
Folate Receptor Beta as a Direct and Indirect Target for Antibody-Based Cancer Immunotherapy


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
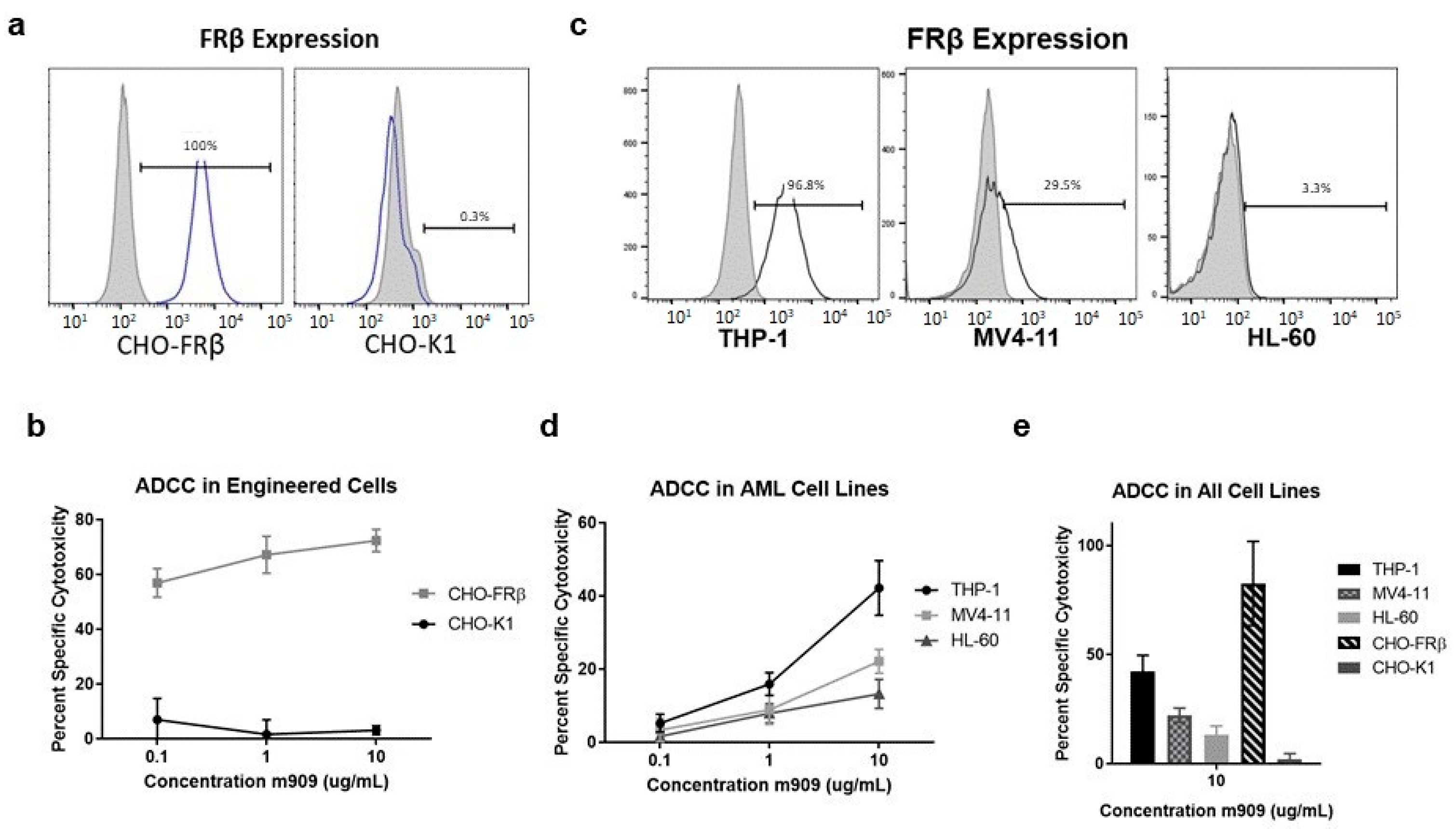
2.1. m909 Detects FRβ Expression and Can Mediate ADCC of AML Cell Lines
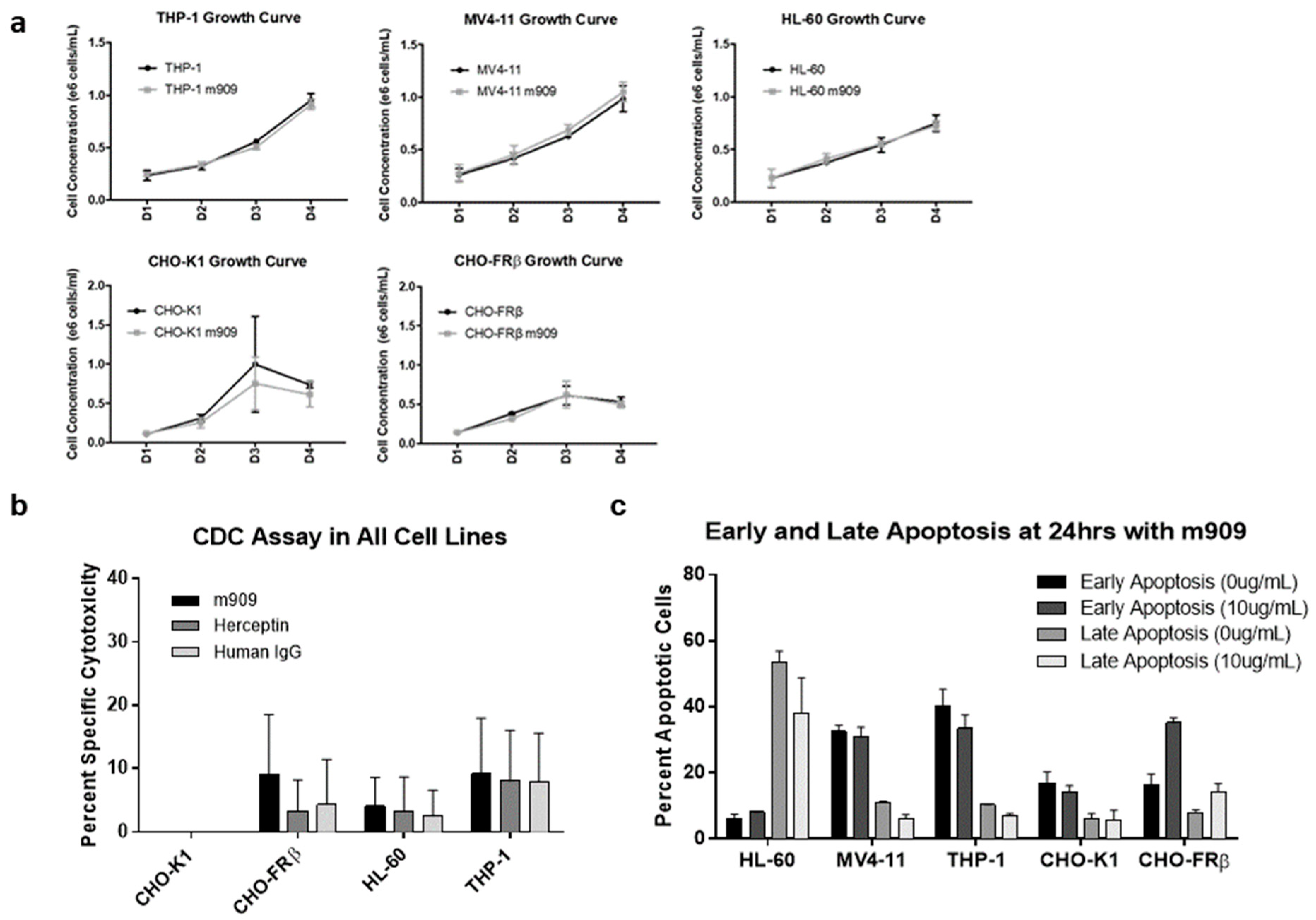
2.2. m909-Mediated Cytotoxicity of AML Cells Relies upon ADCC
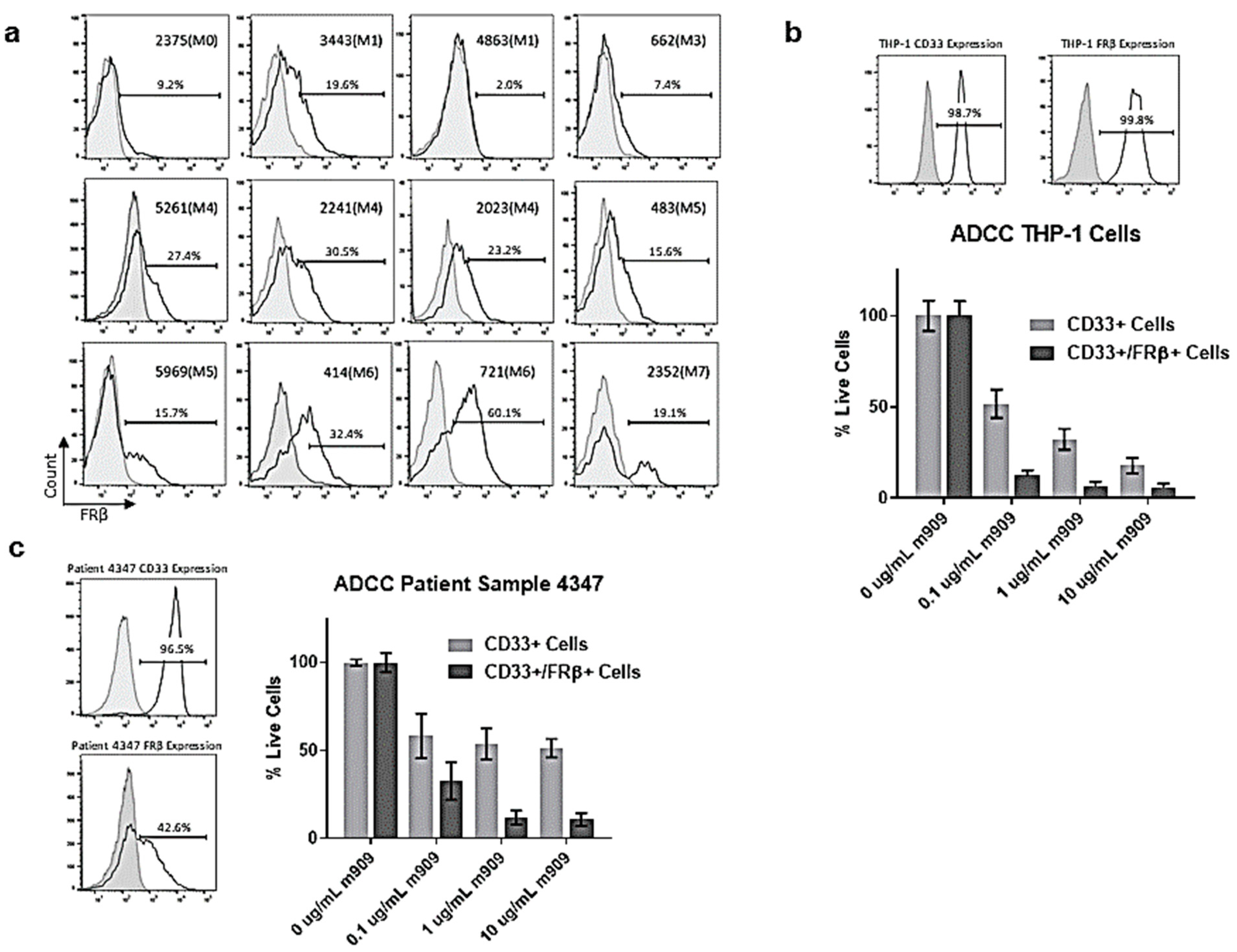
2.3. AML Patient Samples Express FRβ and m909 Can Cause ADCC of These Samples
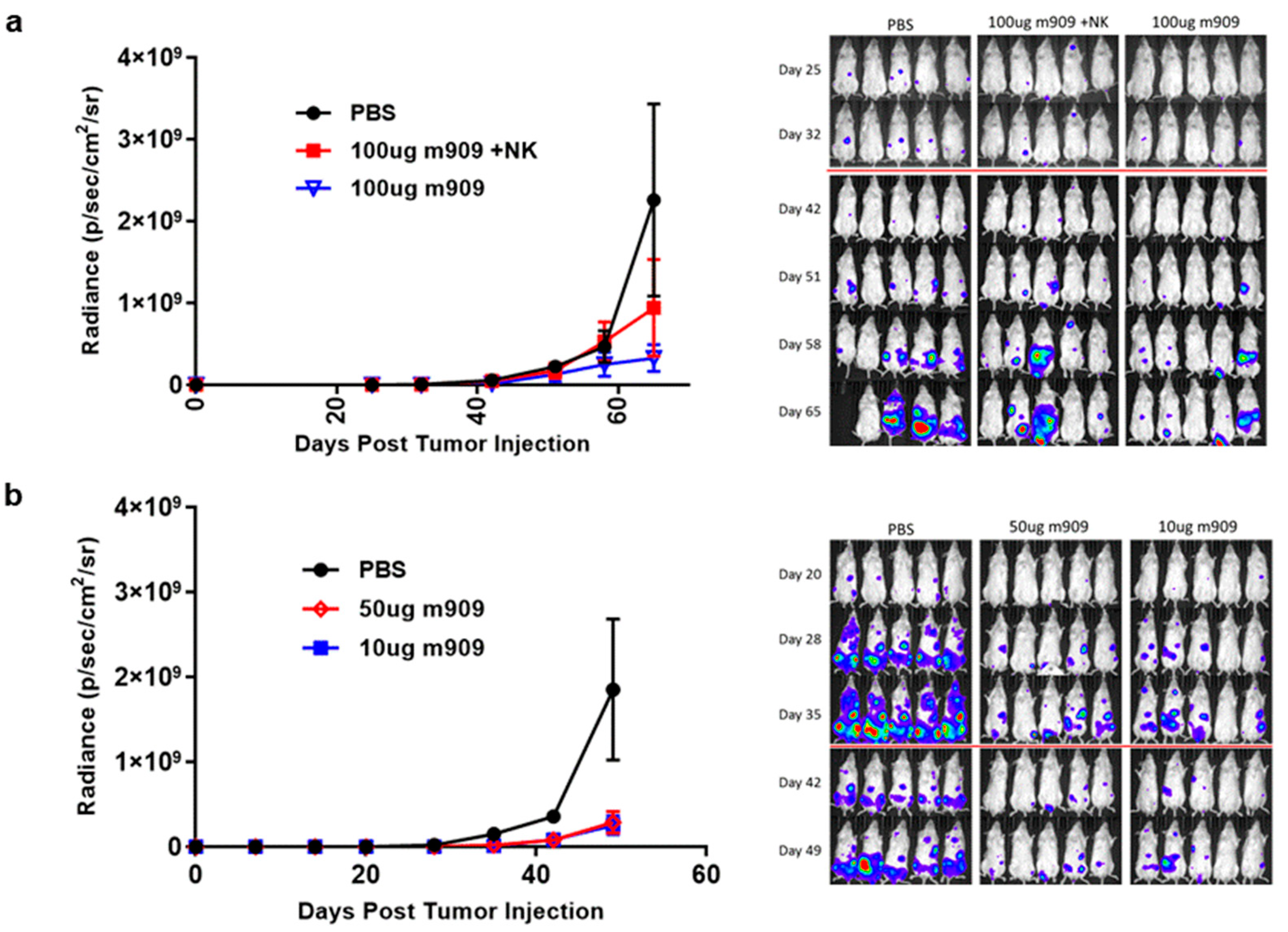
2.4. m909 Reduces Tumor Growth in a Human AML Xenograft Model
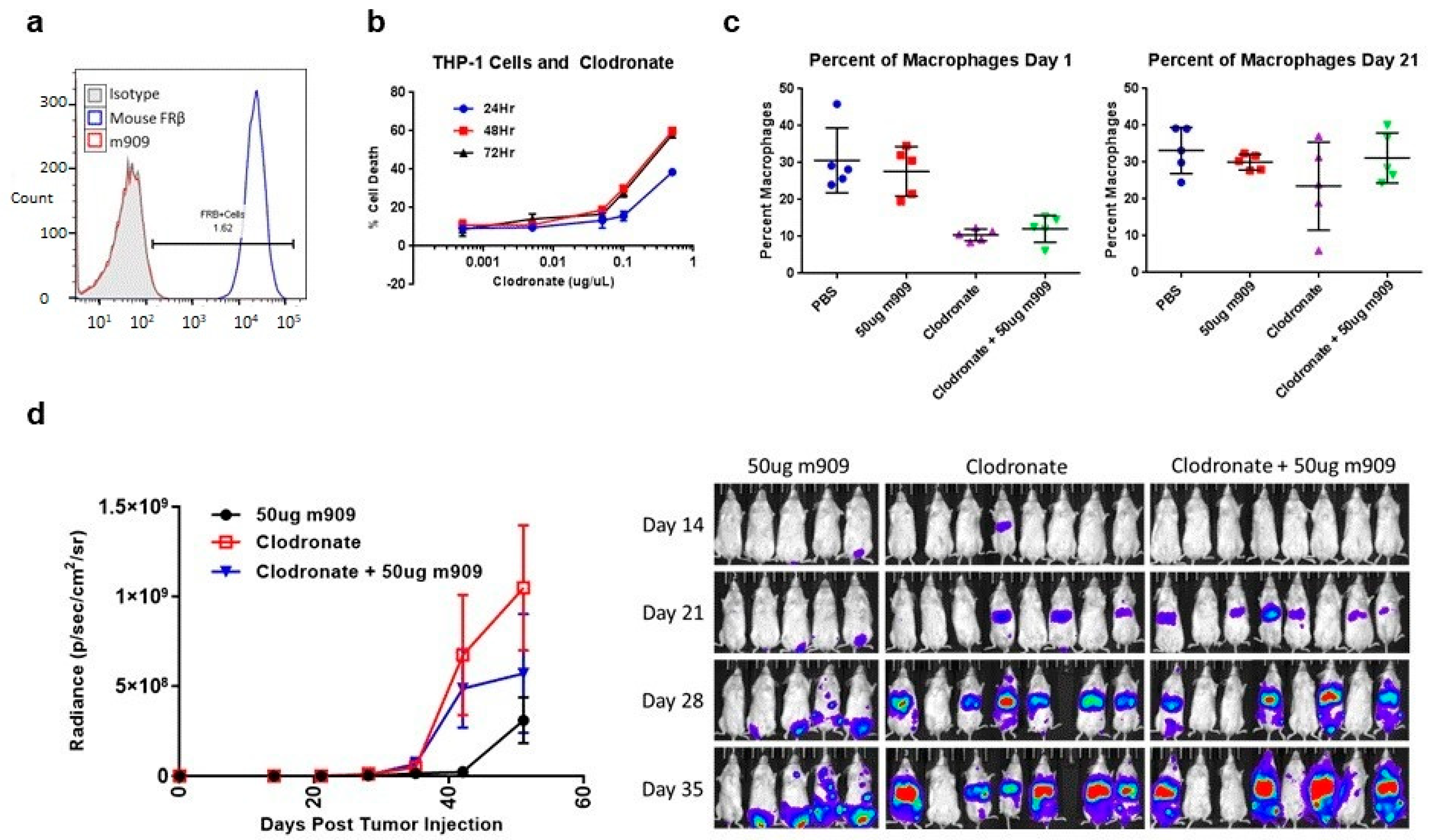
2.5. Mechanisms for In Vivo Reduction of AML Tumor Growth
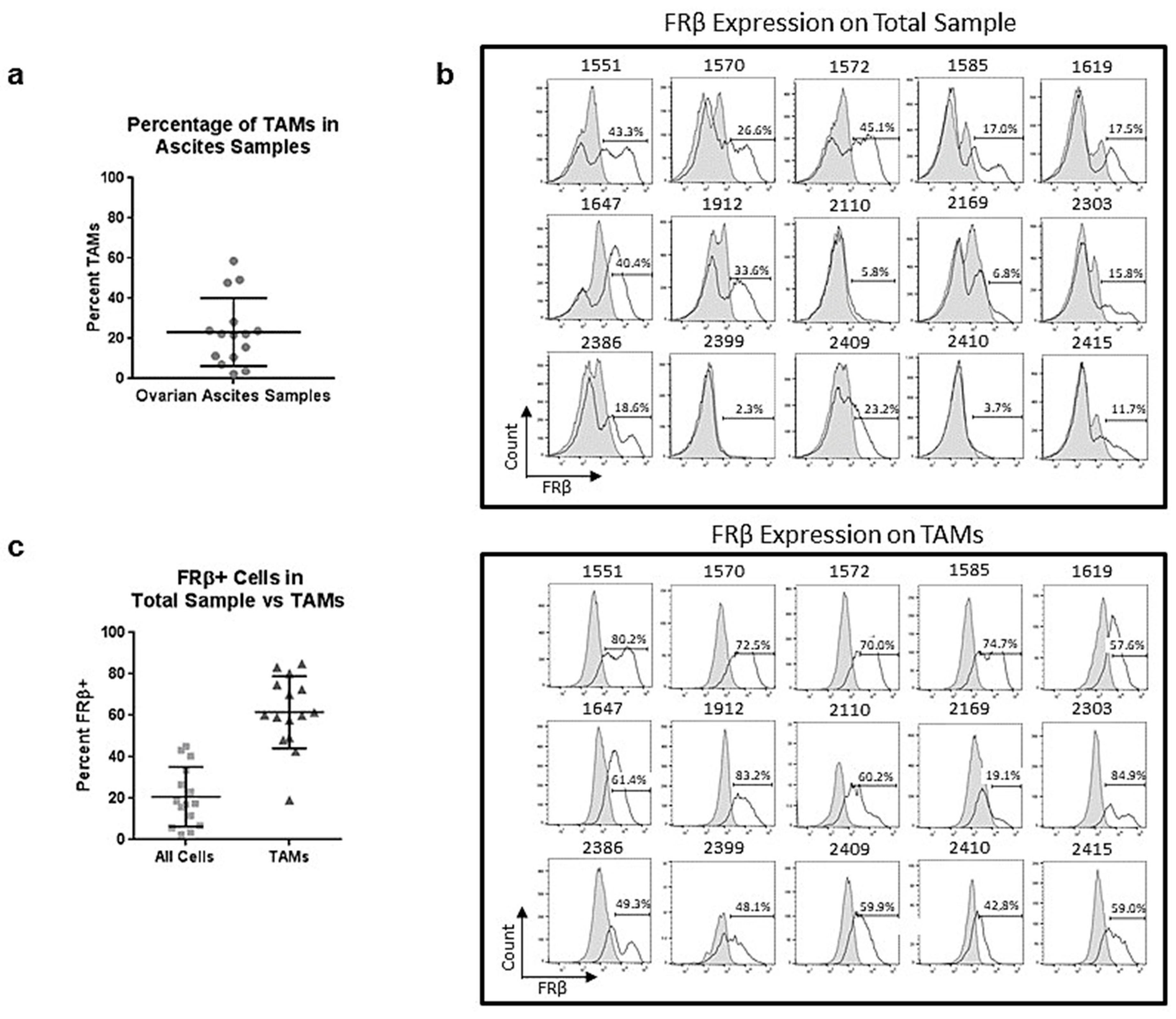
2.6. Tumor-Associated Macrophages in Ovarian Ascites Samples Express FRβ
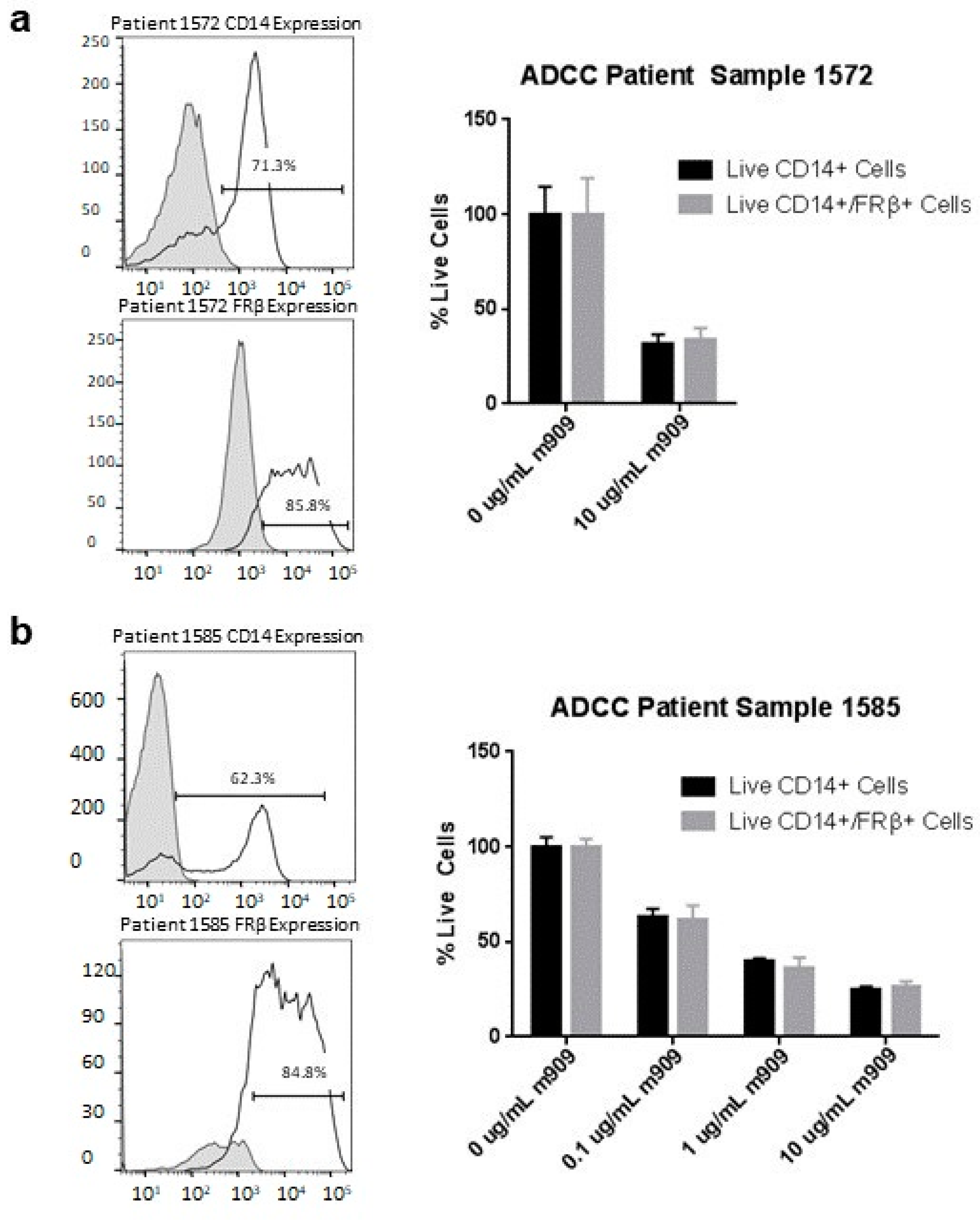
2.7. m909 Can Cause ADCC of TAMs in Primary Ascites Samples
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Lines
4.3. Flow Cytometry
4.4. Antibody-Dependent Cellular Cytotoxicity (ADCC) in Cell Lines
4.5. Cell Growth Assays
4.6. CDC Assays
4.7. Apoptosis Assays
4.8. Antibody-Dependent Cellular Cytotoxicity in AML Patient Samples
4.9. Antibody-Dependent Cellular Cytotoxicity in Ovarian Cancer Patient Ascites Samples
4.10. Mice
4.11. Bioluminescence Imaging
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coney, L.R.; Tomassetti, A.; Carayannopoulos, L.; Frasca, V.; Kamen, B.A.; Colnaghi, M.I.; Zurawski, V.R., Jr. Cloning of a tumor-associated antigen: MOv18 and MOv19 antibodies recognize a folate-binding protein. Cancer Res. 1991, 51, 6125–6132. [Google Scholar] [PubMed]
- Ross, J.F.; Wang, H.; Behm, F.G.; Mathew, P.; Wu, M.; Booth, R.; Ratnam, M. Folate receptor type beta is a neutrophilic lineage marker and is differentially expressed in myeloid leukemia. Cancer 1999, 85, 348–357. [Google Scholar] [CrossRef]
- Ross, J.F.; Chaudhuri, P.K.; Ratnam, M. Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiol. Clin. Implic. Cancer 1994, 73, 2432–2443. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Powell, D.J.; Coukos, G.; Coukos, G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J. Transl. Med. 2012, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Ca-nevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergote, I.; Armstrong, D.; Scambia, G.; Teneriello, M.; Sehouli, J.; Schweizer, C.; Weil, S.C.; Bamias, A.; Fujiwara, K.; Ochiai, K.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase III Study to Assess Efficacy and Safety of Weekly Farletuzumab in Combination With Carboplatin and Taxane in Patients With Ovarian Cancer in First Platinum-Sensitive Relapse. J. Clin. Oncol. 2016, 34, 2271–2278. [Google Scholar] [CrossRef]
- Kim, K.H.; Jelovac, D.; Armstrong, D.K.; Schwartz, B.; Weil, S.C.; Schweizer, C.; Alvarez, R.D. Phase, 1b safety study of farletuzumab, carboplatin and pegylated liposomal doxorubicin in patients with platinum-sensitive epithelial ovarian cancer. Gynecol. Oncol. 2016, 140, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.K.; White, A.J.; Weil, S.C.; Phillips, M.; Coleman, R.L. Farletuzumab (a monoclonal antibody against folate receptor alpha) in relapsed platinum-sensitive ovarian cancer. Gynecol. Oncol. 2013, 129, 452–458. [Google Scholar] [CrossRef]
- Sasaki, Y.; Miwa, K.; Yamashita, K.; Sunakawa, Y.; Shimada, K.; Ishida, H.; Hasegawa, K.; Fujiwara, K.; Kodaira, M.; Fujiwara, Y.; et al. A phase I study of farletuzumab, a humanized anti-folate receptor α monoclonal antibody, in patients with solid tumors. Investig. New Drugs 2015, 33, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Konner, J.A.; Bell-McGuinn, K.M.; Sabbatini, P.; Hensley, M.L.; Tew, W.P.; Pandit-Taskar, N.; Vander Els, N.; Phillips, M.D.; Schweizer, C.; Weil, S.C.; et al. Farletuzumab, a Humanized Monoclonal Antibody against Folate Receptor α, in Epithelial Ovarian Cancer: A Phase I Study. Clin. Cancer Res. 2010, 16, 5288–5295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, C.; Schweizer, C.; Wustner, J.; Weil, S.; Namiki, M.; Nakano, T.; Nakai, K.; Phillips, M.D. Population pharmacokinetics of farletuzumab, a humanized monoclonal antibody against folate receptor alpha, in epithelial ovarian cancer. Cancer Chemother. Pharmacol. 2012, 70, 727–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.P.; Konner, J.A.; Moore, K.N.; Seward, S.M.; Matulonis, U.A.; Perez, R.P.; Su, Y.; Berkenblit, A.; Ruiz-Soto, R.; Birrer, M.J. Characterization of folate receptor alpha (FRα) expression in archival tumor and biopsy samples from relapsed epithelial ovarian cancer patients: A phase I expansion study of the FRα-targeting antibody-drug conjugate mirvetuximab soravtansine. Gynecol. Oncol. 2017, 147, 402–407. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Martin, L.P.; O’Malley, D.M.; Matulonis, U.A.; Konner, J.A.; Perez, R.P.; Bauer, T.M.; Ruiz-Soto, R.; Birrer, M.J. Safety and Activity of Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha-Targeting Antibody-Drug Conjugate, in Platinum-Resistant Ovarian, Fallopian Tube, or Primary Peritoneal Cancer: A Phase I Expansion Study. J. Clin. Oncol. 2017, 35, 1112–1118. [Google Scholar] [CrossRef]
- Moore, K.N.; Borghaei, H.; O’Malley, D.M.; Jeong, W.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Matulonis, U.A.; Running, K.L.; Zhang, X.; et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 2017, 123, 3080–3087. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Ross, J.F.; Wang, X.; Ratnam, M. Identification of a novel folate receptor, a truncated receptor, and receptor type .beta. in hematopoietic cells: cDNA cloning, expression, immunoreactivity, and tissue specificity. Biochemistry 1994, 33, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, X.; Behm, F.G.; Ratnam, M. Differentiation-independent retinoid induction of folate receptor type beta, a potential tumor target in myeloid leukemia. Blood 2000, 96, 3529–3536. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Lan, C.; Huang, X.; Lin, S.; Huang, H.; Cai, Q.; Wan, T.; Lu, J.; Liu, J. Expression of M2-polarized macrophages is associated with poor prognosis for advanced epithelial ovarian cancer. Technol. Cancer Res. Treat. 2013, 12, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Mhawech-Fauceglia, P.; Wang, D.; Ali, L.; Lele, S.; Huba, M.A.; Liu, S.; Odunsi, K. Intraepithelial T cells and tumor-associated macrophages in ovarian cancer patients. Cancer Immun. 2013, 13, 1–6. [Google Scholar] [PubMed]
- Kawamura, K.; Komohara, Y.; Takaishi, K.; Katabuchi, H.; Takeya, M. Detection of M2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathol. Int. 2009, 59, 300–305. [Google Scholar] [CrossRef]
- Wan, T.; Liu, J.H.; Zheng, L.M.; Cai, M.Y.; Ding, T. Prognostic significance of tumor-associated macrophage infiltration in advanced epithelial ovarian carcinoma. Chin. J. Cancer 2009, 28, 268–271. [Google Scholar]
- Puig-Kröger, A.; Sierra-Filardi, E.; Domínguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gómez-Aguado, F.; Ratnam, M.; Sánchez-Mateos, P.; Corbí, A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Shen, J.; Streaker, E.D.; Lockwood, M.; Zhu, Z.; Low, P.S.; Dimitrov, D.S. A folate receptor beta-specific human monoclonal antibody recognizes activated macrophage of rheumatoid patients and mediates antibody-dependent cell-mediated cytotoxicity. Arthritis Res. Ther. 2011, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, R.C.; Feng, Y.; Schutsky, K.; Poussin, M.; Kalota, A.; Dimitrov, D.S.; Powell, D.J., Jr. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia 2016, 30, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Lynn, R.C.; Poussin, M.; Kalota, A.; Feng, Y.; Low, P.S.; Dimitrov, D.S.; Powell, D.J., Jr. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood 2015, 125, 3466–3476. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.Q.; Zheng, X.; Shi, G.; Wang, H.; Ratnam, M.; Lee, R.J. Strategy for the treatment of acute myelogenous leukemia based on folate receptor β-targeted liposomal doxorubicin combined with receptor induction using all-trans retinoic acid. Blood 2002, 100, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Tanaka, M.; Tsuneyoshi, Y.; Xu, B.; Michie, S.A.; Hasui, K.; Hirano, H.; Arita, K.; Matsuyama, T. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor β. Cancer Immunol. Immunother. 2009, 58, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Fan, W.; Zou, J.; Zhang, S.; He, J.; Shu, C.; Zhao, G.; Sun, T.; Hu, Z.; Yang, Y.G. Complement Depletion Improves Human Red Blood Cell Reconstitution in Immunodeficient Mice. Stem Cell Rep. 2017, 9, 1034–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar] [PubMed]
- Van Rooijen, N.; Hendrikx, E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol. Biol. 2010, 605, 189–203. [Google Scholar] [CrossRef]
- Van Rooijen, N.; Sanders, A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 1994, 174, 83–93. [Google Scholar] [CrossRef]
- Moreno, A.; Badell, E.; Van Rooijen, N.; Druilhe, P. Human malaria in immunocompromised mice: New in vivo model for chemotherapy studies. Antimicrob. Agents Chemother. 2001, 45, 1847–1853. [Google Scholar] [CrossRef] [Green Version]
- Badell, E.; Pasquetto, V.; Van Rooijen, N.; Druilhe, P. A mouse model for human malaria erythrocytic stages. Parasitol. Today 1995, 11, 235–237. [Google Scholar] [CrossRef]
- Badell, E.; Oeuvray, C.; Moreno, A.; Soe, S.; van Rooijen, N.; Bouzidi, A.; Druilhe, P. Human malaria in immunocompromised mice: An in vivo model to study defense mechanisms against Plasmodium falciparum. J. Exp. Med. 2000, 192, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Van Rooijen, N.; Yang, Y.-G. Macrophages prevent human red blood cell reconstitution in immunodeficient mice. Blood 2011, 118, 5938–5946. [Google Scholar] [CrossRef] [Green Version]
- Fraser, C.C.; Chen, B.P.; Webb, S.; van Rooijen, N.; Kraal, G. Circulation of human hematopoietic cells in severe combined immunodeficient mice after Cl2MDP-liposome-mediated macrophage depletion. Blood 1995, 86, 183–192. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society Cancer Facts and Figures 2020. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf (accessed on 9 May 2020).
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstegen, M.M.A.; van Hennik, P.B.; Terpstra, W.; van den Bos, C.; Wielenga, J.J.; van Rooijen, N.; Ploemacher, R.E.; Wagemaker, G.; Wognum, A.W. Transplantation of Human Umbilical Cord Blood Cells in Macrophage-Depleted SCID Mice: Evidence for Accessory Cell Involvement in Expansion of Immature CD34+CD38−Cells. Blood 1998, 91, 1966–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekkers, G.; Bentlage, A.E.H.; Stegmann, T.C.; Howie, H.L.; Lissenberg-Thunnissen, S.; Zimring, J.; Rispens, T.; Vidarsson, G. Affinity of human IgG subclasses to mouse Fc gamma receptors. mAbs 2017, 9, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, A.G.; Robinson, J.M.; Sharma, P.; Rodriguez-Garcia, A.; Poussin, M.A.; Nickerson-Nutter, C.; Powell, D.J., Jr. Folate Receptor Beta as a Direct and Indirect Target for Antibody-Based Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 5572. https://doi.org/10.3390/ijms22115572
Roy AG, Robinson JM, Sharma P, Rodriguez-Garcia A, Poussin MA, Nickerson-Nutter C, Powell DJ Jr. Folate Receptor Beta as a Direct and Indirect Target for Antibody-Based Cancer Immunotherapy. International Journal of Molecular Sciences. 2021; 22(11):5572. https://doi.org/10.3390/ijms22115572
Chicago/Turabian StyleRoy, Allison G., J. Michael Robinson, Prannda Sharma, Alba Rodriguez-Garcia, Mathilde A. Poussin, Cheryl Nickerson-Nutter, and Daniel J. Powell, Jr. 2021. "Folate Receptor Beta as a Direct and Indirect Target for Antibody-Based Cancer Immunotherapy" International Journal of Molecular Sciences 22, no. 11: 5572. https://doi.org/10.3390/ijms22115572
APA StyleRoy, A. G., Robinson, J. M., Sharma, P., Rodriguez-Garcia, A., Poussin, M. A., Nickerson-Nutter, C., & Powell, D. J., Jr. (2021). Folate Receptor Beta as a Direct and Indirect Target for Antibody-Based Cancer Immunotherapy. International Journal of Molecular Sciences, 22(11), 5572. https://doi.org/10.3390/ijms22115572