Disord. 2019, 62, 1–14. [Google Scholar] [CrossRef]
- Benz, N.; Hatz, F.; Bousleiman, H.; Ehrensperger, M.M.; Gschwandtner, U.; Hardmeier, M.; Ruegg, S.; Schindler, C.; Zimmermann, R.; Monsch, A.U.; et al. Slowing of EEG Background Activity in Parkinson’s and Alzheimer’s Disease with Early Cognitive Dysfunction. Front. Aging Neurosci. 2014, 6, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voytek, B.; Kramer, M.A.; Case, J.; Lepage, K.Q.; Tempesta, Z.R.; Knight, R.T.; Gazzaley, A. Age-Related Changes in 1/f Neural Electrophysiological Noise. J. Neurosci. 2015, 35, 13257–13265. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.M.; Furlong, S.; Voytek, B.; Donoghue, T.; Boettiger, C.A.; Sheridan, M.A. EEG Power Spectral Slope Differs by ADHD Status and Stimulant Medication Exposure in Early Childhood. J. Neurophysiol. 2019, 122, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Bartos, M.; Vida, I.; Jonas, P. Synaptic Mechanisms of Synchronized Gamma Oscillations in Inhibitory Interneuron Networks. Nat. Rev. Neurosci. 2007, 8, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Vinck, M.; Womelsdorf, T.; Buffalo, E.A.; Desimone, R.; Fries, P. Attentional Modulation of Cell-Class-Specific Gamma-Band Synchronization in Awake Monkey Area V4. Neuron 2013, 80, 1077–1089. [Google Scholar] [CrossRef] [Green Version]
- Hoogenboom, N.; Schoffelen, J.-M.; Oostenveld, R.; Parkes, L.M.; Fries, P. Localizing Human Visual Gamma-Band Activity in Frequency, Time and Space. NeuroImage 2006, 29, 764–773. [Google Scholar] [CrossRef]
- Le Van Quyen, M.; Staba, R.; Bragin, A.; Dickson, C.; Valderrama, M.; Fried, I.; Engel, J. Large-Scale Microelectrode Recordings of High-Frequency Gamma Oscillations in Human Cortex during Sleep. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 7770–7782. [Google Scholar] [CrossRef]
- Zhong, W.; Ciatipis, M.; Wolfenstetter, T.; Jessberger, J.; Müller, C.; Ponsel, S.; Yanovsky, Y.; Brankačk, J.; Tort, A.B.L.; Draguhn, A. Selective Entrainment of Gamma Subbands by Different Slow Network Oscillations. Proc. Natl. Acad. Sci. USA 2017, 114, 4519–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzsáki, G.; Wang, X.-J. Mechanisms of Gamma Oscillations. Annu. Rev. Neurosci. 2012, 35, 203–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colgin, L.L. Do Slow and Fast Gamma Rhythms Correspond to Distinct Functional States in the Hippocampal Network? Brain Res. 2015, 1621, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuval-Greenberg, S.; Tomer, O.; Keren, A.S.; Nelken, I.; Deouell, L.Y. Transient Induced Gamma-Band Response in EEG as a Manifestation of Miniature Saccades. Neuron 2008, 58, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Orekhova, E.V.; Stroganova, T.A.; Nygren, G.; Tsetlin, M.M.; Posikera, I.N.; Gillberg, C.; Elam, M. Excess of High Frequency Electroencephalogram Oscillations in Boys with Autism. Biol. Psychiatry 2007, 62, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Singer, W. Neuronal Dynamics and Neuropsychiatric Disorders: Toward a Translational Paradigm for Dysfunctional Large-Scale Networks. Neuron 2012, 75, 963–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zijlmans, M.; Jiruska, P.; Zelmann, R.; Leijten, F.S.S.; Jefferys, J.G.R.; Gotman, J. High-Frequency Oscillations as a New Biomarker in Epilepsy. Ann. Neurol. 2012, 71, 169–178. [Google Scholar] [CrossRef]
- Verma, N.P.; Nigro, M.A.; Hart, Z.H. Rett Syndrome—A Gray Matter Disease? Electrophysiologic Evidence. Electroencephalogr. Clin. Neurophysiol. 1987, 67, 327–329. [Google Scholar] [CrossRef]
- Glaze, D.G.; Frost, J.D.; Zoghbi, H.Y.; Percy, A.K. Rett’s Syndrome: Characterization of Respiratory Patterns and Sleep. Ann. Neurol. 1987, 21, 377–382. [Google Scholar] [CrossRef]
- Andrade-Valença, L.; Mari, F.; Jacobs, J.; Zijlmans, M.; Olivier, A.; Gotman, J.; Dubeau, F. Interictal High Frequency Oscillations (HFOs) in Patients with Focal Epilepsy and Normal MRI. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2012, 123, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traub, R.D. Fast Oscillations and Epilepsy. Epilepsy Curr. 2003, 3, 77–79. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Yanagita, T.; Kobayashi, H.; Yokoo, H.; Wada, A. Up-Regulation of Sodium Channel Subunit MRNAs and Their Cell Surface Expression by Antiepileptic Valproic Acid: Activation of Calcium Channel and Catecholamine Secretion in Adrenal Chromaffin Cells. J. Neurochem. 1997, 68, 1655–1662. [Google Scholar] [CrossRef]
- Dzirasa, K.; Ramsey, A.J.; Takahashi, D.Y.; Stapleton, J.; Potes, J.M.; Williams, J.K.; Gainetdinov, R.R.; Sameshima, K.; Caron, M.G.; Nicolelis, M.A.L. Hyperdopaminergia and NMDA Receptor Hypofunction Disrupt Neural Phase Signaling. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 8215–8224. [Google Scholar] [CrossRef] [Green Version]
- Bragin, A.; Jandó, G.; Nádasdy, Z.; Hetke, J.; Wise, K.; Buzsáki, G. Gamma (40–100 Hz) Oscillation in the Hippocampus of the Behaving Rat. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 47–60. [Google Scholar] [CrossRef]
- Nomura, Y.; Segawa, M.; Hasegawa, M. Rett Syndrome—Clinical Studies and Pathophysiological Consideration. Brain Dev. 1984, 6, 475–486. [Google Scholar] [CrossRef]
- Nomura, Y.; Honda, K.; Segawa, M. Pathophysiology of Rett Syndrome. Brain Dev. 1987, 9, 506–513. [Google Scholar] [CrossRef]
- Wong, K.; Leonard, H.; Jacoby, P.; Ellaway, C.; Downs, J. The Trajectories of Sleep Disturbances in Rett Syndrome. J. Sleep Res. 2015, 24, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boban, S.; Wong, K.; Epstein, A.; Anderson, B.; Murphy, N.; Downs, J.; Leonard, H. Determinants of Sleep Disturbances in Rett Syndrome: Novel Findings in Relation to Genotype. Am. J. Med. Genet. A 2016, 170, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Piazza, C.C.; Fisher, W.; Kiesewetter, K.; Bowman, L.; Moser, H. Aberrant Sleep Patterns in Children with the Rett Syndrome. Brain Dev. 1990, 12, 488–493. [Google Scholar] [CrossRef]
- Kales, A.; Rechtschaffen, A.; University of California, L.A.; Brain Information Service; National Institute of Neurological Diseases and Blindness (U.S.). A Manual of Standardized Terminology, Techniques and Scoring System for Sleep Stages of Human Subjects; United States Government Printing Office: Washington, DC, USA, 1968.
- Carotenuto, M.; Esposito, M.; D’Aniello, A.; Rippa, C.D.; Precenzano, F.; Pascotto, A.; Bravaccio, C.; Elia, M. Polysomnographic Findings in Rett Syndrome: A Case-Control Study. Sleep Breath. Schlaf Atm. 2013, 17, 93–98. [Google Scholar] [CrossRef]
- Dreyfus-Brisac, C.; Monod, N.; Radvanyi, M.F.; Curzi, L. Interest of Electrophysiological Studies in Human Development. Physiol. Biochem. Basis Perinat. Med. 1981, 337–346. [Google Scholar] [CrossRef]
- Achermann, P.; Dijk, D.J.; Brunner, D.P.; Borbély, A.A. A Model of Human Sleep Homeostasis Based on EEG Slow-Wave Activity: Quantitative Comparison of Data and Simulations. Brain Res. Bull. 1993, 31, 97–113. [Google Scholar] [CrossRef]
- Alhola, P.; Polo-Kantola, P. Sleep Deprivation: Impact on Cognitive Performance. Neuropsychiatr. Dis. Treat. 2007, 3, 553–567. [Google Scholar]
- Ednick, M.; Cohen, A.P.; McPhail, G.L.; Beebe, D.; Simakajornboon, N.; Amin, R.S. A Review of the Effects of Sleep during the First Year of Life on Cognitive, Psychomotor, and Temperament Development. Sleep 2009, 32, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Tham, E.K.; Schneider, N.; Broekman, B.F. Infant Sleep and Its Relation with Cognition and Growth: A Narrative Review. Nat. Sci. Sleep 2017, 9, 135–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Saavedra, M.; Antoun, G.; Yanagiya, A.; Oliva-Hernandez, R.; Cornejo-Palma, D.; Perez-Iratxeta, C.; Sonenberg, N.; Cheng, H.-Y.M. MiRNA-132 Orchestrates Chromatin Remodeling and Translational Control of the Circadian Clock. Hum. Mol. Genet. 2011, 20, 731–751. [Google Scholar] [CrossRef] [Green Version]
- Vosko, A.; van Diepen, H.C.; Kuljis, D.; Chiu, A.M.; Heyer, D.; Terra, H.; Carpenter, E.; Michel, S.; Meijer, J.H.; Colwell, C.S. Role of Vasoactive Intestinal Peptide in the Light Input to the Circadian System. Eur. J. Neurosci. 2015, 42, 1839–1848. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.R.; Simon, T.; Lones, L.; Herzog, E.D. SCN VIP Neurons Are Essential for Normal Light-Mediated Resetting of the Circadian System. J. Neurosci. 2018, 38, 7986–7995. [Google Scholar] [CrossRef]
- Li, Q.; Loh, D.H.; Kudo, T.; Truong, D.; Derakhshesh, M.; Kaswan, Z.M.; Ghiani, C.A.; Tsoa, R.; Cheng, Y.; Sun, Y.E.; et al. Circadian Rhythm Disruption in a Mouse Model of Rett Syndrome Circadian Disruption in RTT. Neurobiol. Dis. 2015, 77, 155–164. [Google Scholar] [CrossRef]
- Bebbington, A.; Anderson, A.; Ravine, D.; Fyfe, S.; Pineda, M.; de Klerk, N.; Ben-Zeev, B.; Yatawara, N.; Percy, A.; Kaufmann, W.E.; et al. Investigating Genotype-Phenotype Relationships in Rett Syndrome Using an International Data Set. Neurology 2008, 70, 868–875. [Google Scholar] [CrossRef]
- Bebbington, A.; Percy, A.; Christodoulou, J.; Ravine, D.; Ho, G.; Jacoby, P.; Anderson, A.; Pineda, M.; Ben Zeev, B.; Bahi-Buisson, N.; et al. Updating the Profile of C-Terminal MECP2 Deletions in Rett Syndrome. J. Med. Genet. 2010, 47, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebbington, A.; Downs, J.; Percy, A.; Pineda, M.; Zeev, B.B.; Bahi-Buisson, N.; Leonard, H. The Phenotype Associated with a Large Deletion on MECP2. Eur. J. Hum. Genet. EJHG 2012, 20, 921–927. [Google Scholar] [CrossRef] [Green Version]
- Archer, H.L.; Whatley, S.D.; Evans, J.C.; Ravine, D.; Huppke, P.; Kerr, A.; Bunyan, D.; Kerr, B.; Sweeney, E.; Davies, S.J.; et al. Gross Rearrangements of the MECP2 Gene Are Found in Both Classical and Atypical Rett Syndrome Patients. J. Med. Genet. 2006, 43, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Jian, L.; Archer, H.; Ravine, D.; Kerr, A.; Klerk, N.; Christodoulou, J.; Bailey, M.; Laurvick, C.; Leonard, H.P. R270X MECP2 Mutation and Mortality in Rett Syndrome. Eur. J. Hum. Genet. EJHG 2005, 13, 1235–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frullanti, E.; Papa, F.T.; Grillo, E.; Clarke, A.; Ben-Zeev, B.; Pineda, M.; Bahi-Buisson, N.; Bienvenu, T.; Armstrong, J.; Roche Martinez, A.; et al. Analysis of the Phenotypes in the Rett Networked Database. Int. J. Genom. 2019, 2019, 6956934. [Google Scholar] [CrossRef]
- Leonard, H.; Cobb, S.; Downs, J. Clinical and Biological Progress over 50 Years in Rett Syndrome. Nat. Rev. Neurol. 2017, 13, 37–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanowicz, A.; Downs, J.; Girdler, S.; Ciccone, N.; Leonard, H. Aspects of Speech-Language Abilities Are Influenced by MECP2 Mutation Type in Girls with Rett Syndrome. Am. J. Med. Genet. A 2015, 167, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-Binding Protein 2 (MECP2) Mutation Type Is Associated with Disease Severity in Rett Syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Nissenkorn, A.; Levy-Drummer, R.S.; Bondi, O.; Renieri, A.; Villard, L.; Mari, F.; Mencarelli, M.A.; Lo Rizzo, C.; Meloni, I.; Pineda, M.; et al. Epilepsy in Rett Syndrome-Lessons from the Rett Networked Database. Epilepsia 2015, 56, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 Duplication Syndrome. Am. J. Med. Genet. A 2010, 152A, 1079–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Esch, H. MECP2 Duplication Syndrome. Mol. Syndromol. 2012, 2, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friez, M.J.; Jones, J.R.; Clarkson, K.; Lubs, H.; Abuelo, D.; Bier, J.-A.B.; Pai, S.; Simensen, R.; Williams, C.; Giampietro, P.F.; et al. Recurrent Infections, Hypotonia, and Mental Retardation Caused by Duplication of MECP2 and Adjacent Region in Xq28. Pediatrics 2006, 118, e1687–e1695. [Google Scholar] [CrossRef] [Green Version]
- Caumes, R.; Boespflug-Tanguy, O.; Villeneuve, N.; Lambert, L.; Delanoe, C.; Leheup, B.; Bahi-Buisson, N.; Auvin, S. Late Onset Epileptic Spasms Is Frequent in MECP2 Gene Duplication: Electroclinical Features and Long-Term Follow-up of 8 Epilepsy Patients. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2014, 18, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Rajaprakash, M.; Richer, J.; Sell, E. Valproic Acid as a Monotherapy in Drug-Resistant Methyl-CpG-Binding Protein 2 Gene (MECP2) Duplication-Related Epilepsy. Epilepsy Behav. Case Rep. 2018, 10, 133–136. [Google Scholar] [CrossRef]
- Marafi, D.; Suter, B.; Schultz, R.; Glaze, D.; Pavlik, V.N.; Goldman, A.M. Spectrum and Time Course of Epilepsy and the Associated Cognitive Decline in MECP2 Duplication Syndrome. Neurology 2019, 92, e108–e114. [Google Scholar] [CrossRef]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, J.H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-like 5 (CDKL5) Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef]
- Demarest, S.; Pestana-Knight, E.M.; Olson, H.E.; Downs, J.; Marsh, E.D.; Kaufmann, W.E.; Partridge, C.-A.; Leonard, H.; Gwadry-Sridhar, F.; Frame, K.E.; et al. Severity Assessment in CDKL5 Deficiency Disorder. Pediatr. Neurol. 2019, 97, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Vegas, N.; Cavallin, M.; Maillard, C.; Boddaert, N.; Toulouse, J.; Schaefer, E.; Lerman-Sagie, T.; Lev, D.; Magalie, B.; Moutton, S.; et al. Delineating FOXG1 Syndrome: From Congenital Microcephaly to Hyperkinetic Encephalopathy. Neurol. Genet. 2018, 4, e281. [Google Scholar] [CrossRef] [Green Version]
- Mitter, D.; Pringsheim, M.; Kaulisch, M.; Plümacher, K.S.; Schröder, S.; Warthemann, R.; Abou Jamra, R.; Baethmann, M.; Bast, T.; Büttel, H.-M.; et al. FOXG1 Syndrome: Genotype-Phenotype Association in 83 Patients with FOXG1 Variants. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.-T.J.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 Disrupts Kinome Profile and Event-Related Potentials Leading to Autistic-like Phenotypes in Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, M.S.; Eubanks, J.H. Seizures in Mouse Models of Rare Neurodevelopmental Disorders. Neuroscience 2020, 445, 50–68. [Google Scholar] [CrossRef]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.L.; Farley, D.; et al. Mapping Pathological Phenotypes in a Mouse Model of CDKL5 Disorder. PLoS ONE 2014, 9, e91613. [Google Scholar] [CrossRef]
- Okuda, K.; Kobayashi, S.; Fukaya, M.; Watanabe, A.; Murakami, T.; Hagiwara, M.; Sato, T.; Ueno, H.; Ogonuki, N.; Komano-Inoue, S.; et al. CDKL5 Controls Postsynaptic Localization of GluN2B-Containing NMDA Receptors in the Hippocampus and Regulates Seizure Susceptibility. Neurobiol. Dis. 2017, 106, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Olimpico, F.; Pancrazi, L.; Borello, U.; Cattaneo, A.; Caleo, M.; Costa, M.; Mainardi, M. Cortical Seizures in FoxG1+/− Mice Are Accompanied by Akt/S6 Overactivation, Excitation/Inhibition Imbalance and Impaired Synaptic Transmission. Int. J. Mol. Sci. 2019, 20, 4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.L.; Levenson, J.M.; Vilaythong, A.P.; Richman, R.; Armstrong, D.L.; Noebels, J.L.; David Sweatt, J.; Zoghbi, H.Y. Mild Overexpression of MeCP2 Causes a Progressive Neurological Disorder in Mice. Hum. Mol. Genet. 2004, 13, 2679–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Li, X.; Zhang, J.-T.; Cai, Y.-J.; Cheng, T.-L.; Cheng, C.; Wang, Y.; Zhang, C.-C.; Nie, Y.-H.; Chen, Z.-F.; et al. Autism-like Behaviours and Germline Transmission in Transgenic Monkeys Overexpressing MeCP2. Nature 2016, 530, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Sztainberg, Y.; Chen, H.; Swann, J.W.; Hao, S.; Tang, B.; Wu, Z.; Tang, J.; Wan, Y.-W.; Liu, Z.; Rigo, F.; et al. Reversal of Phenotypes in MECP2 Duplication Mice Using Genetic Rescue or Antisense Oligonucleotides. Nature 2015. [Google Scholar] [CrossRef]
- Tigani, W.; Rossi, M.P.; Artimagnella, O.; Santo, M.; Rauti, R.; Sorbo, T.; Ulloa Severino, F.P.; Provenzano, G.; Allegra, M.; Caleo, M.; et al. Foxg1 Upregulation Enhances Neocortical Activity. Cereb. Cortex 2020, 30, 5147–5165. [Google Scholar] [CrossRef]
- Ewen, J.B.; Sweeney, J.A.; Potter, W.Z. Conceptual, Regulatory and Strategic Imperatives in the Early Days of EEG-Based Biomarker Validation for Neurodevelopmental Disabilities. Front. Integr. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Specchio, N.; Balestri, M.; Striano, P.; Cilio, M.R.; Nardello, R.; Patanè, S.; Margiotta, M.L.; D’Orsi, G.; Striano, S.; Russo, S.; et al. Efficacy of Levetiracetam in the Treatment of Drug-Resistant Rett Syndrome. Epilepsy Res. 2010, 88, 112–117. [Google Scholar] [CrossRef]
- O’Leary, H.M.; Kaufmann, W.E.; Barnes, K.V.; Rakesh, K.; Kapur, K.; Tarquinio, D.C.; Cantwell, N.G.; Roche, K.J.; Rose, S.A.; Walco, A.C.; et al. Placebo-Controlled Crossover Assessment of Mecasermin for the Treatment of Rett Syndrome. Ann. Clin. Transl. Neurol. 2018, 5, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Pini, G.; Congiu, L.; Benincasa, A.; DiMarco, P.; Bigoni, S.; Dyer, A.H.; Mortimer, N.; Della-Chiesa, A.; O’Leary, S.; McNamara, R.; et al. Illness Severity, Social and Cognitive Ability, and EEG Analysis of Ten Patients with Rett Syndrome Treated with Mecasermin (Recombinant Human IGF-1). Autism Res. Treat. 2016, 2016, 5073078. [Google Scholar] [CrossRef] [Green Version]
- Khwaja, O.S.; Ho, E.; Barnes, K.V.; O’Leary, H.M.; Pereira, L.M.; Finkelstein, Y.; Nelson, C.A.; Vogel-Farley, V.; DeGregorio, G.; Holm, I.A.; et al. Safety, Pharmacokinetics, and Preliminary Assessment of Efficacy of Mecasermin (Recombinant Human IGF-1) for the Treatment of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 4596–4601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Hicks, C.L.; Gupta, S.; Ewen, J.B.; Hong, M.; Kratz, L.; Kelley, R.; Tierney, E.; Vaurio, R.; Bibat, G.; Sanyal, A.; et al. Randomized Open-Label Trial of Dextromethorphan in Rett Syndrome. Neurology 2017, 89, 1684–1690. [Google Scholar] [CrossRef] [PubMed]
- Djukic, A.; Holtzer, R.; Shinnar, S.; Muzumdar, H.; Rose, S.A.; Mowrey, W.; Galanopoulou, A.S.; Shinnar, R.; Jankowski, J.J.; Feldman, J.F.; et al. Pharmacologic Treatment of Rett Syndrome With Glatiramer Acetate. Pediatr. Neurol. 2016, 61, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Fabio, R.A.; Billeci, L.; Crifaci, G.; Troise, E.; Tortorella, G.; Pioggia, G. Cognitive Training Modifies Frequency EEG Bands and Neuropsychological Measures in Rett Syndrome. Res. Dev. Disabil. 2016, 53–54, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Fabio, R.A.; Gangemi, A.; Semino, M.; Vignoli, A.; Canevini, M.P.; Priori, A.; Rosa, G.D.; Caprì, T. Effects of Combined Transcranial Direct Current Stimulation with Cognitive Training in Girls with Rett Syndrome. Brain Sci. 2020, 10, 276. [Google Scholar] [CrossRef]
- Fabio, R.A.; Gangemi, A.; Capri, T.; Budden, S.; Falzone, A. Neurophysiological and Cognitive Effects of Transcranial Direct Current Stimulation in Three Girls with Rett Syndrome with Chronic Language Impairments. Res. Dev. Disabil. 2018, 76, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Mouro, F.M.; Miranda-Lourenço, C.; Sebastião, A.M.; Diógenes, M.J. From Cannabinoids and Neurosteroids to Statins and the Ketogenic Diet: New Therapeutic Avenues in Rett Syndrome? Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Haas, R.H.; Rice, M.A.; Trauner, D.A.; Merritt, T.A.; Opitz, J.M.; Reynolds, J.F. Therapeutic Effects of a Ketogenic Diet in Rett Syndrome. Am. J. Med. Genet. 1986, 25, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Liebhaber, G.M.; Riemann, E.; Baumeister, F.A.M. Ketogenic Diet in Rett Syndrome. J. Child Neurol. 2003, 18, 74–75. [Google Scholar] [CrossRef]
- Wang, J.-F.; Sun, X.; Chen, B.; Young, L.T. Lamotrigine Increases Gene Expression of GABA-A Receptor Β3 Subunit in Primary Cultured Rat Hippocampus Cells. Neuropsychopharmacology 2002, 26, 415–421. [Google Scholar] [CrossRef]
- Huppke, P.; Köhler, K.; Brockmann, K.; Stettner, G.M.; Gärtner, J. Treatment of Epilepsy in Rett Syndrome. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2007, 11, 10–16. [Google Scholar] [CrossRef]
- Henriques, J.B.; Davidson, R.J. Left Frontal Hypoactivation in Depression. J. Abnorm. Psychol. 1991, 100, 535–545. [Google Scholar] [CrossRef]
- Kagan, J.; Snidman, N. Early Childhood Predictors of Adult Anxiety Disorders. Biol. Psychiatry 1999, 46, 1536–1541. [Google Scholar] [CrossRef]
- Bruder, G.E.; Stewart, J.W.; McGrath, P.J. Right Brain, Left Brain in Depressive Disorders: Clinical and Theoretical Implications of Behavioral, Electrophysiological and Neuroimaging Findings. Neurosci. Biobehav. Rev. 2017, 78, 178–191. [Google Scholar] [CrossRef]
- Barnes, K.V.; Coughlin, F.R.; O’Leary, H.M.; Bruck, N.; Bazin, G.A.; Beinecke, E.B.; Walco, A.C.; Cantwell, N.G.; Kaufmann, W.E. Anxiety-like Behavior in Rett Syndrome: Characteristics and Assessment by Anxiety Scales. J. Neurodev. Disord. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- van der Vinne, N.; Vollebregt, M.A.; van Putten, M.J.A.M.; Arns, M. Frontal Alpha Asymmetry as a Diagnostic Marker in Depression: Fact or Fiction? A Meta-Analysis. NeuroImage Clin. 2017, 16, 79–87. [Google Scholar] [CrossRef]
- Ben-Zeev, B.; Aharoni, R.; Nissenkorn, A.; Arnon, R. Glatiramer Acetate (GA, Copolymer-1) an Hypothetical Treatment Option for Rett Syndrome. Med. Hypotheses 2011, 76, 190–193. [Google Scholar] [CrossRef]
- Doppler, E.; Rockenstein, E.; Ubhi, K.; Inglis, C.; Mante, M.; Adame, A.; Crews, L.; Hitzl, M.; Moessler, H.; Masliah, E. Neurotrophic Effects of Cerebrolysin in the Mecp2308/Y Transgenic Model of Rett Syndrome. Acta Neuropathol. (Berl.) 2008, 116, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blue, M.E.; Naidu, S.; Johnston, M.V. Altered Development of Glutamate and GABA Receptors in the Basal Ganglia of Girls with Rett Syndrome. Exp. Neurol. 1999, 156, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Patrizi, A.; Picard, N.; Simon, A.J.; Gunner, G.; Centofante, E.; Andrews, N.A.; Fagiolini, M. Chronic Administration of the N-Methyl-D-Aspartate Receptor Antagonist Ketamine Improves Rett Syndrome Phenotype. Biol. Psychiatry 2016, 79, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivarao, D.V.; Chen, P.; Senapati, A.; Yang, Y.; Fernandes, A.; Benitex, Y.; Whiterock, V.; Li, Y.-W.; Ahlijanian, M.K. 40 Hz Auditory Steady-State Response Is a Pharmacodynamic Biomarker for Cortical NMDA Receptors. Neuropsychopharmacology 2016, 41, 2232–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, S.; Tang, B.; Wu, Z.; Ure, K.; Sun, Y.; Tao, H.; Gao, Y.; Patel, A.J.; Curry, D.J.; Samaco, R.C.; et al. Forniceal Deep Brain Stimulation Rescues Hippocampal Memory in Rett Syndrome Mice. Nature 2015, 526, 430–434. [Google Scholar] [CrossRef]
- Lu, H.; Ash, R.T.; He, L.; Kee, S.E.; Wang, W.; Yu, D.; Hao, S.; Meng, X.; Ure, K.; Ito-Ishida, A.; et al. Loss and Gain of MeCP2 Cause Similar Hippocampal Circuit Dysfunction That Is Rescued by Deep Brain Stimulation in a Rett Syndrome Mouse Model. Neuron 2016, 91, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, R.P.; Simatos, D.; Weston, H.J.; Stanley, T.V. Rett Syndrome: Case Reports and Review. N. Z. Med. J. 1990, 103, 122–125. [Google Scholar]
- Vignoli, A.; Borgatti, R.; Peron, A.; Zucca, C.; Ballarati, L.; Bonaglia, C.; Bellini, M.; Giordano, L.; Romaniello, R.; Bedeschi, M.F.; et al. Electroclinical Pattern in MECP2 Duplication Syndrome: Eight New Reported Cases and Review of Literature. Epilepsia 2012, 53, 1146–1155. [Google Scholar] [CrossRef]
- Faulkner, M.A.; Singh, S.P. Neurogenetic Disorders and Treatment of Associated Seizures. Pharmacotherapy 2013, 33, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; DeLorey, T.M.; Delgado-Escueta, A.; Olsen, R.W. GABRB3, Epilepsy, and Neurodevelopment. In Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Sheth, R.D. Electroencephalogram in Developmental Delay: Specific Electroclinical Syndromes. Semin. Pediatr. Neurol. 1998, 5, 45–51. [Google Scholar] [CrossRef]
- Italiano, D.; Striano, P.; Russo, E.; Leo, A.; Spina, E.; Zara, F.; Striano, S.; Gambardella, A.; Labate, A.; Gasparini, S.; et al. Genetics of Reflex Seizures and Epilepsies in Humans and Animals. Epilepsy Res. 2016, 121, 47–54. [Google Scholar] [CrossRef]
- Khaikin, Y.; Mercimek-Andrews, S. STXBP1 Encephalopathy with Epilepsy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Weng, S.-M.; Bailey, M.E.S.; Cobb, S.R. Rett Syndrome: From Bed to Bench. Pediatr. Neonatol. 2011, 52, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Nomura, Y. Neurophysiology of Rett Syndrome. Brain Dev. 2001, 23 (Suppl 1), S50–S57. [Google Scholar] [CrossRef]
- Niedermeyer, E.; Naidu, S.B. Rett Syndrome, EEG and the Motor Cortex as a Model for Better Understanding of Attention Deficit Hyperactivity Disorder (ADHD). Eur. Child Adolesc. Psychiatry 1998, 7, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Rett Syndrome—An Update. J. Neural Transm. Vienna Austria 1996 2003, 110, 681–701. [Google Scholar] [CrossRef] [PubMed]
- Glaze, D.G. Neurophysiology of Rett Syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 66–71. [Google Scholar] [CrossRef]
- Dunn, H.G.; MacLeod, P.M. Rett Syndrome: Review of Biological Abnormalities. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2001, 28, 16–29. [Google Scholar] [CrossRef]
- Garcés, P.; Vicente, R.; Wibral, M.; Pineda-Pardo, J.Á.; López, M.E.; Aurtenetxe, S.; Marcos, A.; de Andrés, M.E.; Yus, M.; Sancho, M.; et al. Brain-Wide Slowing of Spontaneous Alpha Rhythms in Mild Cognitive Impairment. Front. Aging Neurosci. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Ponomareva, N.; Klyushnikov, S.; Abramycheva, N.; Malina, D.; Scheglova, N.; Fokin, V.; Ivanova-Smolenskaia, I.; Illarioshkin, S. Alpha-Theta Border EEG Abnormalities in Preclinical Huntington’s Disease. J. Neurol. Sci. 2014, 344, 114–120. [Google Scholar] [CrossRef]
- Bhattacharya, B.S.; Coyle, D.; Maguire, L.P. Alpha and Theta Rhythm Abnormality in Alzheimer’s Disease: A Study Using a Computational Model. Adv. Exp. Med. Biol. 2011, 718, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Thomas, A.J.; Peraza, L.R.; Firbank, M.; Cromarty, R.; Hamilton, C.A.; Donaghy, P.C.; O’Brien, J.T.; Taylor, J.-P. EEG Alpha Reactivity and Cholinergic System Integrity in Lewy Body Dementia and Alzheimer’s Disease. Alzheimers Res. Ther. 2020, 12, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Wu, W.; Zhang, Y.; He, H.; Yuan, Z.; Zhu, Z.; Zhao, Z. Selective Preservation of Cholinergic MeCP2 Rescues Specific Rett-Syndrome-like Phenotypes in MeCP2stop Mice. Behav. Brain Res. 2017, 322, 51–59. [Google Scholar] [CrossRef]
- Orekhova, E.V.; Butorina, A.V.; Sysoeva, O.V.; Prokofyev, A.O.; Nikolaeva, A.Y.; Stroganova, T.A. Frequency of Gamma Oscillations in Humans Is Modulated by Velocity of Visual Motion. J. Neurophysiol. 2015, 114, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, C.A.; Gao, R.; Negraes, P.D.; Gu, J.; Buchanan, J.; Preissl, S.; Wang, A.; Wu, W.; Haddad, G.G.; Chaim, I.A.; et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell 2019, 25, 558–569.e7. [Google Scholar] [CrossRef] [PubMed]
- Fair, S.R.; Julian, D.; Hartlaub, A.M.; Pusuluri, S.T.; Malik, G.; Summerfied, T.L.; Zhao, G.; Hester, A.B.; Ackerman, W.E.; Hollingsworth, E.W.; et al. Electrophysiological Maturation of Cerebral Organoids Correlates with Dynamic Morphological and Cellular Development. Stem Cell Rep. 2020, 15, 855–868. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Sample (n: Age) | EEG Settings | EEG Characteristics |
|---|---|---|---|
| [59] | RTT: 1, 4 y o | Video EEG | Needle-like central spikes evoked by contralateral passive finger-tapping drug-resistant |
| [60] | RTT: 1, 5 y o | Video EEG (24 h) | Multifocal bilateral discharges precipitated by right-hand tapping lips (but not cheek or abdomen, or the left-hand tapping lips, or observer’s hand tapping the lips) and immediately disappearing when the movement stopped. |
| [61] | RTT: 1, 12 y o | EEG | Paroxysmal runs of fluctuating 4- to 5-Hz rhythmic frontocentral theta activity at rest that abated with movement or tactile stimulation |
| [62] | RTT: 1, 4 y o | A 32-channel scalp EEG (24 h) | Centrotemporal spikes (CTS) disappeared after hand clapping |
| [63] | 6 RTT among 74 genetic-dysmorphic syndromes | EEG video monitoring with at least one EEG including awake and spontaneous afternoon nap recording | Abnormal EEG in 3 RTT |
| [64] | RTT: 1, 7 y o Lhermitte-Duclos syndrome//Neurofibromatosis: 1, 8 y o | Multiple daytime and sleep EEGs | Continuous spike and wave in slow-wave sleep Focal epileptiform discharges while awake |
| [65] | RTT: 64: 3–9 | Awake and sleep video EEG (24 h) | Unilateral, highly rhythmic hand tapping accompanied by contralateral synchronous centrotemporal spikes and not responsive to drugs (n = 5) |
| [56] | RTT: 8: 7–20 y o | Video-polygraphic, EEG + EMG + EKG recording | – Slowing of the background activity – Epileptiform abnormalities (spike and sharp wave discharges) – Abnormal sleep patterns – Epilepsy: drug-resistant in half//misdiagnosed (n = 6) – Multifocal and asynchronous cortical myoclonus (n = 5), myoclonic seizures (n = 4), myoclonic status (n = 2) |
| [66] | RTT: 3: 14, 18, 22 y o | Video-EEG and polygraphy (with confirmed reflex seizures in n = 1). | Reflex seizures, triggered by food intake (n = 1) or self-provoked by rhythmic pressure on hands (n = 2) or by taking someone’s hand or holding on to a table (n = 1) |
| [50] | Zappella RTT MECP2 high-intermediate performance (HIP): 11: 11–38 y o low-performance (LP): 5: 8–19 y o | 8-channel EEG referenced to linked mastoids continuous awake and sleep (at least 30 min) | Centro-temporal spikes: HIP > LP Multifocal EEG activity: HIP < LP EEG encephalopathy: HIP < LP Spindles and K-complex: HIP > LP Epilepsy (recurrent unproved seizures): HIP < LP (ns) |
| [49] | RTT: 18: 7–21 y o All with seizures | 20–30 min video EEG awake | Correlation between epilepsy and behavior: EEG stage III (moderate to marked slowing of background activity with dominant theta and delta activity) EEG stage IV (no occipital dominant rhythm and marked slowing of background activity). (1) theta slow activity over the frontal and central regions (eight cases) (2) frontocentroparietal spikes (eight cases) (3) generalized spike and waves (one case) (4) diffuse subcontinuous spike and waves suggestive of epileptic encephalopathy (one case) |
| [44] | RTT (165 including Classic (140) Preserved speech variant (PSV) (15) Hanefeld (6) 130 (78%) with epilepsy | Video EEG (not reported) Italian multicenter retrospective study | Epilepsy and RTT variants//mutation type no epilepsy (n = 35), drug-responsive (n = 81) and drug-resistant (n = 49) |
| [67] | RTT: 2: 7, 12 y o | EEG and lower limb EMG during gait | EMG burst were not associated with clinical jerking but EMG burst-locked averaging of the EEG showed contralateral centroparietal spiking preceding the burst by about 35 ms, indicating a cortical reflex myoclonus. |
| [45] | RTT (154 including 65% with seizures) | 8-channel EEG referenced to linked mastoids continuous awake and sleep (at least 30 min) | Epilepsy and RTT variants//mutation type Drug-resistant epilepsy, DRE (n = 16) No relationship between EEG characteristics and DRE |
| [68] | RTT: 11: 1–33 y o | Seizures (n = 8) Epileptiform activity (n = 7) | |
| [69] | RTT: 3: 9.5, 7.4, and 9.4 y o, each with a mutation of the CDKL5 gene. | Video EEGs | Seizure onset 1.5 months |
| [70] | Girl with mutation of MECP2 but no clear RTT phenotype: 8 y o | EEG | 5 and 7 y o EEG: presence of high-amplitude delta waves with a notched appearance and a persistent theta activity over posterior regions (EEG of Angelman Syndrome) |
| [71] | RTT: 50: 1–14 y o | 16-channel EEG |
Descriptive EEG, epilepsy presence is not reported Changes with RTT progression |
| [72] | RTT: 1: MECP2 mutation At age 2 and 6 | EEG | Age 6: rhythmic triphasic 2- to 3-Hz, high voltage (200–500 mkV) activity, mixed with spikes or sharp waves, with a maximum over the frontal regions (EEG of Angelman Syndrome) |
| [73] | RTT: 10: < 5 y o Angelman Syndrome (AS):10 Mental retardation:10 | Central and/or centro-temporal spike-wave complexes as specific to RTT | |
| [74] | RTT: 191 (detailed survey) 78–EEG | Awake and sleep EEG | 76% clinical seizures 78% epileptiform activity that preceded seizure onset |
| [75] | RTT: 13: 2–17 y o | Awake and sleep EEG, SPECT | Epileptiform activity (n = 10) Frontal hypoperfusion that is not correlated with EEG abnormalities |
| [76] | RTT: 10: 2–16 y o The reported 10 cases were selected because of their peculiar slow EEG rhythms | The EEG studies were performed in wakefulness (whenever possible) and sleep, mostly induced with moderate dosages of chloral hydrate. Passive movement carried out whenever spikes or central theta activity occurred. | CTS/abnormal theta reduced after hand movement (n = 4) |
| [77] | RTT: 16: 8 months-20 y o 44 EEG | EEG, respiration | Pseudoperiodic pattern, the short bursts of high-amplitude slow waves tending to be associated with apnea and the lower-amplitude faster rhythms with normal breathing or with hyperventilation (n = 8) |
| [78] | RTT: 14: 6–17, mean 7 y o TD: 12: 6–18, mean 14 y o | Day time video records, respiration | General EEG abnormality with excess of polymorphic slowing and poorly developed (daytime) sleep change (all n = 14) Unreactive Theta (n = 10) Absence of normal slow-wave response to hyperventilation (all n = 14) Attacks of vacancy and staring not associated with significant EEG changes (n = 6) No-epileptic slow-waves mostly during normal breathing (n = 11) |
| [79] | RTT: 4: 4–11 y o | All-night electroencephalograms (EEGs)/polysomnograms on 2 consecutive nights | Epileptiform activity maximum over 1–2 SWS sleep stage and in the morning hours |
| [80] | RTT: 4: 3.5, 6, 11 and 12 y o | Light no-REM sleep or the state lethargy (wake without slight index of awareness) | Epileptiform activity, in particular, CTS, blocked or attenuated by passive finger movements |
| [81] | RTT: 30: 2–22 y o 127 EEG-recordings | Awake and sleep EEG, EMG | Epileptiform activity (n = 26) CTS (n = 10) Pseudoperiodic delta bursts (n = 13) Flat record (n = 2) Developmental changes |
| [82] | RTT: 13: 2–17 y o | Awake and sleep EEG, EMG | Seizures (n = 5) Epileptiform activity (n = 10) Pseudorhythmic flattering (n = 4) |
| [83] | RTT: 8: 2–16 y o | Awake and sleep EEG | Background EEG slowing Epileptiform activity A monotonous theta rhythm (MTR), which was not influenced by either opening or closing of the eyes but attenuated only by a big noise or strong pain stimuli, characteristically dominated the waking tracing EEG through disease progression |
| [84] | RTT: 52: 1–13 y o 83 EEG recordings | Awake and sleep 8-channels EEG, EMG |
Seizures (n = 26 + 3) Epileptiform activity, enhanced during sleep (n = 43) CTS often coincided with stereotyped hand wringing or tapping and facilitated over the contralateral central regions by passive tactile stimulation (9 of 26 girls tested) Excesses of theta and delta activity (n = 12, n = 7) |
| [39] | RTT: 9: 1–6 y o 22 EEG recordings | EEG, video EEG in two patients |
Seizures (n = 7) Epileptiform activity (n = 9, in 3 epileptiform activity preceded development of seizures) Abnormalities increases with age |
| [85] | RTT: 7: 1–7 y o | Awake and sleep EEG | Reactive Theta, Excessed Delta, Flattering EEG, epileptiform activity, atypical sleep EEG CTS (n = 4) including those evoked by tactile stimulation (n = 2) Changes with RTT progression |
| [86] | RTT: 18: 1–17 y o | Awake and sleep EEG, respiration biogenic amine metabolites |
Reduced % stage REM (n = 15) EEG slowing Epileptiform activity Absent vertex transients and spindles during NREM |
| [87] | RTT: 11: 4–14 y o | Awake and sleep EEG |
Slowing of background EEG (n = 11) activity while awake Multifocal spike-waves (centrotemporal regions) (n = 9) Intermittent, high-amplitude discharges followed by relative attenuation of background activity during sleep (n = 6) |
| [88] | RTT: 9: 2–15 y o 35 EEG recordings | EEG |
Seizures (n = 7) Epileptiform activity (n = 7) Slowing of background EEG Progression through disease |
| [89] | RTT: 8: 4–13 y o | Awake and sleep EEG, EMG |
Myoclonic jerks (n = 8) EEG is poorly organized with high amplitude slow waves short, non-periodic bursts; focal or diffuse spike and wave complexes increased in sleep |
| [90] | RTT: 1, 2 y o | EEG | Unspecific modification on EEG, no seizures |
| Study | Genotype (Age) | Electrode Position, Behavioral State | EEG Characteristics | Seizure Characteristics (Prevalence) |
|---|---|---|---|---|
| [113] | Mecp−/+ (from 13 to 104 weeks) | hippocampal CA1 and contralateral somatosensory cortex, free behavior | cortical discharges, behavioral freezing 13 weeks average number per hour: 20.7 ± 11.7 duration: 0.9 ± 0.28 s 104 weeks average number per hour: 116.8 ± 44.2 duration: 1.7 ± 0.23 s aggravated by levetiracetam reduced by valproic acid and acetazolamide suppressed by ethosuximide | |
| [116] | Vglut2-Mecp2flox/y (10 weeks) Vglut2-Mecp2LSL/y (25–30 weeks) | right frontal cortex and somatosensory cortex left hippocampal CA1 and dentate regions | Vglut2-Mecp2flox/y (37.5%) cortical spike-and-wave discharges average number per hour: 3.3 ± 2.1 duration: 4.1 ± 0.4 s Vglut2-Mecp2LSL/y (25%) cortical spike-and-wave discharges average number per hour: 3.3 duration: 4.5 ± 0.8 s | |
| [110] | PV-Mecp2-/y SOM-Mecp2-/y | SOM-Mecp2-/y (50%) epileptic seizures starting at 12 weeks generalized tonic-clonic seizures observed during routine handling PV-Mecp2-/y no seizures | ||
| [111] | Rosa26-Esr/Cre- Mecp2 Stop/+, (around 36 week) Rosa26-Esr/Cre-Mecp2Stop/y, (5.5–8.5 weeks) | hippocampal CA1 and contralateral somatosensory cortex, free behavior | ▼ gamma (35–60 Hz) power (Rosa26-Esr/Cre-Mecp2Stop/+; Rosa26-Esr/Cre-Mecp2Stop/y) rescue in Rosa26-Esr/Cre-Mecp2Stop/y ▼ theta frequency rescue in Rosa26-Esr/Cre-Mecp2Stop/y | Rosa26-Esr/Cre-Mecp2Stop/y cortical discharges average number per hour: 45 ± 8 duration: 2.8 ± 0.7 s frequency: 6.2 ± 0.3 Hz Rosa26-Esr/Cre-Mecp2Stop/y-rescue cortical discharges average number per hour: 21 ± 3 duration: 1.0 ± 0.1 s frequency: 6.8 ± 0.2 Hz Rosa26-Esr/Cre- Mecp2 Stop/+ cortical discharges average number per hour: 76 ± 17 duration: 1.11 ± 0.1 s frequency: 7.9 ± 0.4 Hz Rosa26-Esr/Cre- Mecp2 Stop/ + -rescue cortical discharges average number per hour: 32 ± 4 duration: 1.2 ± 0.1 s frequency: 7.7 ± 0.3 Hz |
| [117] | Mecp2tm1.1Bird (6–7 weeks) | the M1 region of the frontal cortex, free behavior | ▼ delta power during NREM | cortical discharges, behavioral freezing 13 weeks average number per 20.7 ± 11.7 versus 116.8 ± 44.2 104 weeks |
| [98] | Emx1-Mecp2, Dlx6a-Mecp2 (6–8 weeks) | frontal and parietal cortex | Emx1-Mecp2 spike-wave discharges, absence seizures average number per hour: 36 ± 7 duration: 1.3 ± 0.1 s Dlx6a-Mecp2 no discharges | |
| [109] | Mecp22lox/y; Dlx5/6-Cre Mecp22lox/y; Emx1-Cre Mecp2Stop/y; Emx1-Cre (8, 7 weeks) Mecp22lox/y; PV-Cre (13 weeks) Mecp22lox/y; SOM-Cre (13 weeks) | hippocampus | Mecp22lox/y; Dlx5/6-Cre behavioral seizures following handling spikes and wave discharges, behavioral arrest Mecp22lox/y; Emx1-Cre no behavioral or electrographic seizures Mecp2Stop/y; Emx1-Cre behavioral seizures Mecp22lox/y; PV-Cre no behavioral seizures Mecp22lox/y; SOM-Cre no behavioral seizures | |
| [118] | Mecp2−/+, Mecp2−/y, TH-Mecp Stop/+ (around 36 weeks) TH-Mecp2Stop/y (5.5–8.5 weeks) | hippocampal CA1 and contralateral somatosensory cortex, free behavior | ▼ theta frequency partial preservation in TH-Mecp2Stop/y ▼ gamma (35–60 Hz) power partial preservation in TH-Mecp2Stop/y | Mecp2−/y (100%) cortical discharges average number per hour: 42.1 ± 7.9 duration: 2.8 ± 0.7 s frequency: 6.2 ± 0.3 Hz TH-Mecp2Stop/y (100%) cortical discharges average number per hour: 20.5 ± 6.3 duration: 2.2 ± 0.6 s frequency: 6.5 ± 0.36 Hz Mecp2-/+ cortical discharges average number per hour: 69.6 ± 15.5 TH-Mecp Stop/+ cortical discharges average number per hour: 67.1 ± 13.9 |
| [29] | Mecp2T158A/y (4.2 weeks and 12.9 weeks), Mecp2−/y (12.9 weeks) | hippocampal electrode, free behavior | ▲ gamma high (70–140 Hz) power seizure behaviors (unspecified) | |
| [112] | Mecp2−/+ (42–57 weeks) | parietal cortex, free behavior | ▼ the average number of delta cycles over a 24-h period ▼correlation coefficient for delta power and movement | cortical discharges, behavioral freezing average number per hour: 10.7 ± 1.6 duration: 0.76 ± 0.01 s frequency: 8.6 ± 0.02 Hz |
| [119] | Viaat-Mecp2−/y (9 weeks) Dlx5/6-Mecp2−/y (39 weeks) | cortical electrodes, free behavior | Viaat-Mecp2−/y hyperexcitability discharges, no electrographic seizures Dlx5/6-Mecp2−/y no hyperexcitability discharges | |
| [107] | Mecp2−/y (7–10 weeks), Mecp −/+ (35–52 weeks) | hippocampal CA1 and contralateral somatosensory cortex, free behavior | ▼ theta frequency (hippocampus) desynchronized, low-amplitude cortical activity =cortical delta and hippocampal sharp-wave activity during sleep | Mecp −/+ (100%) cortical discharges, immobile animal, no myoclonus average number per hour: 9.1 ± 1.3 duration: 1–2 s frequency: 8.8 ± 1.3 Hz suppressed by ethosuximide |
| [106] | Mecp2308/y (age of recording not specified) | frontal and parietal cortex, free behavior | spike-and-wave discharges, behavioral arrest, generalized myoclonic jerks |
| Study | Sample (n: Age) | EEG Settings | EEG Characteristics |
|---|---|---|---|
| [139] | RTT (57 including 20 in active regression (AR), 29 post-regression (PR)): 2–11) TD (37: 2–11) | 128-channel Hydrocel Geodesic Sensor Net System, 5–10 min while watching movies | Spectral power (2–50 Hz) ▼ beta (13–30 Hz) at frontal and central sites (absolute and relative) ▼ beta (13–30 Hz) at temporal sites (relative) ▼ high alpha (9–13Hz) widespread (absolute and relative) ▼ low alpha (6–9 Hz) at temporal and occipital sites (relative) ▼ low alpha (6–9 Hz) at central sites (absolute and relative) ▲ delta (2–4 Hz) widespread (relative) ▼ 1/f slope (more negative, in general and in PR) for AR: ▼ theta (4–6 Hz) at frontal sites (absolute) ▼ high alpha and beta (9–30 Hz) at frontal and central (absolute and relative) ▼ high alpha (9–13 Hz) at occipital (absolute) Abnormal development of delta and theta |
| [140] | RTT, MECP2 (32) PSV, MECP2 (4) RTT, CDKL5 (4) including Resistant Epilepsy (8), Treatment Responsive Epilepsy (16), without Epilepsy (18) Mean age: 7.69 +/− 5.22 | 8-electrode EEG, minimum of 20 min recordings during resting state | =spectral power (1–50 Hz) ▼ network activity (principal component of inter-electrode coherence) in CDKL5 compared to MECP2 ▲ left-hemispheric predominance in the occipital region Resistant > Epilepsy>no Epilepsy within RTT * ▲ left-sided parieto-occipital coherence in PSV compared to classic RTT * |
| [141] | RTT (10: 2–9) TD (15: 2–9) | Overnight (F3, C3, and O1), slow wave sleep (SWS) | Spectral power (0.5–45 Hz), ▲ delta Absence of developmental decrease of gamma (35–45 Hz) * ▼ %SWS |
| [138] | RTT (38(20 with disease onset <1 year and 18 > 1 year): 2–8) TD (60: 2–8) | 16 channels; >15.4-s epochs | Spectral power (1.5–25 Hz) ▼ alpha ▲ theta activity in RTT ▼ central and occipital alpha ▲central and frontal theta activity in the group 1 (early disease onset). |
| [137] | RTT (9:2–8, stage 3) TD (17: 2–8) | 16 channels: eyes open (n= 6) and eyes close (n= 5); >8 4-s epochs | Relative Power (1–20 Hz) ▼ alpha and beta ▲ theta band |
| [142] | RTT (1:10) TD (10:10) | 16 channels: 20 artifact-free 5-s epochs | Spectral power (1.5–30 Hz) ▲ delta and left anterior theta ▼ posterior alpha 1 |
| [143] | RTT (9: 10–22) Compared to unspecified normative data | n = 11: 16–20 channels; 5–10 min of recording | Spectral power (1.0–16 Hz): ▲ low frequency activity (unspecified, statistical values not provided) |
| Study | Sample (n: Age) | Treatment Schema | EEG Settings | EEG Effects | Other Treatment Effects |
|---|---|---|---|---|---|
| [57] | RTT 75% MECP2 (12: 2–11, 20, 30) | Folinic acid: 2-year prospective randomized, double blinded, placebo-controlled crossover | Video 16-channel EEG (10–20 system) | Improvement: ▼ Seizure rate (only in 2 out of 8 patients who had seizures at baseline) ▼ Frequency of epileptiform discharges (only in 3 patients) Worsening: 3 patients developed seizure during treatment ▲ Frequency of epileptiform discharges (2 patients) | N/A |
| [214] | RTT 100% MECP2 with drug-resistant epilepsy (8:8–18) | levetiracetam (LEV) prospective, pragmatic, OLE: mean dose achieved was 44.84 mg/kg/day SD 18.02 | EEG at baseline and 6-months after beginning of treatment | Improvement: ▼ Seizure rate, background EEG, epileptiform abnormalities ▼ Frequency of epileptiform discharges (in 6 out of 8 patients) | N/A |
| [215] | RTT 100% MECP2 post-regression (17:2–10) | rhIGF-1: 20 wk, double-blind cross over: 0.12 mg/kg/twice a day | 128 channels; 5–10 min calm recording when girls watched movie of their choice | = Frontal alpha asymmetry (FA) Worsening: ▲ Frontal delta power ▼ Beta relative power ▼Gamma relative power | Worsening: ▲ The Kerr severity scale, ▲ ADAMS Depressed Mood subscale, ▲ Visual Analog Scale ▲Hyperventilation Improvement: ▼ ABC-C stereotypy ▲ CSBS-DP Social |
| [216] | RTT 100% MECP2 stage III (10: 3–11) RTT untreated (9: 2–12) | rhIGF-1: 20–24 wk: 0.1 mg/kg/twice a day | 10 channels; 1–2 h EEG | = Frontal alpha asymmetry (FA) Improvement: ▲ Theta frequency ▼ Delta amplitude | Improvement: ▼ International Scoring System: ISS), ▲ RSS: Rett Severity Score ▲ Endurance to RSS testing |
| [217] | RTT 100% MECP2 (6 out of 10:2–10) | rhIGF1: 4 wk MAD + 20 wk OLE: 0.120 mg/kg) | Frontal and parietal channels | Frontal alpha asymmetry (FA) (Pre-OLE: R > L with trend to reverse Post-OLE) | Worsening: ▼ Behavioral subtotal (MBA) Improvement: ▼ Apnea index |
| [218] | RTT 100% MECP2 with epileptiform activity 33 (13 + 12 + 8 dose groups): 2–14.5) | Dextromethorph
an:
25 wk OLE 3 dose groups: 0.25 mg/kg/day; 2.5 mg/kg/day; 5.0 mg/kg/day; |
Neuroscan EEG system; Overnight recordings | =Epileptiform spikes and sharp waves during non-REM sleep Improvement: Seizure rate (parental report): low dose group (in all? small number of patients who had seizures before OLE n = 5/4/2 at each dose group) | Improvement: ▲ Receptive Language (5.0 mg/kg/day vs 0.25 mg/kg/day) ▲ * Behavioral hyperactivity (2.5 mg/kg dose) |
| [219] | RTT 100% MECP2 (10:10–21) ambulatory | Glatiramer acetate (GA): 24 wk of 20 mg/day | A 21-channel 1-h video EEG (wakefulness (n = 10), drowsiness (n = 5), sleep (n = 3)) | =Background EEG abnormalities =Seizure rate Improvement: ▼ Frequency of epileptiform discharges (in all 4 patients who had them at baseline) | Improvement: ▲ Gait velocity ▲ Memory Novelty Score % ▼ Breath holding index/% |
| [137] | RTT N/S (9:2–8, stage 3) 17 TD aged-matched | Cerebrolyzin (CL): 20 days (2.0–2.5 mL daily, i.m.). | 16 channels: eyes open (n= 6) and eyes close (n= 5); >8.4-s epochs | Improvement: ▲beta activity in the parietal region and occipital alpha in the 8–9 Hz narrow band ▼ central and frontal theta the after CL | Improvement: Qualitative assessment by clinicians and parents ▲ the behavioral activity ▲ attention level ▲ motor functions ▲ non-verbal social communication |
| [220] | RTT (33: 21 + 13: 5–36) | Visual discrimination training: Short-term, 30 min (STT: n = 13 + 21) Long-term, 5 days, (LTT: n = 21 vs. Control: n = 13) | 19 dry electrodes (wireless): visual discrimination task, at rest eyes open | After STT: ▼ Alpha and Beta Brain Symmetry Index (shift from L > R into R > L) After LTT: =Brain Symmetry Index Improvement Theta relative power (frontal left and parietal) ▲ Beta relative power (frontal right and parietal left) | After STT: No changes Improvement after LTT: ▲ Length of fixation (FL) ▼ Time before first fixation (TFF) |
| [221] | RTT (31: 13–35) non-sham tDCS (n = 18) vs. sham tDCS (n = 13) | TDCS stimulation on Broca’s area together with linguistic training for 20 min/day over 10 days | 21 EEG channels | ▲ Frequency and Power of Alpha and Beta =Frequency and Power of Theta | Improvement: ▲ Attention ▲ Language abilities (the number of phonemes produced) |
| [222] | RTT MECP2 mutations: C468G, R133C, R306C (3: 29, 30, 31) with chronic language impairments | TDCS stimulation on Broca’s area together with linguistic training, case-study design for 20 min/day over 10 days | 19-channels EEG (20 min wit eyes closed) | ▲ Frequency and Power of Alpha, Beta and Theta | Improvement: ▲ language abilities (the number of vowel/consonant sounds and words and the production and comprehension through discrimination) ▲ motor coordination (functional movements) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnov, K.; Stroganova, T.; Molholm, S.; Sysoeva, O. Reviewing Evidence for the Relationship of EEG Abnormalities and RTT Phenotype Paralleled by Insights from Animal Studies. Int. J. Mol. Sci. 2021, 22, 5308. https://doi.org/10.3390/ijms22105308
Smirnov K, Stroganova T, Molholm S, Sysoeva O. Reviewing Evidence for the Relationship of EEG Abnormalities and RTT Phenotype Paralleled by Insights from Animal Studies. International Journal of Molecular Sciences. 2021; 22(10):5308. https://doi.org/10.3390/ijms22105308
Chicago/Turabian StyleSmirnov, Kirill, Tatiana Stroganova, Sophie Molholm, and Olga Sysoeva. 2021. "Reviewing Evidence for the Relationship of EEG Abnormalities and RTT Phenotype Paralleled by Insights from Animal Studies" International Journal of Molecular Sciences 22, no. 10: 5308. https://doi.org/10.3390/ijms22105308