
Rapamycin Ameliorates Defects in Mitochondrial Fission and Mitophagy in Glioblastoma Cells

 ,
,  , , , ,
, , , , 
Abstract
1. Introduction
2. Results
2.1. Dose–Response and Time-Course of Rapamycin-Induced Mitochondrial Number and Alterations
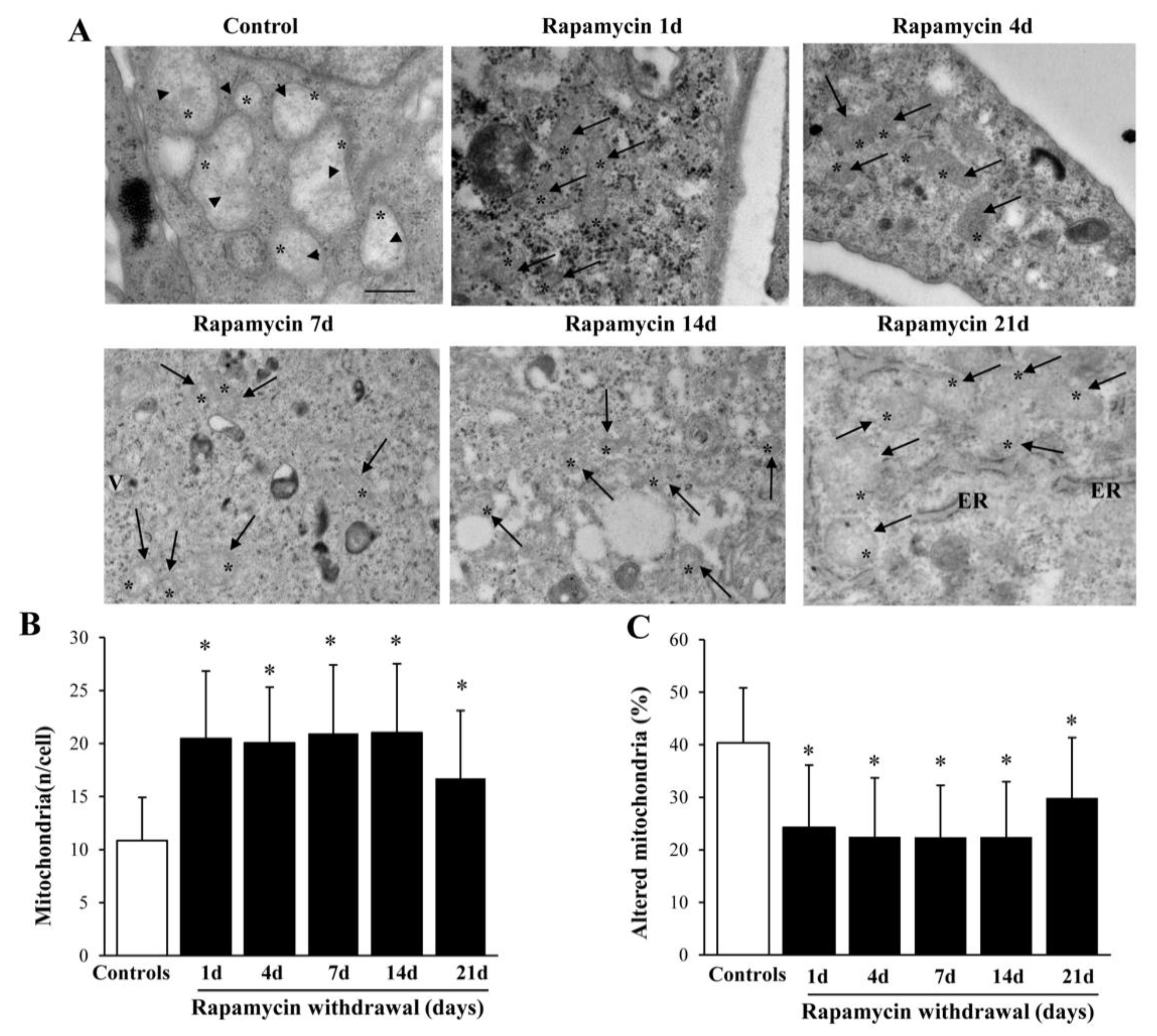
2.2. Long-Lasting Increase of Mitochondria and Clearance of Altered Mitochondria
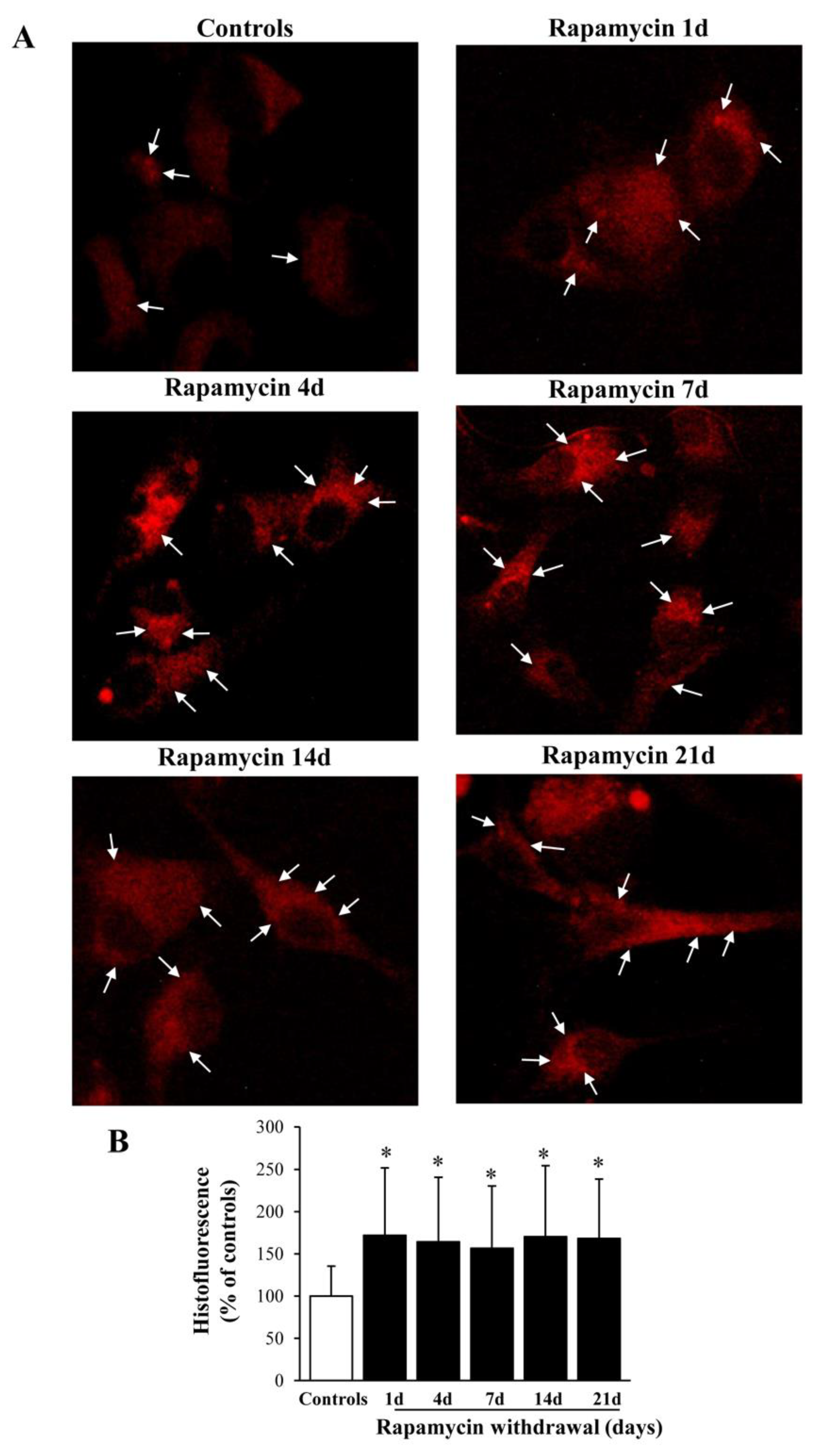
2.3. Rapamycin Increases the Histofluorescence from Healthy Mitochondria
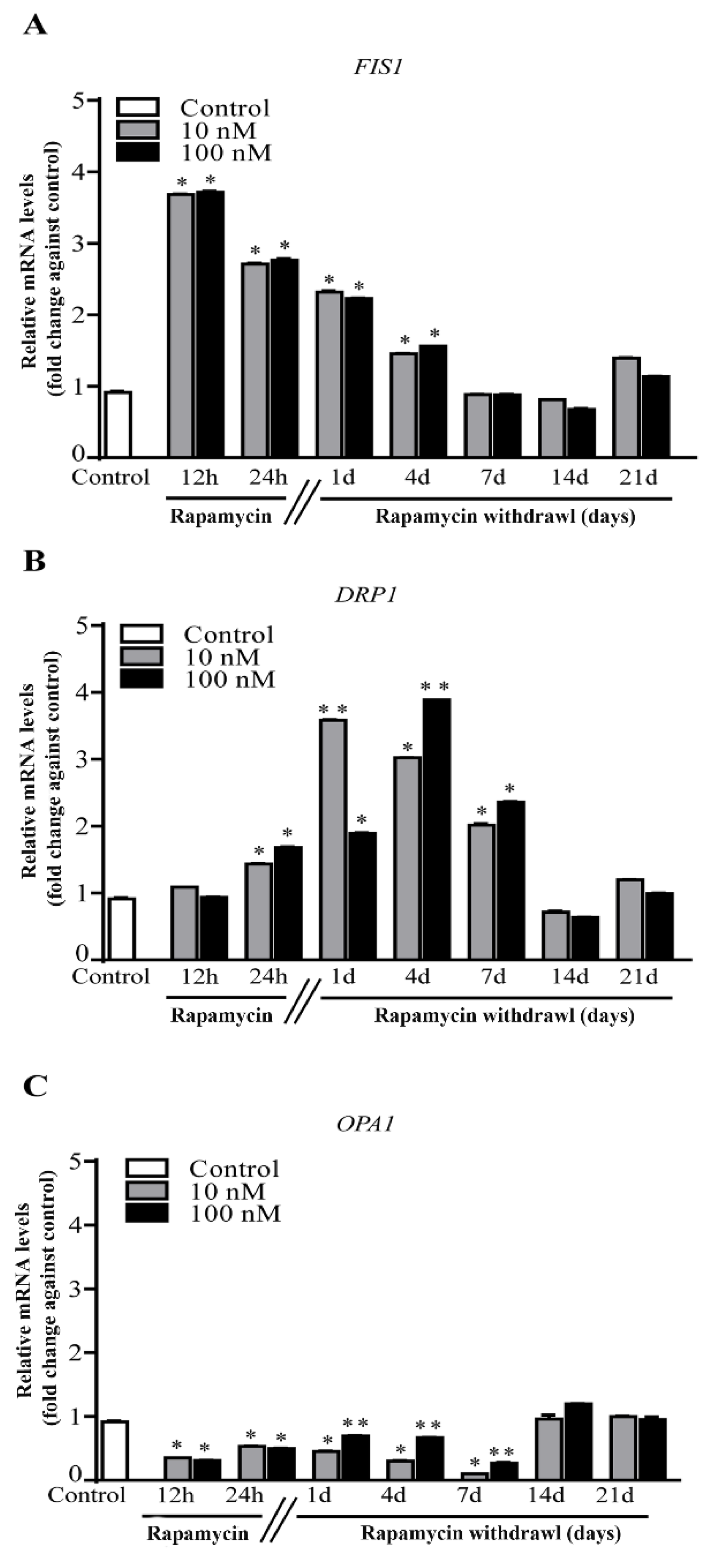
2.4. Rapamycin Increases Mitochondrial Fission Genes FIS1 and DRP1, While Decreases Fusion Gene OPA1
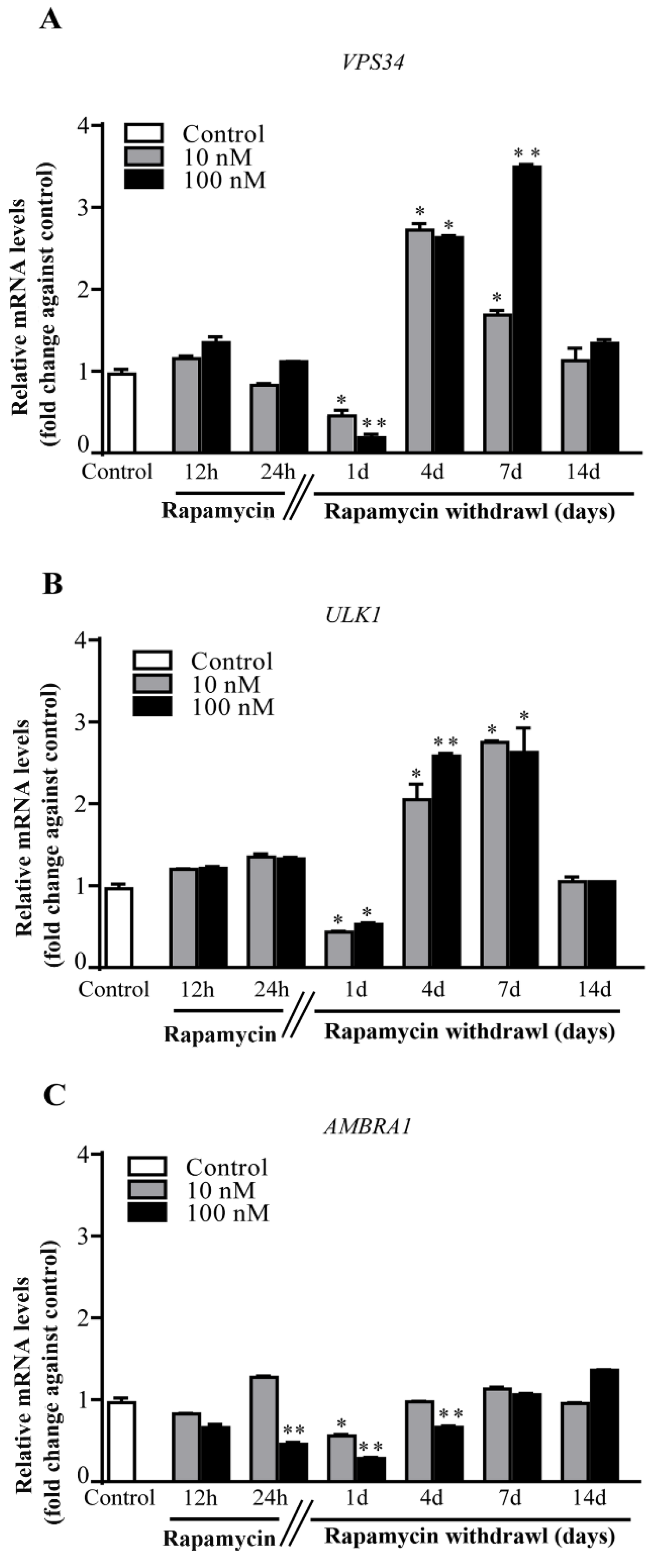
2.5. Rapamycin Differently Alters the Autophagy Genes VPS34, ULK1 and AMBRA1
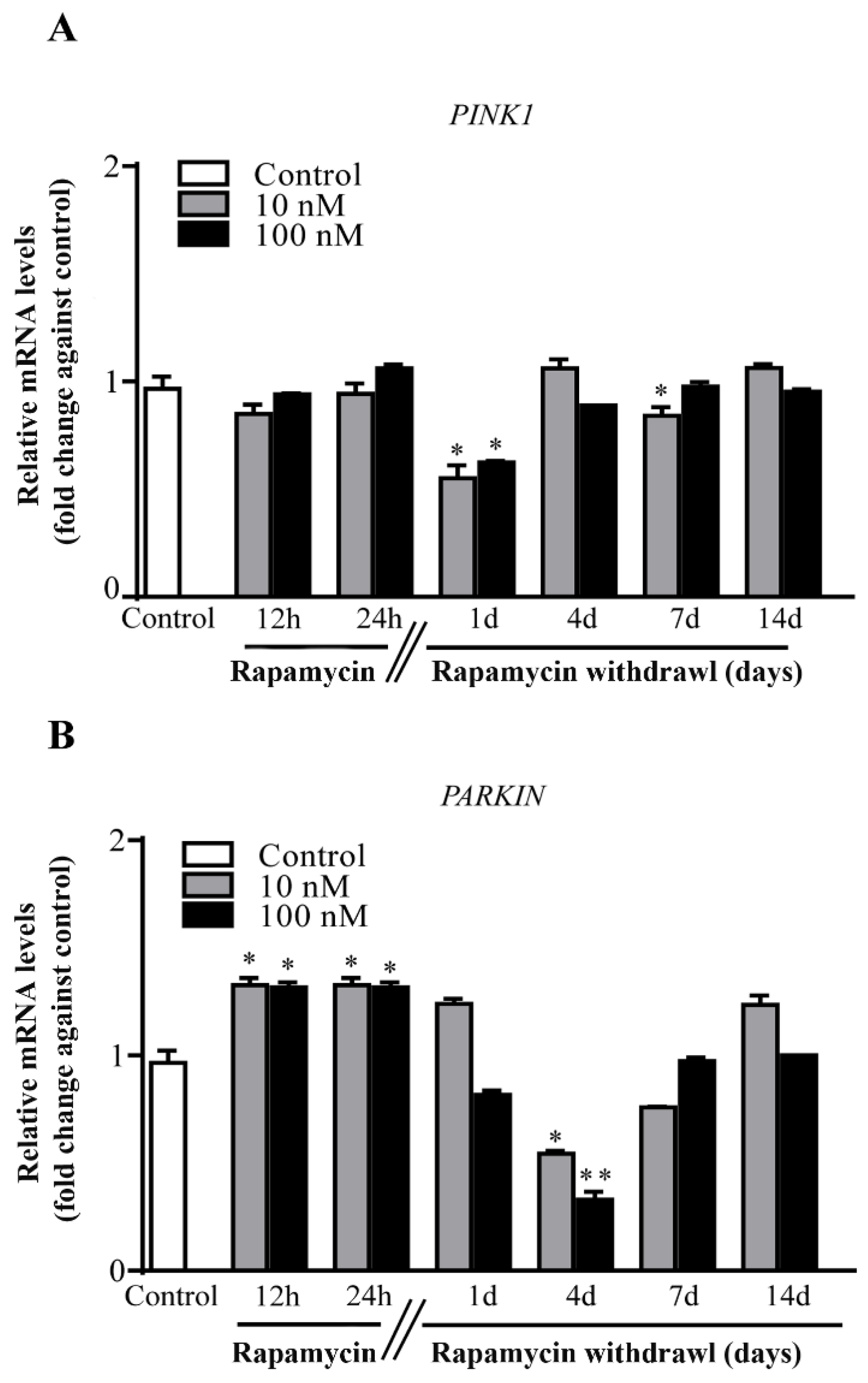
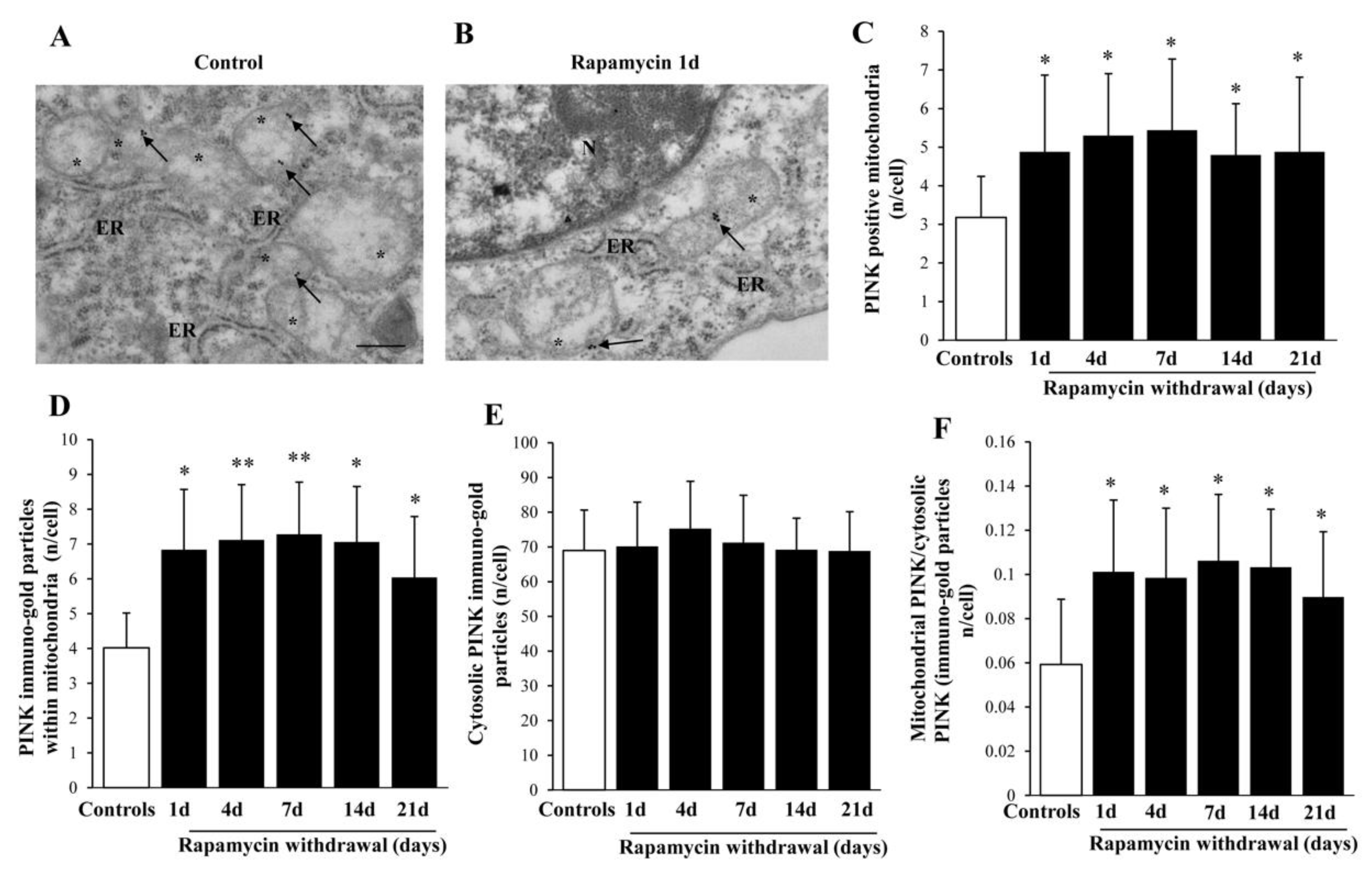
2.6. Rapamycin Withdrawal Modulates PINK1 and PARKIN Genes
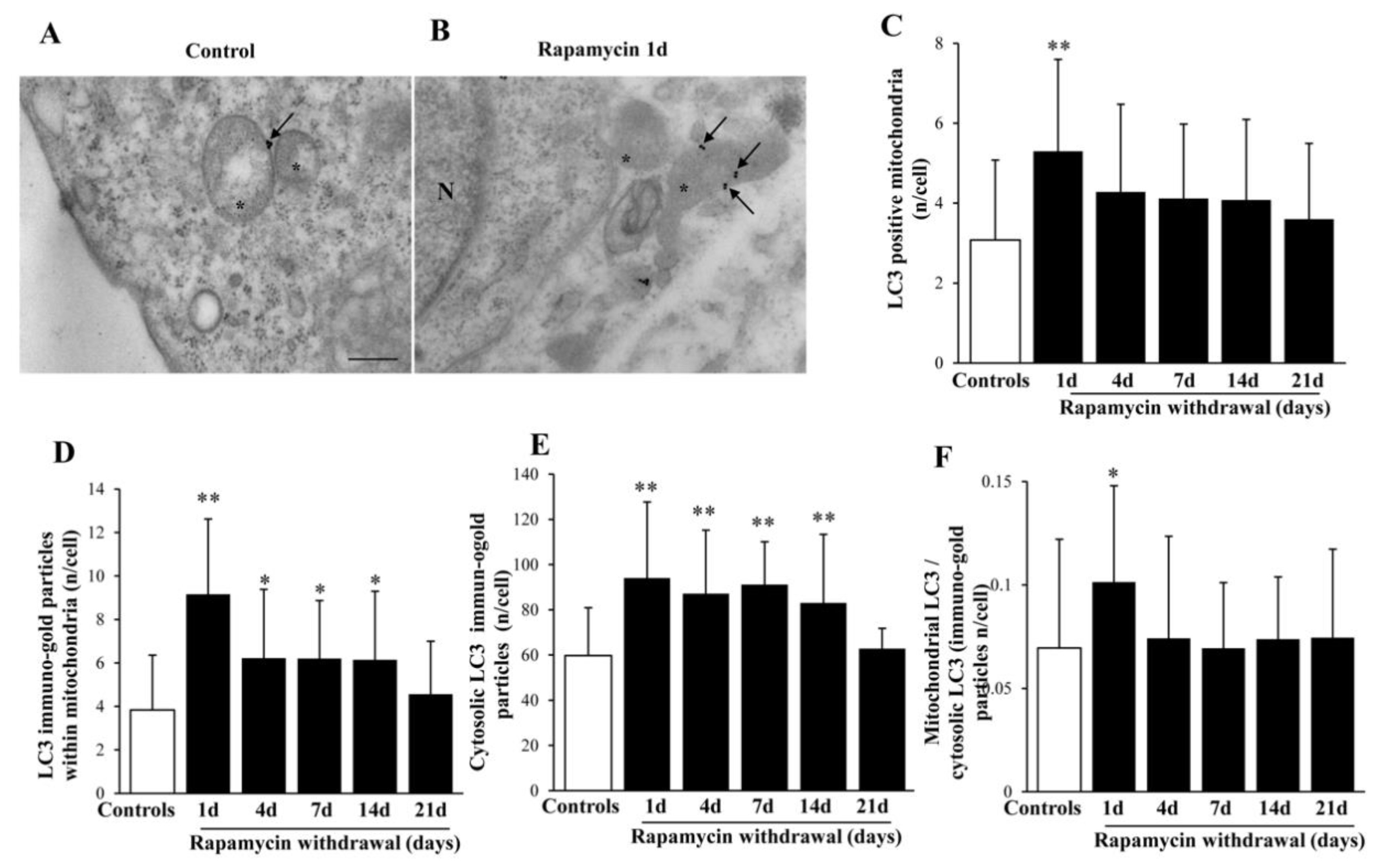
2.7. Rapamycin Polarizes Increased LC3 within Mitochondria
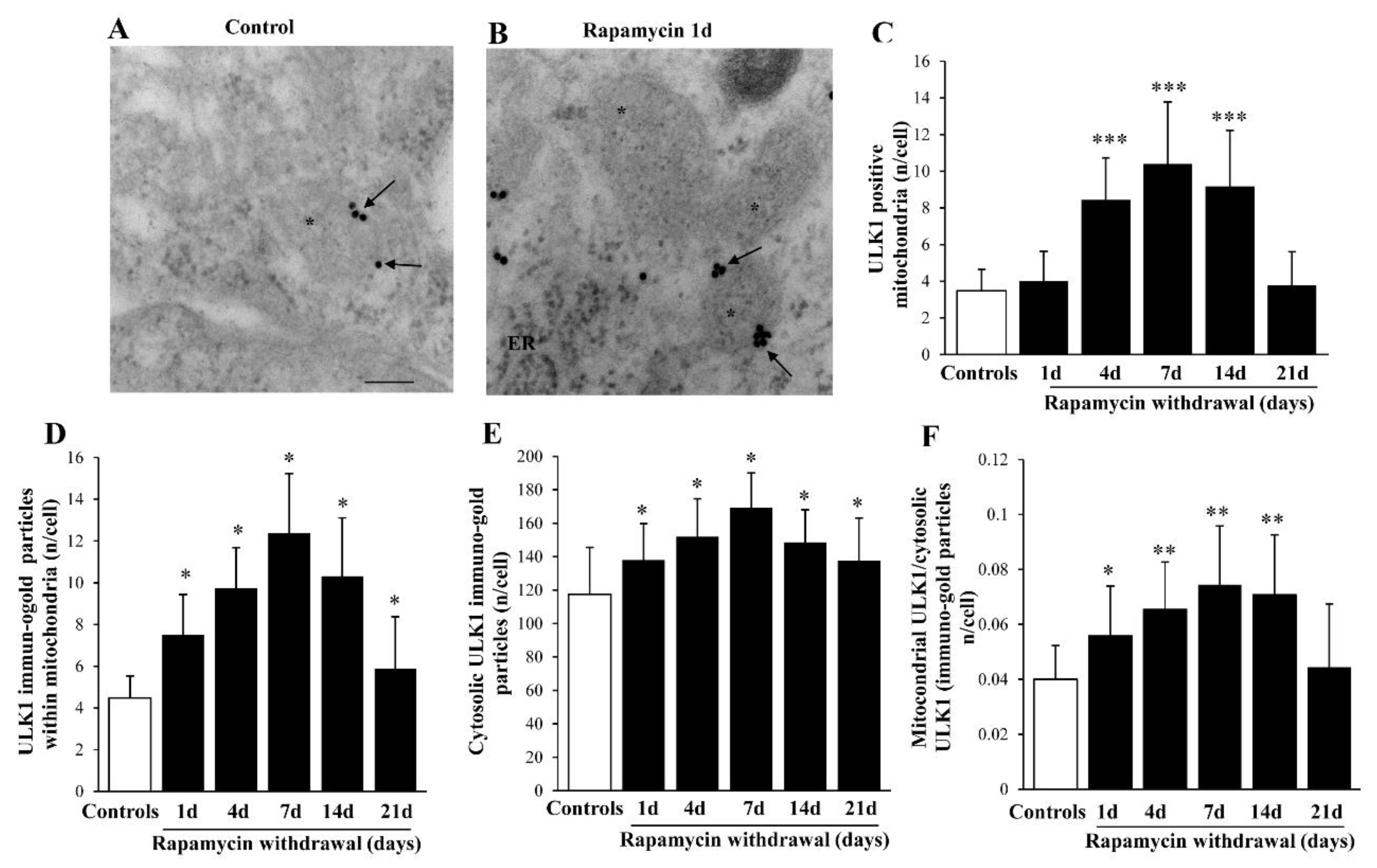
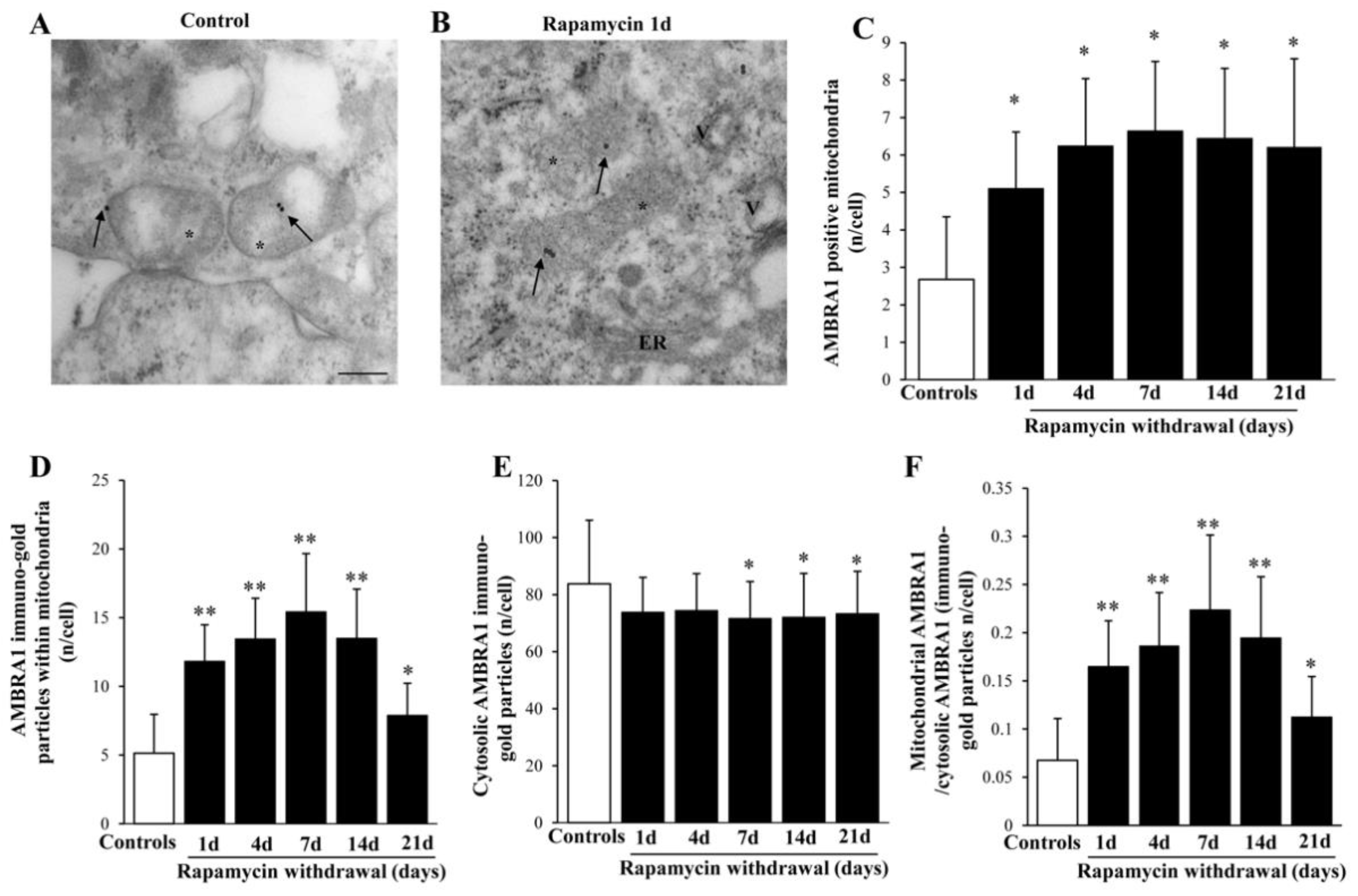
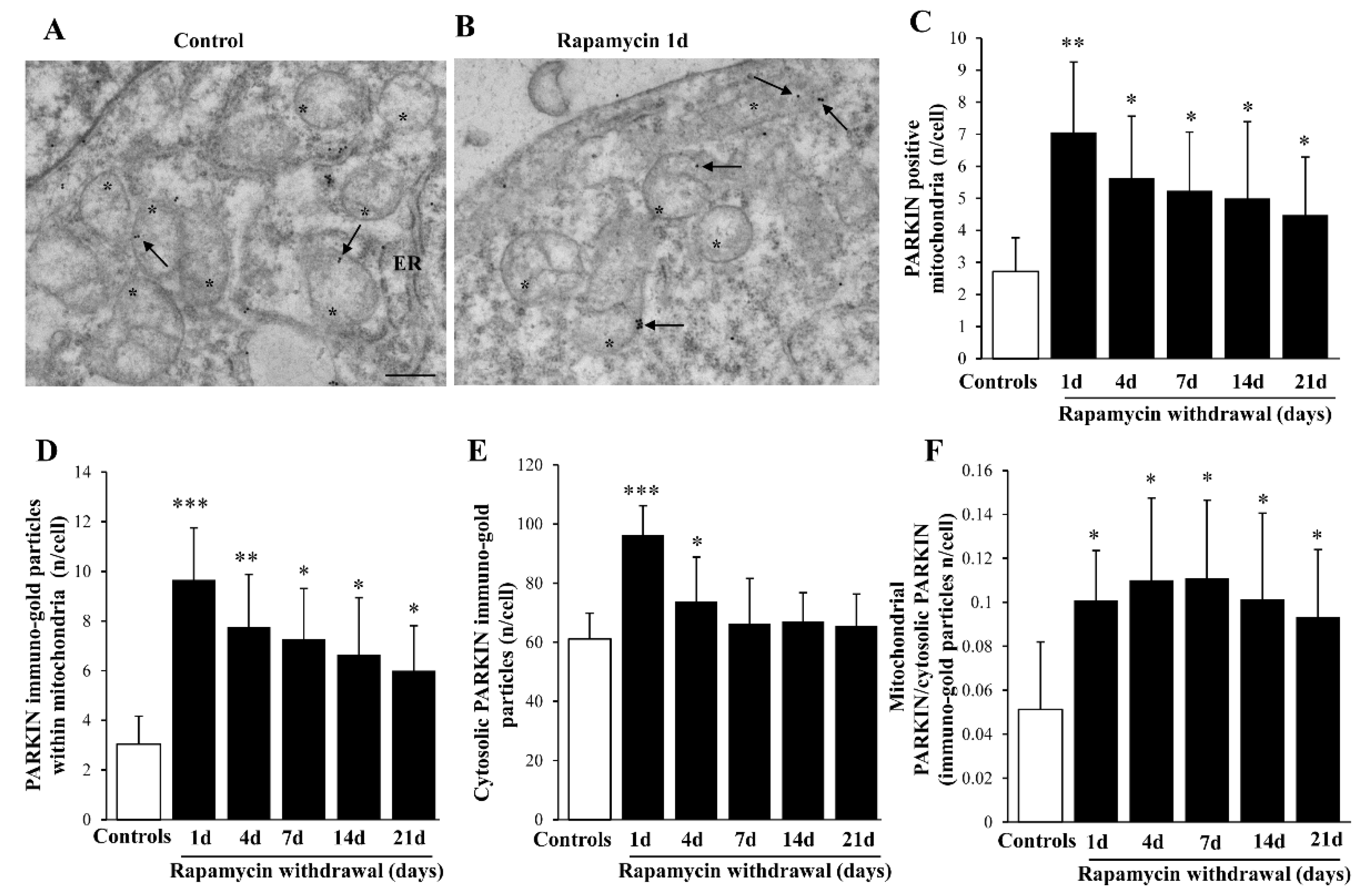
2.8. Rapamycin Increases Mitophagy-Related Proteins within Mitochondria
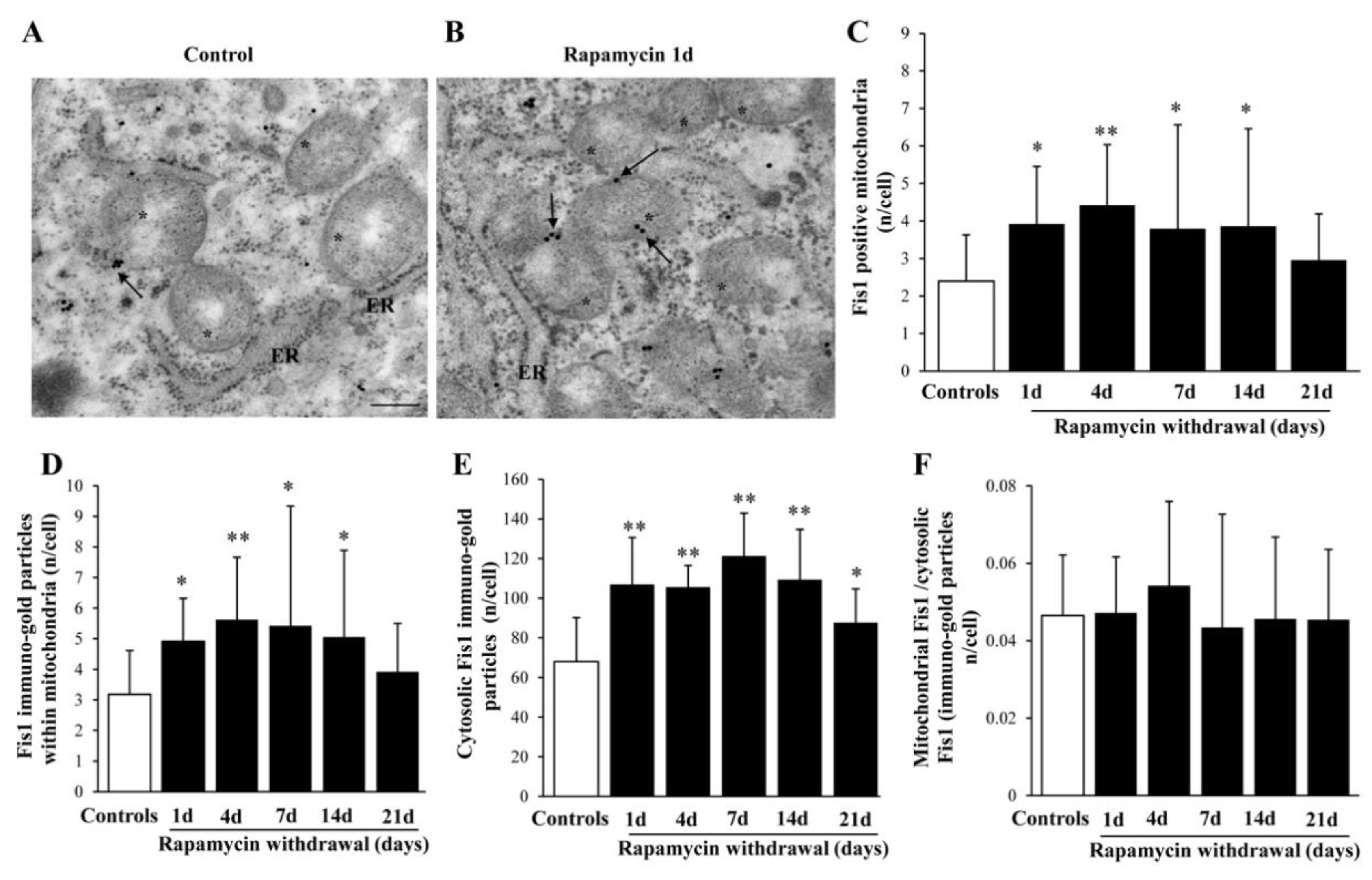
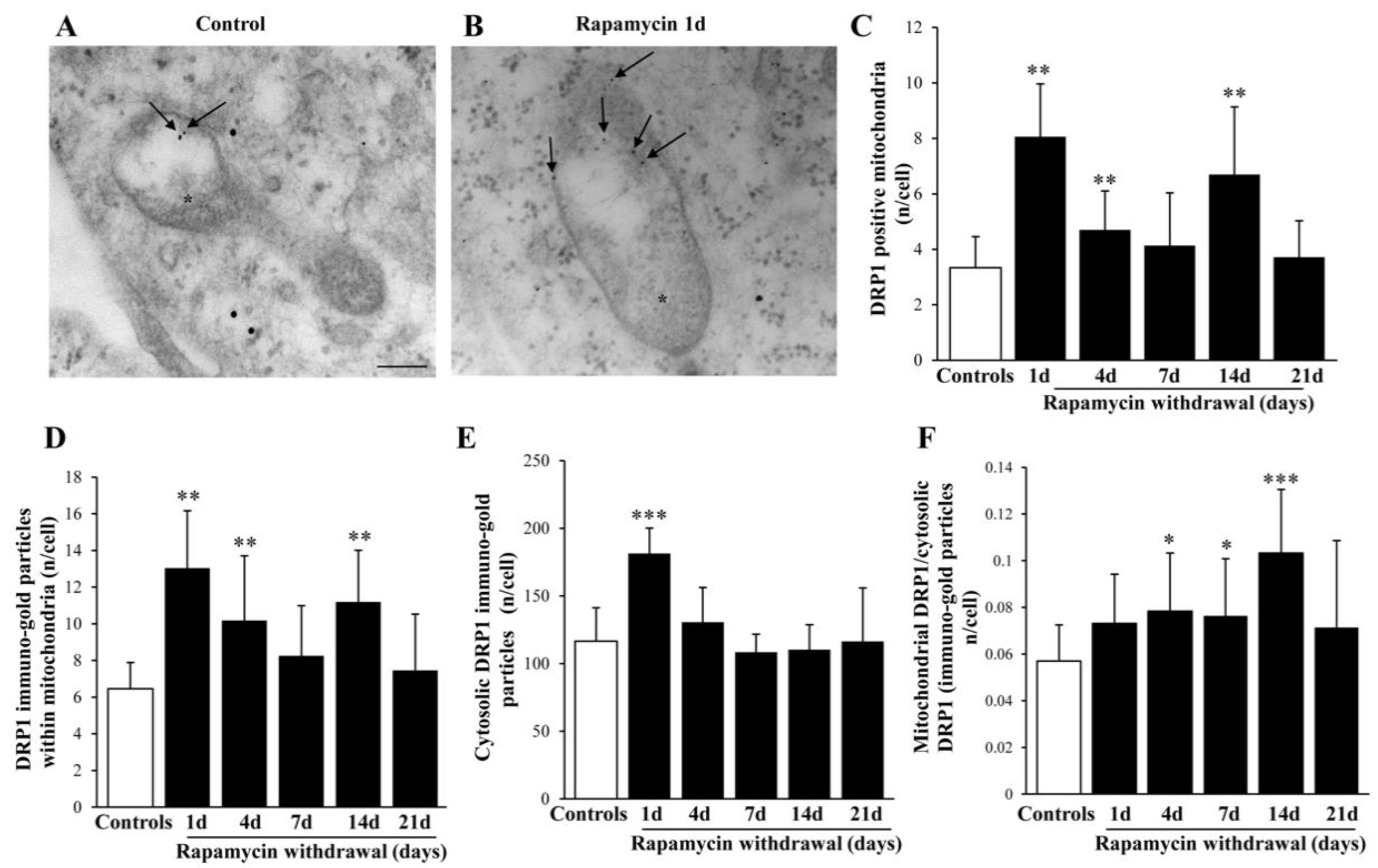
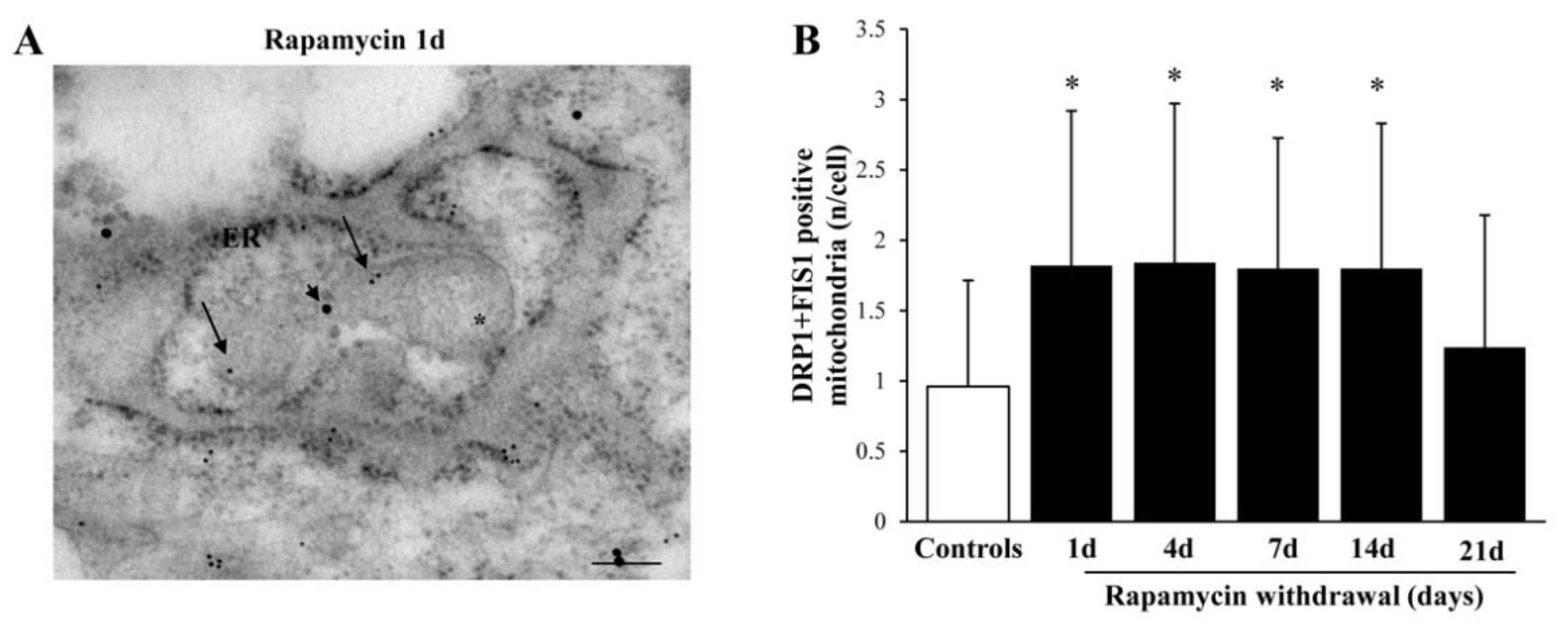
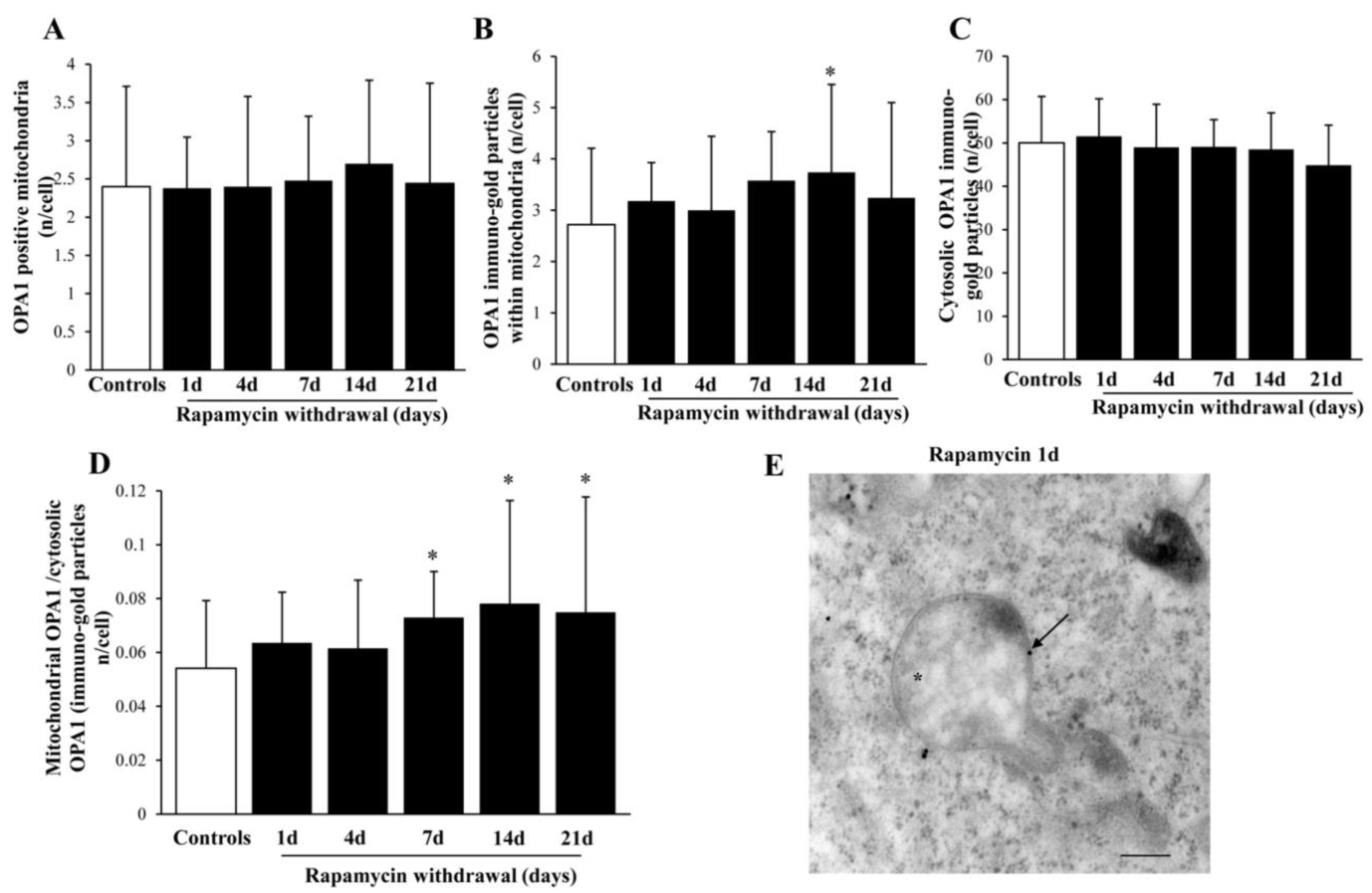
2.9. Within Mitochondria, Rapamycin Increases the Fission Proteins FIS1 and DRP1, While the Fusion Protein OPA1 Is not Changed
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. RNA Extraction
4.3. qReal-Time-PCR
4.4. MitoTracker-Red
4.5. Transmission Electron Microscopy (TEM)
4.5.1. Post-Embedding Immuno-Electron Microscopy
4.5.2. Ultra-Structural Analysis of Mitochondria
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jiang, H.; White, E.J.; Conrad, C.; Gomez-Manzano, C.; Fueyo, J. Autophagy pathways in glioblastoma. Methods Enzymol. 2009, 453, 273–286. [Google Scholar]
- Zhao, Y.; Huang, Q.; Yang, J.; Lou, M.; Wang, A.; Dong, J.; Qin, Z.; Zhang, T. Autophagy impairment inhibits differentiation of glioma stem/progenitor cells. Brain Res. 2010, 1313, 250–258. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [PubMed]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef] [PubMed]
- Arcella, A.; Biagioni, F.; Oliva, M.A.; Bucci, D.; Frati, A.; Esposito, V.; Cantore, G.; Giangaspero, F.; Fornai, F. Rapamycin inhibits the growth of glioblastoma. Brain Res. 2013, 1495, 37–51. [Google Scholar] [CrossRef]
- Ferrucci, M.; Biagioni, F.; Lenzi, P.; Gambardella, S.; Ferese, R.; Calierno, M.T.; Falleni, A.; Grimaldi, A.; Frati, A.; Esposito, V.; et al. Rapamycin promotes differentiation increasing βIII-tubulin, NeuN, and NeuroD while suppressing nestin expression in glioblastoma cells. Oncotarget 2017, 8, 29574–29599. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed. Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The emerging role of m-TOR up-regulation in brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar] [PubMed]
- Ferese, R.; Lenzi, P.; Fulceri, F.; Biagioni, F.; Fabrizi, C.; Gambardella, S.; Familiari, P.; Frati, A.; Limanaqi, F.; Fornai, F. Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells. Int. J. Mol. Sci. 2020, 21, 4570. [Google Scholar] [CrossRef]
- Ryskalin, L.; Biagioni, F.; Lenzi, P.; Frati, A.; Fornai, F. mTOR Modulates Intercellular Signals for Enlargement and Infiltration in Glioblastoma Multiforme. Cancers 2020, 12, 2486. [Google Scholar] [CrossRef] [PubMed]
- Arcella, A.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Oliva, M.A.; Storto, M.; Fanelli, M.; Gambardella, S.; Fornai, F. Dissecting Molecular Features of Gliomas: Genetic Loci and Validated Biomarkers. Int. J. Mol. Sci. 2020, 21, 685. [Google Scholar] [CrossRef]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in gliomas and their vascular microenvironment. Biochim. Biophys. Acta 2011, 1807, 602–608. [Google Scholar] [CrossRef]
- Arismendi-Morillo, G.; Castellano-Ramírez, A.; Seyfried, T.N. Ultrastructural characterization of the Mitochondria-associated membranes abnormalities in human astrocytomas: Functional and therapeutics implications. Ultrastruct. Pathol. 2017, 41, 234–244. [Google Scholar] [CrossRef]
- Deighton, R.F.; Le Bihan, T.; Martin, S.F.; Gerth, A.M.J.; McCulloch, M.; Edgar, J.M.; Kerr, L.E.; Whittle, I.R.; McCulloch, J. Interactions among mitochondrial proteins altered in glioblastoma. J. Neurooncol. 2014, 118, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.C.; Grootveld, M. The importance of mitochondria in the tumourigenic phenotype: Gliomasas the paradigm. Int. J. Mol. Med. 2011, 27, 159–171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Epple, L.M.; Dodd, R.D.; Merz, A.L.; Dechkovskaia, A.M.; Herring, M.; Winston, B.A.; Lencioni, A.M.; Russell, R.L.; Madsen, H.; Nega, M.; et al. Induction of the Unfolded Protein Response Drives Enhanced Metabolism and Chemoresistance in Glioma Cells. PLoS ONE. 2013, 8, e73267. [Google Scholar] [CrossRef] [PubMed]
- Redzic, J.S.; Gomez, J.D.; Hellwinkel, J.E.; Anchordoquy, T.J.; Graner, M.W. Proteomic analyses of brain tumor cell lines amidst the unfolded protein response. Oncotarget 2016, 7, 47831–47847. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget 2016, 7, 38638–38657. [Google Scholar] [CrossRef]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. Mitochondrial Fission Protein 1: Emerging Roles in Organellar Form and Function in Health and Disease. Front. Endocrinol. 2021, 12, 660095. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell. Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Dorn, G.W., 2nd. Evolving and Expanding the Roles of Mitophagy as a Homeostatic and Pathogenic Process. Physiol. Rev. 2019, 99, 853–892. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Zhai, G.; Suzuki, Y.; Sarkesh, S.; Black, P.M.; Muzikansky, A.; Loeffler, J.S. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Sato, A.; Matsuda, K.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro. Oncol. 2010, 12, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Zhang, L.Q.; Zhang, X.G.; Li, X.; Ren, Y.B.; Ma, X.Y.; Li, X.G.; Wang, L.X. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J. Neuroncol. 2011, 103, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Weiss, W.A. Autophagy and Akt promote survival in glioma. Autophagy 2011, 7, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Galan-Moya, E.M.; Le Guelte, A.; Lima Fernandes, E.; Thirant, C.; Dwyer, J.; Bidere, N.; Couraud, P.O.; Scott, M.G.; Junier, M.P.; Chneiweiss, H.; et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011, 12, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of glioblastoma multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Suppression of the invasive potential of glioblastoma cells by mTOR inhibitors involves modulation of NFκB and PKC-α signaling. Sci. Rep. 2016, 6, 22455. [Google Scholar] [CrossRef] [PubMed]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, L.; Jia, R. The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar] [PubMed]
- Gautam, N.; Sankaran, S.; Yason, J.A.; Tan, K.S.W.; Gascoigne, N.R.J. A high content imaging flow cytometry approach to study mitochondria in T cells: MitoTracker Green FM dye concentration optimization. Methods 2018, 134–135, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Carinci, M.; Valacca, C.; Bielli, P.; Strappazzon, F.; Antonioli, M.; Ciccosanti, F.; Rodolfo, C.; Campello, S.; Fimia, G.M.; et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J. Cell. Biol. 2016, 215, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell. Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Marongiu, R.; Falleni, A.; Gelmetti, V.; Busceti, C.L.; Michiorri, S.; Valente, E.M.; Fornai, F. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 2012, 150, 194–217. [Google Scholar] [PubMed]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar]
- Archer, S.L. Mitochondrial dynamics-Mitochondrial fission and fusion in Human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Jin, S.-B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Song, Y.; Ren, Z.; Cheng, X.; Li, P.; Song, H.; Jia, L. Glioma cells are resistant to inflammation-induced alterations of mitochondrial dynamics. Int. J. Oncol. 2020, 57, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Pinto, G.; Saenz-de-Santa-Maria, I.; Chastagner, P.; Perthame, E.; Delmas, C.; Toulas, C.; Moyal-Jonathan-Cohen, E.; Brou, C.; Zurzolo, C. Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem. J. 2021, 478, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Salaud, C.; Alvarez-Arenas, A.; Geraldo, F.; Belmonte-Beitia, J.; Calvo, G.F.; Gratas, C.; Pecqueur, C.; Garnier, D.; Pérez-Garcià, V.; Vallette, F.M.; et al. Mitochondria transfer from tumor-activated stromal cells (TASC) to primary Glioblastoma cells. Biochem. Biophys. Res. Commun. 2020, 533, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, B.; Tullet, J.M.A.; Hartman, J.L.; Mitra, K. Interplay of mitochondrial fission-fusion with cell cycle regulation: Possible impacts on stem cell and organismal aging. Exp. Gerontol. 2020, 135, 110919. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Nazio, F.; Cecconi, F. Connecting autophagy: AMBRA1 and its network of regulation. Mol. Cell Oncol. 2015, 2, e970059. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, T.; Okamoto, K.; Omiya, S.; Taneike, M.; Yamaguchi, O.; Otsu, K. A Mammalian Mitophagy Receptor, Bcl2-L-13, Recruits the ULK1 Complex to Induce Mitophagy. Cell Rep. 2019, 26, 338–345. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell. Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and Neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef]
- Kirchner, G.I.; Meier-Wiedenbach, I.; Manns, M.P. Clinical pharmacokinetics of everolimus. Clin. Pharmacokinet. 2004, 43, 83–95. [Google Scholar] [CrossRef]
- Supko, J.G.; Malspeis, L. Dose-dependent pharmacokinetics of rapamycin-28-N,N-dimethylglycinate in the mouse. Cancer Chemother. Pharmacol. 1994, 33, 325–330. [Google Scholar] [CrossRef]
- Schefe, J.H.; Lehmann, K.E.; Buschmann, I.R.; Unger, T.; Funke-Kaiser, H. Quantitative real-time RT-PCR data analysis: Current concepts and the novel “gene expression’s CT difference” formula. J. Mol. Med. 2006, 84, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.S.; Reed, A.; Chen, F.; Stewart, C.N., Jr. Statistical analysis of real-time PCR data. BMC Bioinformatics 2006, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell. Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef]
- Bendayan, M.; Zollinger, M. Ultrastructural localization of antigenic sites on osmium-fixed tissues applying the protein A-gold technique. J. Histochem. Cytochem. 1983, 31, 101–109. [Google Scholar] [CrossRef]
- Lucocq, J.M.; Habermann, A.; Watt, S.; Backer, J.M.; Mayhew, T.M.; Griffiths, G. A rapid method for assessing the distribution of gold labeling on thin sections. J. Histochem. Cytochem. 2004, 52, 991–1000. [Google Scholar] [CrossRef]
- Stirling, J.W.; Graff, P.S. Antigen unmasking for immunoelectron microscopy: Labeling is improved by treating with sodium ethoxide or sodium metaperiodate, then heating on retrieval medium. J. Histochem. Cytochem. 1995, 43, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Ghadially, F.N. Ultrastructural Pathology of the Cell and Matrix, 3rd ed.; Wellington Butterworths: London, UK, 1988; pp. 191–328. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Target Sequence Number | Primer Sequences |
|---|---|---|
| DRP1 | NM_001330380.1 | FW 5′-AGCTGCTCAGTATCAGTCTC-3′ RW 5′-GGCAGTCAAAATGTCAATAGT-3′ |
| FIS1 | NM_016068.3 | FW 5′-TCAGTCTGAGAAGGCAGCA-3′ RW 5′-CGCTGTTCCTCCTTGCT-3′ |
| OPA1 | NM_130837.2 | FW 5′-TTCAGTATCAGCAAAGCT-3′ RW 5′-GAGGGTCCATTTGACTGAC-3′ |
| ULK1 | NM_003565.3 | FW 5′-CAGACGACTTCGTCATGGTC-3′ RW 5′-AGCTCCCACTGCACATCAG-3′ |
| VPS34 | NM_002647.4 | FW 5′-GGGGAAGCAGAGAAGTTTCA-3′ RW 5′-TCTTCCCTTCCAAGCTTCCT-3′ |
| AMBRA1 | NM_001367468.1 | FW 5′-GAGCACCCAATTTACCCAGA-3′ RW 5′-GATCATCCTCTGGGCGTAGTA-3′ |
| PARKIN | NM_004562.3 | FW 5′-TGAGAAGCTGGATTACCATC-3′ RW 5′-TTGAGAGTGACACAGATGACCT-3′ |
| PINK1 | NM_032409.3 | FW 5′-GGCTTTCGGCTGGAGGAGTA-3′ RW 5′-GCTCGCTGGGACCAGCTCC-3′ |
| β–Actin | NM_001101.3 | FW 5′-GTGCGTGACATTAAGGAG-3′ RW 5′-GCCAGACAGCACTGTGT-3′ |
| β-Globin | NM_000518.4 | FW 5′-CTAAGGTGAAGGCTCATG-3′ RW 5′-GATAGGCAGCCTGCACT-3 |
| Antibodies | Distributors | Cod. N° | RRID |
|---|---|---|---|
| Rabbit anti-LC3 | Abcam | ab128025 | AB_11143008 |
| Rabbit anti-FIS1 | GeneTex | GTX111010 | AB_10731288 |
| Mouse anti-DRP1 | Abcam | ab56788 | AB_941306 |
| Rabbit anti-OPA1 | Abcam | ab90857 | AB_2050139 |
| Mouse anti-PINK1 | Abcam | ab186303 | AB_2827698 |
| Rabbit anti-PARKIN | Millipore | AB5112 | AB_2283497 |
| Rabbit anti-ULK1 | Cell Signaling Technology | D8H5 | AB_11178668 |
| Rabbit anti-AMBRA1 | Abcam | ab 59141 | AB_941614 |
| Rabbit anti-Vps34 | Thermo Fisher Scientific | PA1-46456 | AB_2283904 |
| EM Goat anti-Rabbit IgG, gold conjugated antibody | BBInternational | EM.GAR 20 | AB_1769136 |
| EM Goat anti-Rabbit IgG, gold conjugate antibody | BBInternational | EM.GAR 10 | AB_1769128 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenzi, P.; Ferese, R.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Falleni, A.; Gambardella, S.; Frati, A.; Fornai, F. Rapamycin Ameliorates Defects in Mitochondrial Fission and Mitophagy in Glioblastoma Cells. Int. J. Mol. Sci. 2021, 22, 5379. https://doi.org/10.3390/ijms22105379
Lenzi P, Ferese R, Biagioni F, Fulceri F, Busceti CL, Falleni A, Gambardella S, Frati A, Fornai F. Rapamycin Ameliorates Defects in Mitochondrial Fission and Mitophagy in Glioblastoma Cells. International Journal of Molecular Sciences. 2021; 22(10):5379. https://doi.org/10.3390/ijms22105379
Chicago/Turabian StyleLenzi, Paola, Rosangela Ferese, Francesca Biagioni, Federica Fulceri, Carla L. Busceti, Alessandra Falleni, Stefano Gambardella, Alessandro Frati, and Francesco Fornai. 2021. "Rapamycin Ameliorates Defects in Mitochondrial Fission and Mitophagy in Glioblastoma Cells" International Journal of Molecular Sciences 22, no. 10: 5379. https://doi.org/10.3390/ijms22105379
APA StyleLenzi, P., Ferese, R., Biagioni, F., Fulceri, F., Busceti, C. L., Falleni, A., Gambardella, S., Frati, A., & Fornai, F. (2021). Rapamycin Ameliorates Defects in Mitochondrial Fission and Mitophagy in Glioblastoma Cells. International Journal of Molecular Sciences, 22(10), 5379. https://doi.org/10.3390/ijms22105379