Abstract
Hydrocarbon stapling is a useful tool for stabilizing the secondary structure of peptides. Among several methods, hydrocarbon stapling at i,i + 1 positions was not extensively studied, and their secondary structures are not clarified. In this study, we investigate i,i + 1 hydrocarbon stapling between cis-4-allyloxy-l-proline and various olefin-tethered amino acids. Depending on the ring size of the stapled side chains and structure of the olefin-tethered amino acids, E- or Z-selectivities were observed during the ring-closing metathesis reaction (E/Z was up to 8.5:1 for 17–14-membered rings and up to 1:20 for 13-membered rings). We performed X-ray crystallographic analysis of hydrocarbon stapled peptide at i,i + 1 positions. The X-ray crystallographic structure suggested that the i,i + 1 staple stabilizes the peptide secondary structure to the right-handed α-helix. These findings are especially important for short oligopeptides because the employed stapling method uses two minimal amino acid residues adjacent to each other.
1. Introduction
Introducing hydrocarbon stapling on the side chains of peptides is a promising technique for stabilizing the secondary structure of peptides and enhancing their functionalities [1,2,3,4,5]. Hydrocarbon stapling can be easily obtained by ring-closing metathesis reactions between olefin-bearing amino acid residues using Ru catalysts [6,7]. After the report on α-helicity-inducing all-hydrocarbon stapled peptides at i,i + 4 and i,i + 7 positions by Verdine et al. [8], several studies focused on the approach (as illustrated in Figure 1a) [9,10,11]. Currently, all-hydrocarbon stapled peptides are very important in drug development targeting protein–protein interactions because the pharmacophores interact via α-helical motifs [12]. Hydrocarbon stapling at i,i + 3 positions are reported in the literature [13,14,15]. For example, O’Leary et al. reported E-selective ring-closing metathesis between O-allyl-tethered l-serines at i,i + 3 positions to produce 310-helical peptides [13]. Other hydrocarbon staples, such as i,i + 1 and i,i + 2, were not well researched, and their 3D structures are unknown (as illustrated in Figure 1b) [16,17,18,19]. In general, hydrocarbon stapling sacrifices two amino acid residues for the crosslinking motif, and those residues should not include essential residues for their biological activities. Based on this, the development of a large variety of hydrocarbon stapling at different positions can be achieved. Herein, we report hydrocarbon stapling of peptides at i,i + 1 positions by ring-closing metathesis reactions and the X-ray crystallographic structure of the right-handed α-helical octapeptide stabilized by i,i + 1 stapling.
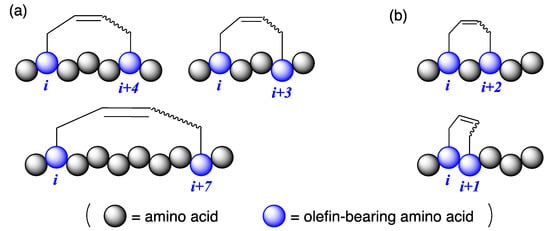
Figure 1.
Peptides with hydrocarbon stapling at different positions. (a) Commonly used hydrocarbon stapling (at i,i + 4, i,i + 3, and i,i + 7), and (b) rarely investigated hydrocarbon stapling (at i,i + 2 and i,i + 1).
2. Results and Discussion
Our previous report suggests the usefulness of cis-4-hydroxy-l-proline as an olefin-bearing amino acid for peptide stapling [19]. Thus, in this study, we started by optimizing the reaction conditions for i,i + 1 peptide stapling using cis-4-hydroxy-l-proline. We screened the ring-closing metathesis reaction at i,i + 1 positions using dipeptide 1 as the cyclization precursor (as illustrated in Table 1). The reaction catalyzed by 20 mol% of second-generation Grubbs catalyst in CH2Cl2 (20 mM) produced the desired 1′ in 55% yield as a mixture of E/Z-isomers (E/Z = 1.0:5.6; Entry 1). A comparable result was obtained using the first-generation Grubbs catalyst (Entry 2). Replacing the reaction solvents, such as toluene, 1,2-dichloroethane (DCE) and tetrahydrofuran (THF), decreased the yields and Z-selectivities (Entries 3–5). The reaction under diluted condition (5 mM in CH2Cl2) afforded the best yield at 76% (Entry 6). The reactions in refluxing CH2Cl2 resulted in insufficient yields due to the degradation of the desired product (Entries 8 and 9).

Table 1.
Screening of reaction conditions for ring-closing metathesis of dipeptide 1.
Further, we investigated the substrate scope for the ring-closing metathesis of peptides at i,i + 1 positions using the optimized reaction conditions (as illustrated in Scheme 1). As the ring size of the stapled peptides increased from 13- to 15-membered rings, the yields and E-selectivities increased (Entries 1–3). l-Tyrosine and D-serine-derived unstapled peptides 4 and 5 produced the desired stapled peptides 4′ and 5′ in 23% and 21% yields, respectively, with large amounts of unreacted starting material (Entries 4 and 5). Surprisingly, high Z-selectivities were observed for the reaction of dipeptides 6 and 7, which were composed of either O-allyl-tethered l-threonine or (S)-α-(4-pentenyl)alanine (Entries 6 and 7; E/Z = 1: >20 for 6′ and 1:14 for 7′). These results suggest that α-methyl or β-methyl groups of i + 1 residue strongly affect the transition state of the ring-closing metathesis to yield Z-isomers.
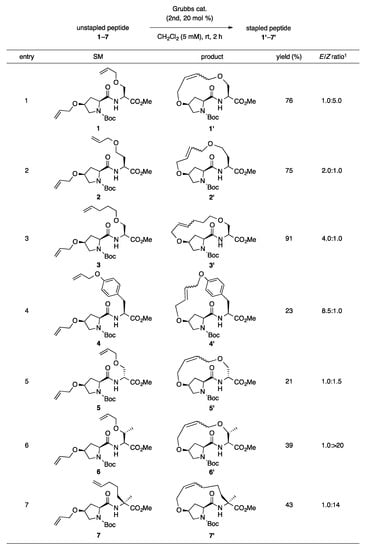
Scheme 1.
Substrate scope for ring-closing metathesis of peptides 1–7 at i,i + 1 positions. 1 Determined by 1H NMR.
The i,i + 1 hydrocarbon-stapling reaction of octapeptide 8, in possession of 1-aminocycloalkane-1-carboxylic acid [20,21,22,23,24,25,26,27,28,29,30,31,32,33], was investigated under the optimized reaction conditions for the ring-closing metathesis (Scheme 2). In contrast with the moderate Z-selectivity of 1 (E/Z = 1:5), much higher Z-selectivity was observed for the ring-closing metathesis reaction of 8 (E/Z = 1: >20). The Z-selectivity could be influenced by their secondary structure. Hydrogenation of 9 afforded saturated stapled peptide 10 in high yield. The high Z-selectivities (E/Z was up to 1: >20) of the i,i + 1 hydrocarbon stapling is advantageous for peptide staples compared to those reported for i,i + 4 and i,i + 7 hydrocarbon stapling (E/Z was up to 1: >9) [15].
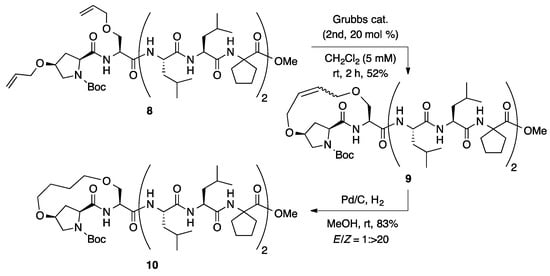
Scheme 2.
Ring-closing metathesis of octapeptide 8 at i,i + 1 positions.
Crystals suitable for X-ray crystallographic analyses were successfully obtained by slow evaporation of the solution of 10 in N,N-dimethylformamide (DMF)/water at room temperature (20–30 °C) [34]. The structure was solved in the orthorhombic P212121 space group to give an α-helical structure with a DMF molecule in the asymmetric unit (as illustrated in Figure 2 and Figure S1 and Table 2 and Table 3, and Table S1). To the best of our knowledge, this is the first X-ray crystallographic structure of α-helical stapled peptides at i and i + 1 positions. In the crystal state of the (i,i + 1)-stapled peptide 10, four consecutive intramolecular hydrogen bonds of the i←i + 4 type, N(4)H···O = C(0) (N···O, 3.09 Å; N–H···O, 163.6°), N(5)H···O = C(1) (N···O, 2.98 Å; N–H···O, 168.6°), N(6)H···O = C(2) (N···O, 2.91 Å; N–H···O, 157.2°), and N(7)H···O = C(3) (N···O, 3.14 Å; N–H···O, 139.7°) were observed. These hydrogen bonds indicate the existence of the α-helical secondary structure in 10. The average torsion angles of 10 at the N-terminus [avg.(ϕ1–ϕ5) = −62.4° and avg.(Ψ1–Ψ5) = −46.5°] were much closer to the ideal values of a right-handed α-helix [ϕ = −57° and Ψ = −47°] [35]. Therefore, the crosslinkage of the i,i + 1 staples at the N-terminus could affect the stabilization of the α-helical structure of 10. On the C-terminus, weak intramolecular hydrogen bonds of the i←i + 3 type were observed, N(7)H···O = C(4) (N···O, 3.37 Å; N–H···O, 136.7°) and N(8)H···O = C(5) (N···O, 3.40 Å; N–H···O, 162.7°), while the N(8)–H···O(4) angle of i←i + 4 type was too small for a hydrogen bond. These bifurcated hydrogen bonds suggest that the conformation of the C-terminus exists as a mixture of α- and 310-helix. Another intramolecular hydrogen bond between the N(2)–H of the main chain and ethereal oxygen of cis-4-hydroxyproline, N(2)H···O = C(Hyp4) (N···O, 2.93 Å; N–H···O, 137.6°), was observed. Such hydrogen bond stabilizes the secondary structures of peptides [30,36,37]. On the other hand, no intermolecular hydrogen bonds between peptides were observed in the packing mode (Figure S2). These results suggest that packing contacts have a small or no influence on the secondary structure of right-handed α-helix in this case. Thus, introducing hydrocarbon stapling at i,i + 1 positions using cis-4-hydroxyproline could be used for the stabilization of α-helical peptides likewise i,i + 4 and i,i + 7 staples. In our previous study, we reported asymmetric Michael addition of 1-methylindole to α,β-unsaturated aldehydes catalyzed by Boc-deprotected 10 [19]. We hypothesized that the reactive iminium ion intermediate between cis-4-hydroxy-l-proline and α,β-unsaturated aldehyde was formed inside the helical pipe with a rigid conformation caused by i,i + 1 staple. The X-ray crystallographic structure of 10 supports this observed conformation of the intermediate.
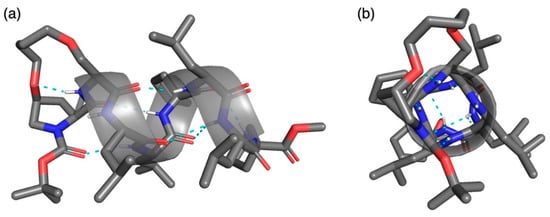
Figure 2.
X-ray crystallographic structure of (i,i + 1)-stapled peptide 10: a view (a) perpendicular to α-helical axis and (b) along helical axis from N-terminus.
Table 2.
Crystal and diffraction parameters of peptide 10.
Table 3.
Intra and intermolecular H-bond parameters for peptide 10.
In summary, we developed i,i + 1 peptide stapling between cis-4-allyloxy-l-proline and various olefin-tethered amino acids. Depending on the ring size of the stapled peptides, E- or Z-selectivities were observed. The E-configured stapled product was preferred when the product was greater than a 14-membered ring, whereas the Z-configured isomer was preferred when the product was a 13-membered ring. The α-or β-methyl substituent of the i + 1 residue improved the Z-selectivities of the ring-closing metathesis (E:Z = 1: >20). X-ray crystallographic analysis of the octapeptide 10 revealed a stabilized α-helical structure. These results are useful for developing peptide-based organocatalysts [38,39,40] (i.e., considering mechanistic insights and structural modification of peptide catalysts based on the X-ray crystal structure), fluorinated peptides [41] (e.g., stabilization effects of using intramolecular hydrogen bonds beside main chain hydrogen bonds), and peptide-based drug delivery systems [42,43,44,45,46] (e.g., introducing i,i + 1 hydrocarbon stapling with essential residues for their biological activities remained intact).
3. Materials and Methods
3.1. General Procedure and Method
Melting points were taken on an AS ONE melting point apparatus ATM-01 (AS ONE Corporation, Osaka, Japan) and were uncorrected. Optical rotations were measured on a JASCO DIP-370 polarimeter (JASCO Corporation, Tokyo, Japan) using CHCl3 as a solvent. 1H NMR and 13C NMR spectra were recorded on the JEOL JNM-AL-400 (400 MHz), a Varian NMR System 500PS SN (500 MHz and 125 MHz) spectrometer (Agilent Inc., Santa Clara, CA, USA). Chemical shifts (δ) are reported in parts per million (ppm). For the 1H NMR spectra (CDCl3), tetramethylsilane was used as the internal reference (0.00 ppm), while the central solvent peak was used as the reference (77.0 ppm in CDCl3) for the 13C NMR spectra. The IR spectra were recorded on a Shimadzu IRAffinity-1 FT-IR spectrophotometer (Shimadzu Corporation, Kyoto, Japan). High-resolution mass spectra (HRMS) were obtained on a JEOL JMS-T100TD using electrospray ionization (ESI) (JEOL Ltd., Tokyo, Japan) or direct analysis in the realtime (DART) ionization in time-of-flight TOF mode. Analytical and semipreparative thin layer chromatography (TLC) was performed with Merck Millipore precoated TLC plates (MilliporeSigma, Burlington, VT, USA), silica gel 60 F254, and layer thicknesses of 0.25 and 0.50 mm, respectively. Compounds were observed in UV light at 254 nm and then visualized by staining with iodine, p-anisaldehyde, or phosphomolybdic acid stain. Flash and gravity column chromatography separations were performed on Kanto Chemical silica gel 60N, spherical neutral, with particle sizes of 63–210 μm and 40–50 μm, respectively. All moisture-sensitive reactions were conducted under an inert atmosphere. Reagents and solvents were of commercial grade and were used as supplied, unless otherwise noted. Compounds 1, 8 [19], S-1 [47,48], S-2 [49,50], S-3 [51], and S-5 [52] were prepared according to the reported procedures. Copies of NMR Spectra are given in the Supplementary Materials.
3.2. Synthesis of Unstapled Dipeptides 2–7

Boc-l-HypOAll-l-HseOAll-OMe (2): to a solution of N-tert-butoxycarbonyl 4-O-allyl-cis-4-hydroxy-l-proline (Boc-L-HypOAll-OH, S-1 [47,48]; 88.1 mg, 0.325 mmol) in CH2Cl2 (2 mL) were added N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDCI·HCl, 67.9 mg, 0.354 mmol) and 1-hydroxybenzotriazole hydrate (HOBt·H2O; 54.2 mg, 0.354 mmol) at 0 °C, and the solution was stirred for 30 min at 0 °C. Then, a solution of O-allyl-l-homoserine methyl ester (H-l-HseOAll-OMe, S-2 [49,50], 51.1 mg, 0.295 mmol) in CH2Cl2 (1 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring for three days, CH2Cl2 was removed, and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (40% EtOAc in n-hexane) to give 2 (72.1 mg, 58%) as a pale yellow oil. Rf = 0.58 (EtOAc). –11.0 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.38–7.17 (m, 1H), 5.98–5.76 (m, 2H), 5.34–5.09 (m, 4H), 4.72–4.56 (m, 1H), 4.42–4.25 (m, 1H), 4.11–4.05 (m, 1H), 4.05–3.84 (m, 4H), 3.73 (s, 0.6H), 3.72 (s, 2.4H), 3.63–3.39 (m, 4H), 2.65–2.41 (m, 1H), 2.27–1.96 (m, 3H), 1.48 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.5, 172.0, 171.0, 154.7, 134.5, 134.4, 134.34, 134.26, 117.3, 117.2, 117.1, 117.0, 80.9, 76.3, 72.04, 71.98, 69.6, 66.3, 66.0, 60.1, 52.7, 52.3, 52.1, 50.6, 50.4, 36.9, 35.6, 31.6, 28.3, 28.1. IR (film): 3385 (br), 2978, 2868, 1744, 1690 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd. for C21H34N2O7Na, 449.2264; found, 449.2262.

Boc-l-HypOAll-l-SerOPte-OMe (3): to a solution of carboxylic acid S-3 [51] (135 mg, 0.495 mmol) in MeOH (5 mL), thionyl chloride (0.143 mL, 1.98 mmol) was added dropwise at 0 °C. The reaction mixture was stirred at room temperature for 2 h and was concentrated to give H-L-SerOPte-OMe·HCl (S-4, Rf = 0.57 with 0.5% AcOH in EtOAc), which was used for the next step without further purification. To a mixture of H-l-SerOPte-OMe·HCl (S-4, 0.495 mmol) and Boc-l-HypOAll-OH (S-1, 148 mg, 0.545 mmol) in CH2Cl2 (5 mL) were added EDCI·HCl (114 mg, 0.594 mmol), HOBt·H2O (91.0 mg, 0.594 mmol), and DIPEA (0.253 mL, 1.49 mmol) at 0 °C, and the mixture was gradually warmed to room temperature. After stirring for 17 h, CH2Cl2 was removed under vacuum, and the residue was diluted with EtOAc. The resultant solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (40% EtOAc in n-hexane) to give 3 (90.1 mg, 41% in 2 steps) as a pale yellow oil. Rf = 0.71 (EtOAc). –2.6 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.29 (br s, 1H), 7.11–6.90 (m, 1H), 5.98–5.71 (m, 2H), 5.34–5.22 (m, 1H), 5.21–5.11 (m, 1H), 5.05–4.92 (m, 2H), 4.77–4.63 (m, 1H), 4.44–4.28 (m, 1H), 4.11–3.92 (m, 2H), 3.92–3.78 (m, 2H), 3.75 (s, 3H), 3.66–3.48 (m, 3H), 3.47–3.35 (m, 2H), 2.67–2.45 (m, 1H), 2.24–2.02 (m, 3H), 1.67–1.57 (m, 2H), 1.49 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.1, 171.2, 170.7, 170.4, 154.7, 138.0, 134.4, 134.2, 117.2, 116.9, 114.79, 114.75, 81.0, 76.1, 72.0, 70.7, 70.6, 70.31, 70.27, 69.4, 65.9, 60.0, 52.8, 52.51, 52.45, 52.39, 52.2, 36.8, 35.3, 30.01, 29.99, 28.4, 28.2, 28.1. IR (film): 3428 (br), 2978, 2918, 1753, 1692 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd. for C22H36N2O7Na, 463.2420; found, 463.2418.

Boc-l-HypOAll-l-TyrOAll-OMe (4): to a solution of Boc-L-HypOAll-OH (S-1, 445 mg, 1.64 mmol) in CH2Cl2 (8 mL) were added EDCI·HCl (314 mg, 1.64 mmol) and HOBt·H2O (301 mg, 1.97 mmol) at 0 °C, and the reaction mixture was stirred for 30 min at 0 °C. Then, a solution of O-allyl-l-tyrosine methyl ester (H-l-TyrOAll-OMe, S-5 [52], 386 mg, 1.64 mmol) in CH2Cl2 (3 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring for 35 h, CH2Cl2 was removed in vacuo, and the residue was diluted with EtOAc. The resultant solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (50% EtOAc in n-hexane) to give 4 (562 mg, 70%) as a pale yellow oil. Rf = 0.75 (EtOAc). +0.90 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.10–6.98 (m, 2H), 6.87–6.70 (m, 3H), 6.10–5.99 (m, 1H), 5.90–5.77 (m, 1H), 5.40 (dp, J = 17.2, 1.7 Hz, 1H), 5.31–5.20 (m, 2H), 5.19–5.10 (m, 1H), 4.89–4.76 (m, 1H), 4.54–4.45 (m, 2H), 4.42–4.21 (m, 1H), 4.12–4.02 (m, 1H), 4.00–3.91 (m, 1H), 3.91–3.83 (m, 1H), 3.65 (s, 3H), 3.55 (br s, 2H), 3.13–2.99 (m, 1H), 2.94 (br s, 1H), 2.53–2.41 (m, 1H), 2.21–1.95 (m, 1H), 1.38 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 171.9, 171.6, 171.5, 171.1, 171.0, 157.6, 155.4, 154.5, 134.3, 134.1, 133.22, 133.18, 130.5, 130.2, 128.0, 127.8, 117.6, 117.5, 117.3, 117.2, 114.7, 114.6, 114.4, 81.0, 76.1, 72.0, 69.5, 68.69, 68.67, 65.9, 60.1, 59.3, 53.7, 53.3, 53.1, 52.9, 52.2, 52.0, 37.3, 37.2, 36.9, 36.8, 35.0, 32.5, 28.2, 28.0. IR (film): 3424 (br), 2978, 2934, 1744, 1665 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H36N2O7Na, 511.2420; found, 511.2422.

Boc-l-HypOAll-d-SerOAll-OMe (5): to a solution of Boc-d-Ser-OH (S-6, 2.05 g, 10.0 mmol) in DMF (35 mL) was added sodium hydride (60% in mineral oil, 880 mg, 22.0 mmol) portionwise at −15 °C, and the reaction mixture was stirred at the same temperature for 2 h. To the above suspension, allyl bromide (0.952 mL, 11.0 mmol) was added dropwise at −15 °C, and the reaction mixture was stirred at room temperature for 14 h. The reaction mixture was quenched by adding water and washed twice with Et2O. The aqueous phase was acidified with 1 M of HCl, which was extracted with EtOAc three times. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by flash column chromatography on silica gel (40% EtOAc in n-hexane) to give Boc-d-SerOAll-OH (S-7, 1.68 g, 69%, Rf = 0.28 with 10% MeOH in EtOAc) as a pale yellow oil. To a solution of S-7 (123 mg, 0.500 mmol) in MeOH (5 mL) was added thionyl chloride (0.145 mL, 2.00 mmol) dropwise at 0 °C. The reaction mixture was stirred at room temperature for 2 h and was concentrated under vacuum to give crude H-d-SerOAll-OMe·HCl (S-8, Rf = 0.57 with 0.5% AcOH in EtOAc), which was used for the next step without further purification. To a mixture of H-d-SerOAll-OMe·HCl (S-8, 0.500 mmol) and Boc-l-HypOAll-OH (S-1, 149 mg, 0.550 mmol) in CH2Cl2 (5 mL) were added EDCI·HCl (115 mg, 0.600 mmol), HOBt·H2O (91.9 mg, 0.600 mmol), and DIPEA (0.255 mL, 1.50 mmol) at 0 °C, and the reaction mixture was gradually warmed to room temperature. After stirring for 17 h, CH2Cl2 was removed under vacuum, and the residue was diluted with EtOAc. The organic solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (40% EtOAc in n-hexane) to give 5 (84.6 mg, 41% in 2 steps) as a pale yellow oil. Rf = 0.66 (EtOAc). –22.6 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.43–7.06 (m, 1H), 5.99–5.75 (m, 2H), 5.35–5.08 (m, 4H), 4.80–4.60 (m, 1H), 4.42–4.22 (m, 1H), 4.14–3.81 (m, 6H), 3.75 (s, 3H), 3.72–3.41 (m, 3H), 2.60–2.36 (m, 1H), 2.32–2.07 (m, 1H), 1.65–1.24 (m, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.6, 171.2, 170.7, 170.3, 154.6, 134.4, 134.2, 134.0, 133.9, 117.4, 117.3, 117.2, 116.9, 80.7, 75.8, 72.14, 72.12, 72.0, 69.61, 69.59, 69.4, 65.9, 60.3, 59.7, 53.2, 52.6, 52.52, 52.46, 52.3, 36.8, 35.2, 33.4, 28.2. IR (film): 3325 (br), 2978, 2932, 1753, 1692 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd. for C20H32N2O7Na, 435.2107; found, 435.2106.

Boc-l-HypOAll-l-ThrOAll-OMe (6): to a solution of N-tert-butoxycarbonyl O-allyl-l-threonine (Boc-l-ThrOAll-OH, S-9; 130 mg, 0.500 mmol) in MeOH (2.5 mL) was added thionyl chloride (0.144 mL, 2.00 mmol) dropwise at 0 °C. The reaction mixture was stirred at room temperature for 3 h prior to the addition of sat. NaHCO3 aq. After removal of MeOH by evaporation, the aqueous residue was extracted with CHCl3 (five times) and the combined organics were dried over anhydrous Na2SO4. Concentration of the solution gave H-L-ThrOAll-OMe (S-10, 41.9 mg, 48%), which was used for the next step without further purification. To a solution of Boc-l-HypOAll-OH (S-1, 72.1 mg, 0.266 mmol) in CH2Cl2 (0.8 mL) were added EDCI·HCl (51.0 mg, 0.266 mmol) and HOBt·H2O (48.2 mg, 0.315 mmol) at 0 °C, and the solution was stirred for 30 min at 0 °C. Then, a solution of H-l-ThrOAll-OMe (S-10, 41.9 mg, 0.242 mmol) in CH2Cl2 (0.8 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring for 42 h, CH2Cl2 was removed, and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (40% EtOAc in n-hexane) to give 6 (57.6 mg, 56%) as a pale yellow oil. Rf = 0.58 (EtOAc). –11.2 (c 1.00, CHCl3). 1H NMR (400 MHz, CDCl3) δ: 6.92 (s, 1H), 5.98–5.70 (m, 2H), 5.35–5.05 (m, 4H), 4.71–4.55 (m, 1H), 4.45–4.29 (m, 1H), 4.15–3.79 (m, 6H), 3.74 (s, 0.6H), 3.73 (s, 2.4H), 3.63–3.46 (m, 2H), 2.70–2.40 (m, 1H), 2.30–2.10 (m, 1H), 1.49 (s, 9H), 1.18 (d, J = 6.3 Hz, 0.6H), 1.13 (d, J = 6.4 Hz, 2.4H). 13C NMR (100 MHz, CDCl3) δ: 172.5, 171.8, 171.1, 170.6, 154.9, 134.5, 134.2, 117.2, 117.0, 116.8, 81.0, 76.0, 74.4, 74.2, 72.0, 69.8, 69.7, 66.0, 60.2, 56.4, 52.7, 52.2, 52.1, 36.9, 35.6, 28.1, 16.3, 16.1. IR (film): 3441 (br), 2978, 2934, 1753, 1703 cm−1. HRMS (DART) m/z: [M + H]+ calcd. for C21H35N2O7, 427.2444; found, 427.2437.

Boc-l-HypOAll-(S)-Ala(4-Pte)-OMe (7): to a solution of p-nitrobenzoic acid salt of (S)-(4-pentenyl)alanine tert-butyl ester (H-(S)-Ala(4-Pte)-OtBu·p-NO2C6H4CO2H, S-11; 100 mg, 0.263 mmol) in MeOH (3 mL) was added thionyl chloride (0.152 mL, 2.10 mmol) dropwise at 0 °C. The reaction mixture was stirred at 65 °C for 69 h prior to the addition of sat. NaHCO3 aq. After removal of MeOH by evaporation, the aqueous residue was extracted with CHCl3 (five times) and the combined organics were dried over anhydrous Na2SO4. Concentration of the solution gave H-(S)-Ala(4-Pte)-OMe (S-12) contaminated with p-NO2C6H4CO2Me, which was used for the next step without further purification. To a solution of Boc-l-HypOAll-OH (S-1, 60.5 mg, 0.223 mmol) in CH2Cl2 (1.5 mL) were added EDCI·HCl (42.8 mg, 0.223 mmol) and HOBt·H2O (40.4 mg, 0.264 mmol) at 0 °C, and the solution was stirred for 30 min at 0 °C. Then, a solution of H-(S)-Ala(4-Pte)-OMe (S-12) in CH2Cl2 (0.5 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring at room temperature for 3 d, CH2Cl2 was removed, and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (40% EtOAc in n-hexane) to give 7 (46.4 mg, 42% in 2 steps) as a pale yellow oil. Rf = 0.58 (EtOAc). –16.2 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.28–6.82 (m, 1H), 5.99–5.79 (m, 1H), 5.79–5.67 (m, 1H), 5.35–5.22 (m, 1H), 5.22–5.13 (m, 1H), 5.03–4.90 (m, 2H), 4.33–4.13 (m, 1H), 4.10–3.84 (m, 3H), 3.77–3.71 (m, 3H), 3.71–3.61 (m, 1H), 3.56–3.46 (m, 1H), 2.50–2.32 (m, 1H), 2.30–2.06 (m, 2H), 2.05–1.98 (m, 2H), 1.83–1.74 (m, 1H), 1.58 (s, 1H), 1.53 (s, 2H), 1.48 (s, 9H), 1.43–1.34 (m, 1H), 1.23–1.13 (m, 1H). 13C NMR (125 MHz, CDCl3) δ: 174.8, 174.4, 171.4, 170.6, 154.9, 138.11, 138.06, 134.4, 134.2, 117.31, 117.25, 114.9, 114.8, 80.8, 80.6, 76.1, 72.1, 69.7, 66.2, 60.7, 60.1, 59.7, 52.9, 52.6, 52.4, 37.6, 36.5, 36.1, 35.5, 33.5, 33.4, 28.2, 23.4, 23.1, 23.0, 22.6. IR (film): 3393 (br), 2978, 2936, 1740, 1692 cm−1. HRMS (DART) m/z: [M + H]+ calcd. for C22H37N2O6, 425.2652; found, 425.2651.
3.3. Synthesis of Stapled Dipeptides 1′–7′
Boc-l-HypOX-l-SerOX-OMe (1′; X = n-but-2-enyl tether): to a solution of unstapled peptide 1 [20] (20.5 mg, 0.0500 mmol) in degassed CH2Cl2 (10 mL) was added second-generation Grubbs catalyst (8.5 mg, 0.010 mmol) at room temperature under an argon atmosphere. The reaction mixture was stirred at the same temperature for 2 h and then passed through a short plug of amino silica gel/silica gel, which was eluted with EtOAc. After removal of the solvent, the residue was purified by flash column chromatography on silica gel (70% EtOAc in n-hexane) to give 1′ (14.6 mg, 76%) as a colorless oil. Rf = 0.32 (EtOAc). 1H NMR (500 MHz, CDCl3) δ: 7.23–7.00 (m, 1H), 5.86–5.73 (m, 1H), 5.68 (dt, J = 11.9, 6.5 Hz, 1H), 4.87–4.57 (m, 1H), 4.45–4.15 (m, 2H), 4.10–3.78 (m, 6H), 3.76 (s, 3H), 3.72–3.62 (m, 1H), 3.45 (dd, J = 12.1, 3.9 Hz, 1H), 2.66–2.47 (m, 1H), 2.26–2.11 (m, 1H), 1.60–1.39 (m, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.2, 171.4, 170.2, 154.9, 130.4, 130.0, 129.4, 81.0, 80.8, 78.6, 77.9, 77.3, 67.7, 67.2, 66.4, 66.0, 65.1, 60.2, 59.8, 54.0, 53.2, 52.5, 52.5, 35.7, 34.2, 30.9, 29.7, 28.2. HRMS (DART) m/z: [M + H]+ calcd. for C18H29N2O7, 385.1975; found, 385.1970.
Boc-l-HypOX-l-HseOX-OMe (2′; X = n-but-2-enyl tether): compound 2′ (15.0 mg, 75%) was obtained from compound 2 (21.3 mg, 0.0500 mmol) in a similar manner to that described for the synthesis of 1′. Colorless oil. Eluent for column: 70% EtOAc/n-hexane. Rf = 0.34 (EtOAc). 1H NMR (500 MHz, CDCl3) δ: 6.78 (s, 1/3H), 6.41 (s, 2/3H), 5.86 (ddd, J = 10.8, 8.6, 6.6 Hz, 1/3H), 5.75 (dt, J = 15.7, 5.9 Hz, 2/3H), 5.74–5.59 (m, 1H), 4.78–4.66 (m, 1H), 4.35 (d, J = 10.0 Hz, 1H), 4.24–3.95 (m, 3H), 3.89–3.74 (m, 2H), 3.73 (s, 1H), 3.72 (s, 2H), 3.69–3.58 (m, 1H), 3.57–3.36 (m, 3H), 2.28–1.99 (m, 2H), 1.87–1.64 (m, 2H), 1.49 (s, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.8, 172.2, 131.9, 131.7, 131.3, 128.7, 81.2, 75.9, 69.5, 68.5, 66.4, 63.5, 60.6, 60.2, 53.6, 52.3, 52.2, 49.4, 48.8, 34.7, 32.3, 28.2. HRMS (ESI) m/z: [M + Na]+ calcd. for C19H30N2O7Na, 421.1951; found, 421.1954.
Boc-l-HypOX-l-SerOX-OMe (3′; X = n-hex-2-enyl tether): compound 3′ (18.8 mg, 91%) was obtained from compound 3 (22.0 mg, 0.0500 mmol) in a manner similar to that described for the synthesis of 1′. Colorless oil. Eluent for column: 60% EtOAc/n-hexane. Rf = 0.42 (EtOAc). 1H NMR (500 MHz, CDCl3) δ: 7.85 (d, J = 8.1 Hz, 0.2H), 7.26–7.09 (m, 0.8H), 5.93–5.81 (m, 0.8H), 5.66 (dt, J = 14.9, 5.9 Hz, 0.1H), 5.60 (ddd, J = 9.1, 7.4, 6.2 Hz, 0.2H), 5.47 (dt, J = 15.3, 4.2 Hz, 0.7H), 5.42 (td, J = 10.1, 10.0, 5.1 Hz, 0.2H), 4.83–4.64 (m, 1H), 4.43–4.24 (m, 1H), 4.12 (t, J = 4.4 Hz, 0.2H), 4.04 (t, J = 3.8 Hz, 0.8H), 3.92–3.77 (m, 3H), 3.76 (s, 0.6H), 3.74 (s, 2.4H), 3.72–3.53 (m, 3H), 3.46–3.36 (m, 1H), 3.30 (td, J = 9.6, 3.4 Hz, 1H), 2.61–1.99 (m, 4H), 1.78–1.56 (m, 2H), 1.55–1.37 (m, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.7, 172.1, 170.8, 170.1, 155.2, 136.4, 133.8, 132.9, 131.9, 125.5, 124.3, 124.1, 80.9, 77.3, 72.7, 72.2, 70.7, 70.4, 70.3, 68.6, 68.1, 65.7, 60.5, 53.0, 52.8, 52.6, 52.4, 52.3, 36.4, 35.8, 32.3, 31.7, 28.6, 28.3, 28.1, 28.0, 27.9, 22.7. HRMS (ESI) m/z: [M + Na]+ calcd. for C20H32N2O7Na, 435.2107; found, 435.2117.
Boc-l-HypOX-l-TyrOX-OMe (4′; X = n-but-2-enyl tether): compound 4′ (5.4 mg, 23%) was obtained from compound 4 (24.4 mg, 0.0500 mmol) in a similar manner to that described for the synthesis of 1′. White solid. Eluent for column: 50% EtOAc/n-hexane. Rf = 0.61 (EtOAc). 1H NMR (500 MHz, CDCl3) δ: 7.17 (d, J = 8.5 Hz, 1H), 6.96–6.82 (m, 2H), 6.76 (s, 1H), 6.25 (s, 1H), 5.58 (dt, J = 15.1, 4.9 Hz, 1H), 5.48 (dt, J = 15.1, 6.6, 5.6 Hz, 1H), 4.94 (ddd, J = 10.9, 8.9, 4.5 Hz, 1H), 4.63 (d, J = 5.1 Hz, 2H), 4.18–3.82 (m, 3H), 3.79 (s, 3H), 3.77–3.54 (m, 2H), 3.37 (dd, J = 14.1, 4.5 Hz, 1H), 3.09 (s, 1H), 2.67 (t, J = 12.4 Hz, 1H), 2.39–1.82 (m, 2H), 1.46 (s, 9H). HRMS (ESI) m/z: [M + Na]+ calcd. for C24H32N2O7Na, 483.2107; found, 483.2105.
Boc-l-HypOX-d-SerOX-OMe (5′; X = n-but-2-enyl tether): compound 5′ (4.4 mg, 21%) was obtained from compound 2 (20.6 mg, 0.0500 mmol) in a similar manner to that described for the synthesis of 1′. Colorless oil. Eluent for column: 70% EtOAc/n-hexane. Rf = 0.39 (EtOAc). 1H NMR (500 MHz, CDCl3) δ: 7.41 (br s, 0.6H), 7.17 (br s, 0.4H), 5.95–5.85 (m, 1H), 5.85–5.66 (m, 1H), 4.55–4.18 (m, 4H), 4.14 (t, J = 4.1 Hz, 1H), 4.02–3.94 (m, 1H), 3.93–3.84 (m, 2H), 3.82 (m, 1.2H), 3.77 (s, 1.8H), 3.73–3.56 (m, 2H), 3.45 (dd, J = 12.2, 4.1 Hz, 1H), 2.62 (d, J = 15.2 Hz, 0.6H), 2.50 (d, J = 14.0 Hz, 0.4H), 2.29–2.12 (m, 1H), 1.45 (d, J = 8.9 Hz, 9H). HRMS (ESI) m/z: [M + Na]+ calcd. for C18H28N2O7Na, 407.1794; found, 407.1790.
Boc-l-HypOX-l-ThrOX-OMe (6′; X = n-but-2-enyl tether): compound 6′ (7.8 mg, 39%) was obtained from compound 6 (21.3 mg, 0.0500 mmol) in a similar manner to that described for the synthesis of 1′. Colorless oil. Eluent for column: 70% EtOAc/n-hexane. Rf = 0.32 (EtOAc). 1H NMR (500 MHz, CDCl3) δ: 7.02 (d, J = 8.2 Hz, 1H), 5.86 (dt, J = 11.5, 6.7 Hz, 1H), 5.78 (dt, J = 11.5, 6.2 Hz, 1H), 4.72–4.54 (m, 1H), 4.39–4.21 (m, 2H), 4.16 (dd, J = 11.8, 6.5 Hz, 1H), 4.12–4.06 (m, 1H), 4.01 (dd, J = 11.8, 6.2 Hz, 1H), 3.89–3.76 (m, 2H), 3.72 (s, 3H), 3.76–3.62 (m, 1H), 3.45 (dd, J = 12.0, 3.3 Hz, 1H), 2.51 (d, J = 14.8 Hz, 1H), 2.27–2.14 (m, 1H), 1.43 (s, 9H), 1.21 (d, J = 6.3 Hz, 3H). HRMS (ESI) m/z: [M + Na]+ calcd. for C19H30N2O7Na, 421.1951; found, 421.1958.
Boc-l-HypOX-(S)-Ala(EtX)-OMe (7′; X = n-but-2-enyl tether): compound 7′ (8.5 mg, 43%) was obtained from compound 7 (21.2 mg, 0.0500 mmol) in a similar manner to that described for the synthesis of 1′. Colorless oil. Eluent for column: 5% MeOH in CHCl3. Rf = 0.52 (10% MeOH in CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.52 (s, 1H), 5.90–5.77 (m, 1H), 5.65 (dt, J = 10.5, 7.3 Hz, 1H), 4.42–4.22 (m, 1H), 4.15–3.92 (m, 2H), 3.74 (s, 3H), 3.70–3.43 (m, 3H), 2.83–2.64 (m, 1H), 2.62–2.48 (m, 1H), 2.22–1.86 (m, 4H), 1.64 (s, 3H), 1.52–1.43 (m, 9H), 1.51–1.43 (m, 1H), 1.22–1.12 (m, 1H). HRMS (ESI) m/z: [M + Na]+ calcd. for C20H32N2O6Na, 419.2158; found, 419.2166.
3.4. Synthesis of Stapled Octapeptides 9 and 10
Boc-l-HypOX-l-SerOX-[(l-Leu)2-Ac5c]2-OMe (9; X = n-but-2-enyl tether): compound 9 (10.2 mg, 52%) was obtained from compound 8 [19] (20.0 mg, 0.0184 mmol) in a similar manner to that described for the synthesis of 1′. Eluent for column: 80% EtOAc/n-hexane. White amorphous. Rf = 0.26 (EtOAc). 1H NMR (500 MHz, CDCl3) δ: 7.73 (d, J = 2.3 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.37 (d, J = 4.8 Hz, 1H), 7.27–7.24 (m, 1H), 7.24–7.18 (m, 3H), 6.08 (dd, J = 10.9, 6.6 Hz, 1H), 6.04 (dd, J = 10.9, 5.8 Hz, 1H), 4.46 (ddd, J = 11.7, 5.3, 2.3 Hz, 1H), 4.39–4.30 (m, 2H), 4.26–4.15 (m, 5H), 4.01 (dd, J = 11.4, 5.3 Hz, 1H), 3.96–3.88 (m, 2H), 3.74 (dd, J = 10.0, 5.3 Hz, 1H), 3.71–3.68 (m, 1H), 3.67 (s, 3H), 3.66–3.60 (m, 1H), 3.46 (dd, J = 12.1, 3.4 Hz, 1H), 2.66 (dt, J = 13.5, 8.1 Hz, 1H), 2.45–2.30 (m, 2H), 2.26 (ddd, J = 13.6, 8.5, 6.7 Hz, 1H), 2.22–2.10 (m, 3H), 2.10–2.02 (m, 1H), 1.97–1.55 (m, 28H), 1.49 (s, 9H), 1.00–0.83 (m, 24H). HRMS (ESI) m/z: [M + Na]+ calcd. for C54H90N8O13Na, 1081.6525; found, 1081.6536.
Boc-l-HypOX-l-SerOX-[(l-Leu)2-Ac5c]2-OMe (10; X = n-butyl tether): to a solution of peptide 9 (10.2 mg, 0.00963 mmol) in MeOH (2 mL) was added 10% Pd/C (10 mg) at room temperature and the reaction mixture was stirred at room temperature overnight. The resultant dark suspension was filtered through a short plug of celite (MeOH), and the organics were concentrated under vacuum. The crude material was purified by preparative TLC (EtOAc) to give 10 (8.5 mg, 83%) as white amorphous. Rf = 0.31 (EtOAc). 1H NMR (500 MHz, CDCl3) δ: 7.70 (s, 1H), 7.46 (d, J = 5.0 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.26–7.24 (m, 2H), 7.23 (d, J = 5.6 Hz, 2H), 4.35 (ddd, J = 11.4, 8.1, 3.0 Hz, 1H), 4.28 (ddd, J = 11.0, 4.8, 1.5 Hz, 1H), 4.24–4.16 (m, 2H), 4.14 (d, J = 10.9 Hz, 1H), 4.04 (t, J = 3.5 Hz, 1H), 3.98 (dd, J = 11.1, 5.1 Hz, 1H), 3.93 (dt, J = 9.6, 4.5 Hz, 1H), 3.83 (dd, J = 11.9, 2.2 Hz, 1H), 3.70 (dd, J = 9.3, 1.6 Hz, 1H), 3.67 (s, 3H), 3.63 (dd, J = 9.3, 2.7 Hz, 1H), 3.58 (dt, J = 9.3, 3.2 Hz, 1H), 3.54 (t, J = 11.2 Hz, 1H), 3.44–3.36 (m, 2H), 2.65 (dt, J = 13.6, 8.3 Hz, 1H), 2.38 (ddd, J = 15.1, 11.1, 4.3 Hz, 1H), 2.27 (dd, J = 13.8, 7.5 Hz, 1H), 2.24–2.03 (m, 5H), 1.96–1.66 (m, 22H), 1.66–1.57 (m, 4H), 1.52 (s, 9H), 0.99–0.93 (m, 9H), 0.92–0.85 (m, 15H). X-ray crystallographic data and CIF file of compound 10 are provided in the Supplementary Materials.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ijms22105364/s1: 1H and 13C NMR spectra of compounds 2–7, 1′–7′, 9, and 10; X-ray crystallographic data of compound 10, and CIF file of compound 10.
Author Contributions
Conceptualization, A.U. and M.T.; methodology, A.U. and M.T.; validation, Y.M., A.U. and T.K.; formal analysis, Y.M., A.U., T.K., A.I., M.H., M.D. and M.T.; investigation, Y.M., A.U., T.K., A.I. and M.H.; writing—original draft preparation, A.U. and M.T.; writing—review and editing, Y.M., A.U., T.K., A.I., M.H., M.D. and M.T.; visualization, Y.M., A.U. and T.K.; supervision, M.D. and M.T.; project administration, A.U.; funding acquisition, A.U. and M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI Grant Numbers JP17H03998 (M.T.), JP18K14870 (A.U.), and JP20K06967 (A.U.), the Ube Industries Foundation (A.U.), and Shionogi Award in Synthetic Organic Chemistry, Japan (A.U.).
Institutional Review Board Statement
The study did not involve humans or animals.
Informed Consent Statement
The study did not involve humans.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Y.M. is grateful for a fellowship from the Tokyo Biochemical Research Foundation. This work was the result of using research equipment shared in MEXT Project for promoting the public utilization of advanced research infrastructure (program for supporting introduction of the new sharing system), grant number JPMXS0422500320.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moretto, A.; Crisma, M.; Formaggio, F.; Toniolo, C. Building a bridge between peptide chemistry and organic chemistry: Intramolecular macrocyclization reactions and supramolecular chemistry with helical peptide substrates. Biopolymers 2010, 94, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Verdine, G.L.; Hilinski, G.J. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012, 503, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Cromm, P.M.; Spiegel, J.; Grossmann, T.N. Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol. 2015, 10, 1362–1375. [Google Scholar] [CrossRef] [PubMed]
- Moiola, M.; Memeo, M.G.; Quadrelli, P. Stapled Peptides—A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24, 3654. [Google Scholar] [CrossRef] [PubMed]
- Bozovičar, K.; Bratkovič, T. Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties. Int. J. Mol. Sci. 2021, 22, 1611. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, H.E.; Grubbs, R.H. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew. Chem. Int. Ed. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Blackwell, H.E.; Sadowsky, J.D.; Howard, R.J.; Sampson, J.N.; Chao, J.A.; Steinmetz, W.E.; O’Leary, D.J.; Grubbs, R.H. Ring-closing metathesis of olefinic peptides: Design, synthesis, and structural characterization of macrocyclic helical peptides. J. Org. Chem. 2001, 66, 5291–5302. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Sawyer, T.K.; Partridge, A.W.; Kaan, H.Y.K.; Juang, Y.C.; Lim, S.; Johannes, C.; Yuen, T.Y.; Verma, C.; Kannan, S.; Aronica, P.; et al. Macrocyclic alpha helical peptide therapeutic modality: A perspective of learnings and challenges. Bioorg. Med. Chem. 2018, 26, 2807–2815. [Google Scholar] [CrossRef]
- Ali, A.M.; Atmaj, J.; Oosterwijk, N.V.; Groves, M.R.; Dömling, A. Stapled peptides inhibitors: A new window for target drug discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Saito, C.; Yokoo, H.; Goto, C.; Kawano, R.; Misawa, T.; Demizu, Y. Development of Antimicrobial Stapled Peptides Based on Magainin 2 Sequence. Molecules 2021, 26, 444. [Google Scholar] [CrossRef] [PubMed]
- Jochim, A.L.; Arora, P.S. Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors. ACS Chem. Biol. 2010, 5, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Boal, A.K.; Guryanov, I.; Moretto, A.; Crisma, M.; Lanni, E.L.; Toniolo, C.; Grubbs, R.H.; O’Leary, D.J. Facile and E-selective intramolecular ring-closing metathesis reactions in 310-helical peptides: A 3D structural study. J. Am. Chem. Soc. 2007, 129, 6986–6987. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-W.; Kutchukian, P.S.; Verdine, G.L. Introduction of all-hydrocarbon i,i+3 staples into α-helices via ring-closing olefin metathesis. Org. Lett. 2010, 12, 3046–3049. [Google Scholar] [CrossRef] [PubMed]
- Mangold, S.L.; O’Leary, D.J.; Grubbs, R.H. Z-Selective olefin metathesis on peptides: Investigation of side-chain influence, preorganization, and guidelines in substrate selection. J. Am. Chem. Soc. 2014, 136, 12469–12478. [Google Scholar] [CrossRef]
- Mangold, S.L.; Grubbs, R.H. Stereoselective synthesis of macrocyclic peptides via a dual olefin metathesis and ethenolysis approach. Chem. Sci. 2015, 6, 4561–4569. [Google Scholar] [CrossRef]
- Creighton, C.J.; Reitz, A.B. Synthesis of an eight-membered cyclic pseudo-dipeptide using ring closing metathesis. Org. Lett. 2001, 3, 893–895. [Google Scholar] [CrossRef]
- Islam, N.M.; Islam, M.S.; Hoque, M.A.; Kato, T.; Nishino, N.; Ito, A.; Yoshida, M. Bicyclic tetrapeptides as potent HDAC inhibitors: Effect of aliphatic loop position and hydrophobicity on inhibitory activity. Bioorg. Med. Chem. 2014, 22, 3862–3870. [Google Scholar] [CrossRef]
- Ueda, A.; Higuchi, M.; Sato, K.; Umeno, T.; Tanaka, M. Design and synthesis of helical N-terminal L-prolyl oligopeptides possessing hydrocarbon stapling. Molecules 2020, 25, 4667. [Google Scholar] [CrossRef]
- Hill, T.A.; Shepherd, N.E.; Diness, F.; Fairlie, D.P. Constraining cyclic peptides to mimic protein structure motifs. Angew. Chem. Int. Ed. 2014, 53, 13020–13041. [Google Scholar] [CrossRef]
- Paul, P.K.C.; Sukumar, M.; Bardi, R.; Piazzesi, A.M.; Valle, G.; Toniolo, C.; BaIaram, P. Stereochemically constrained peptides. Theoretical and experimental studies on the conformations of peptides containing 1-aminocyclohexanecarboxylic acid. J. Am. Chem. Soc. 1986, 108, 6363–6370. [Google Scholar] [CrossRef]
- Royo, S.; de Borggraeve, W.M.; Peggion, C.; Formaggio, F.; Crisma, M.; Jimeéez, A.I.; Cativiela, C.; Toniolo, C. Turn and helical peptide handedness governed exclusively by side-chain chiral centers. J. Am. Chem. Soc. 2005, 127, 2036–2037. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Demizu, Y.; Doi, M.; Kurihara, M.; Suemune, H. Chiral centers in the side chains of α-amino acids control the helical screw sense of peptides. Angew. Chem. Int. Ed. 2004, 43, 5360–5363. [Google Scholar] [CrossRef] [PubMed]
- Nagano, M.; Tanaka, M.; Doi, M.; Demizu, Y.; Kurihara, M.; Suemune, H. Helical-screw directions of diastereoisomeric cyclic α-amino acid oligomers. Org. Lett. 2009, 11, 1135–1137. [Google Scholar] [CrossRef]
- Oba, M.; Ishikawa, N.; Demizu, Y.; Kurihara, M.; Suemune, H.; Tanaka, M. Helical oligomers with a changeable chiral acetal moiety. Eur. J. Org. Chem. 2013, 7679–7682. [Google Scholar] [CrossRef]
- Hirata, T.; Ueda, A.; Oba, M.; Doi, M.; Demizu, Y.; Kurihara, M.; Nagano, M.; Suemune, H.; Tanaka, M. Amino equatorial effect of a six-membered ring amino acid on its peptide 310- and α-helices. Tetrahedron 2015, 71, 2409–2420. [Google Scholar] [CrossRef]
- Crisma, M.; Toniolo, C. Helical screw-sense preferences of peptides based on chiral, Cα-tetrasubstituted α-amino acids. Biopolymers 2015, 104, 46. [Google Scholar] [CrossRef]
- Koba, Y.; Hirata, Y.; Ueda, A.; Oba, M.; Doi, M.; Demizu, Y.; Kurihara, M.; Tanaka, M. Synthesis of chiral five-membered carbocyclic ring amino acids with an acetal moiety and helical conformations of its homo-chiral homopeptides. Biopolymers 2016, 106, 555–562. [Google Scholar] [CrossRef]
- Eto, R.; Oba, M.; Ueda, A.; Uku, T.; Doi, M.; Matsuo, Y.; Tanaka, T.; Demizu, Y.; Kurihara, M.; Tanaka, M. Diastereomeric right- and left-handed helical structures with fourteen (R)-chiral centers. Chem. Eur. J. 2017, 23, 18120–18124. [Google Scholar] [CrossRef]
- Koba, Y.; Ueda, A.; Oba, M.; Doi, M.; Kato, T.; Demizu, Y.; Tanaka, M. Left-handed helix of three-membered ring amino acid homopeptide interrupted by an N–H·ethereal O-type hydrogen bond. Org. Lett. 2018, 20, 7830–7834. [Google Scholar] [CrossRef]
- Demizu, Y.; Tanaka, M.; Nagano, M.; Kurihara, M.; Doi, M.; Maruyama, T.; Suemune, H. Controlling 310-helix and α-helix of short peptides in the solid state. Chem. Pharm. Bull. 2007, 55, 840–842. [Google Scholar] [CrossRef][Green Version]
- Umeno, T.; Ueda, A.; Oba, M.; Doi, M.; Hirata, T.; Suemune, H.; Tanaka, M. Helical structures of L-Leu-based peptides having chiral six-membered ring amino acids. Tetrahedron 2016, 72, 3124–3131. [Google Scholar] [CrossRef]
- Koba, Y.; Ueda, A.; Oba, M.; Doi, M.; Demizu, Y.; Kurihara, M.; Tanaka, M. Helical L-Leu-based peptides having chiral five-membered carbocyclic ring amino acids with an ethylene acetal moiety. Chem. Select. 2017, 2, 8108–8114. [Google Scholar] [CrossRef]
- The Cambridge Crystallographic Data Centre (CCDC) Home Page. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 18 May 2021).
- Tanaka, M. Design and synthesis of chiral α,α-disubstituted amino acids and conformational study of their oligopeptides. Chem. Pharm. Bull. 2007, 55, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Wolf, W.M.; Stasiak, M.; Leplawy, M.T.; Bianco, A.; Formaggio, F.; Crisma, M.; Toniolo, C. Destabilization of the 310-helix in peptides based on Cα-tetrasubstituted α-amino acids by main-chain to side-chain hydrogen bonds. J. Am. Chem. Soc. 1998, 120, 11558–11566. [Google Scholar] [CrossRef]
- Tanda, K.; Eto, R.; Kato, K.; Oba, M.; Ueda, A.; Suemune, H.; Doi, M.; Demizu, Y.; Kurihara, M.; Tanaka, M. Peptide foldamers composed of six-membered ring α, α-disubstituted α-amino acids with two changeable chiral acetal moieties. Tetrahedron 2015, 71, 3909–3914. [Google Scholar] [CrossRef]
- Ueda, A.; Umeno, T.; Doi, M.; Akagawa, K.; Kudo, K.; Tanaka, M. Helical-peptide-catalyzed enantioselective Michael addition reactions and their mechanistic insights. J. Org. Chem. 2016, 81, 5864–5871. [Google Scholar] [CrossRef]
- Ueda, A.; Higuchi, M.; Umeno, T.; Tanaka, M. Enantioselective synthesis of 2,4,5-trisubstituted tetrahydropyrans via peptide-catalyzed Michael addition followed by Kishi’s reductive cyclization. Heterocycles 2019, 99, 989–1002. [Google Scholar] [CrossRef]
- Umeno, T.; Ueda, A.; Doi, M.; Kato, T.; Oba, M.; Tanaka, M. Helical foldamer-catalyzed enantioselective 1,4-addition reaction of dialkyl malonates to cyclic enones. Tetrahedron Lett. 2019, 60, 151301. [Google Scholar] [CrossRef]
- Ueda, A.; Ikeda, M.; Kasae, T.; Doi, M.; Demizu, Y.; Oba, M.; Tanaka, M. Synthesis of chiral α-trifluoromethyl α, α-disubstituted α-amino acids and conformational analysis of L-Leu-based peptides with (R)- or (S)-α-trifluoromethylalanine. Chem. Select. 2020, 5, 10882–10886. [Google Scholar] [CrossRef]
- Oba, M. Cell-penetrating peptide foldamers: Drug-delivery tools. ChemBioChem 2019, 20, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Kunitake, M.; Kato, T.; Ueda, A.; Tanaka, M. Enhanced and prolonged cell-penetrating abilities of arginine-rich peptides by introducing cyclic α, α-disubstituted α-amino acids with stapling. Bioconj. Chem. 2017, 28, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Oba, M.; Nishida, K.; Tanaka, M. Cell-penetrating peptides using cyclic α, α-disubstituted α-amino acids with basic functional groups. ACS Biomater. Sci. Eng. 2018, 4, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Tanaka, M.; Oba, M. siRNA delivery using amphipathic cell-penetrating peptides into human hepatoma cells. Bioorg. Med. Chem. 2020, 28, 115402. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kita, Y.; Iwanari, K.; Asano, A.; Oba, M.; Tanaka, M.; Doi, M. Synthesis of six-membered carbocyclic ring α, α-disubstituted amino acids and arginine-rich peptides to investigate the effect of ring size on the properties of the peptide. Bioorg. Med. Chem. 2021, 38, 116111. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Bacher, M.; Buenemann, C.L.; Kricek, F.; Rondeau, J.-M.; Weigand, K. Conformationally constrained mimics of the membrane-proximal domain of FcεRIα. ChemBioChem 2007, 8, 1785–1789. [Google Scholar] [CrossRef]
- Bortolini, O.; Cavazzini, A.; Giovannini, P.P.; Greco, R.; Marchetti, N.; Massi, A.; Pasti, L. A combined kinetic and thermodynamic approach for the interpretation of continuous-flow heterogeneous catalytic processes. Chem. Eur. J. 2013, 19, 7802–7808. [Google Scholar] [CrossRef]
- Yamagata, N.; Demizu, Y.; Sato, Y.; Doi, M.; Tanaka, M.; Nagasawa, K.; Okuda, H.; Kurihara, M. Design of a stabilized short helical peptide and its application to catalytic enantioselective epoxidation of (E)-chalcone. Tetrahedron Lett. 2011, 52, 798–801. [Google Scholar] [CrossRef]
- Demizu, Y.; Yamagata, N.; Nagoya, S.; Sato, Y.; Doi, M.; Tanaka, M.; Nagasawa, K.; Okuda, H.; Kurihara, M. Enantioselective epoxidation of α, β-unsaturated ketones catalyzed by stapled helical L-Leu-based peptides. Tetrahedron 2011, 67, 6155–6165. [Google Scholar] [CrossRef]
- Tang, H.; Yin, L.; Lu, H.; Cheng, J. Water-soluble poly(L-serine)s with elongated and charged side- chains: Synthesis, conformations, and cell-penetrating properties. Biomacromolecules 2012, 13, 2609–2615. [Google Scholar] [CrossRef]
- Bayardon, J.; Sinou, D. Synthesis of two new chiral fluorous bis(oxazolines) and their applications as ligands in catalytic asymmetric reactions. Tetrahedron Asymmetry 2005, 16, 2965–2972. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).