
Induction of the Proinflammatory Chemokine Interleukin-8 Is Regulated by Integrated Stress Response and AP-1 Family Proteins Activated during Coronavirus Infection

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
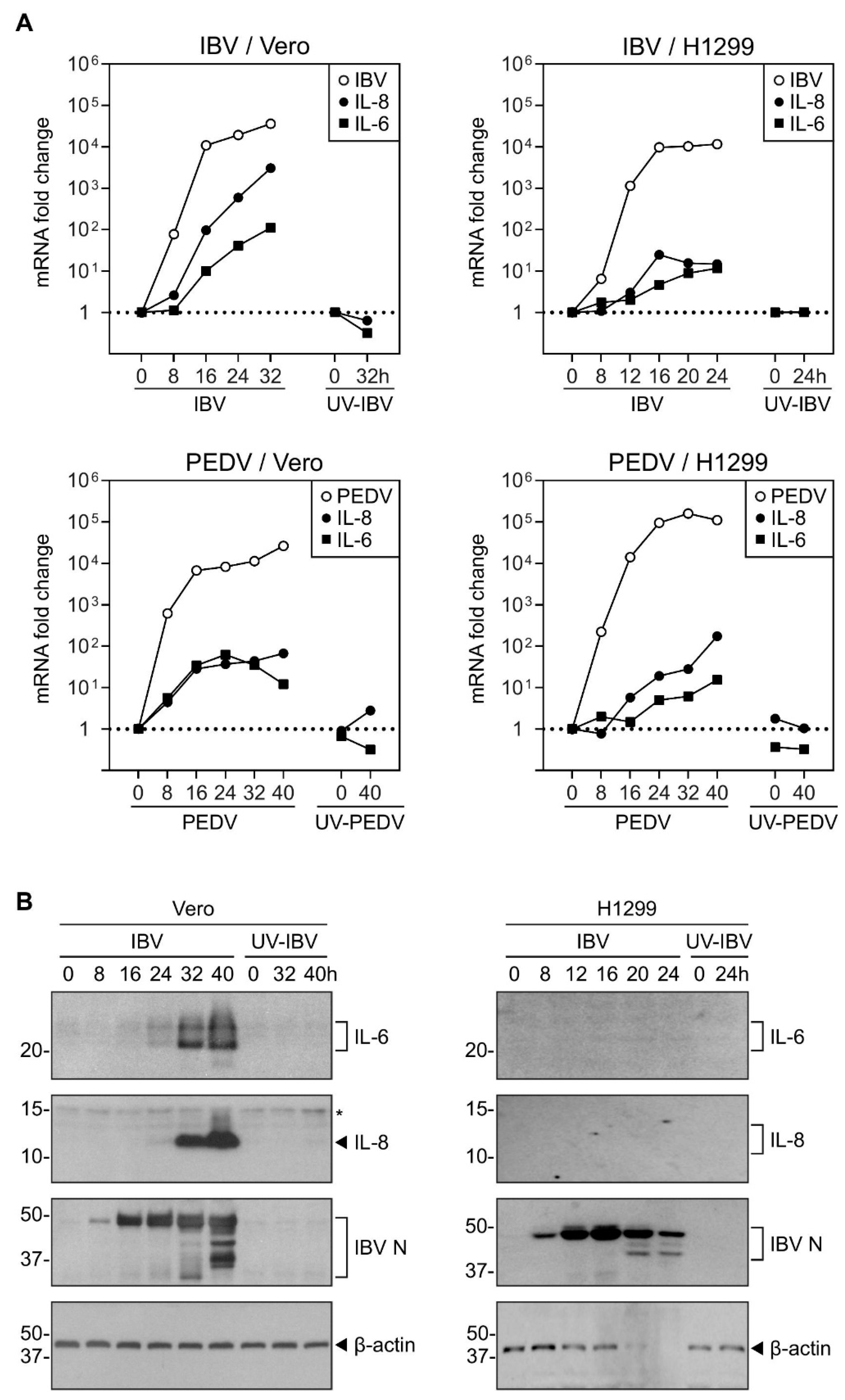
2.1. Induction of IL-8 and IL-6 Was Detected in Cells Infected with Gammacoronavirus IBV and Alphacoronavirus PEDV
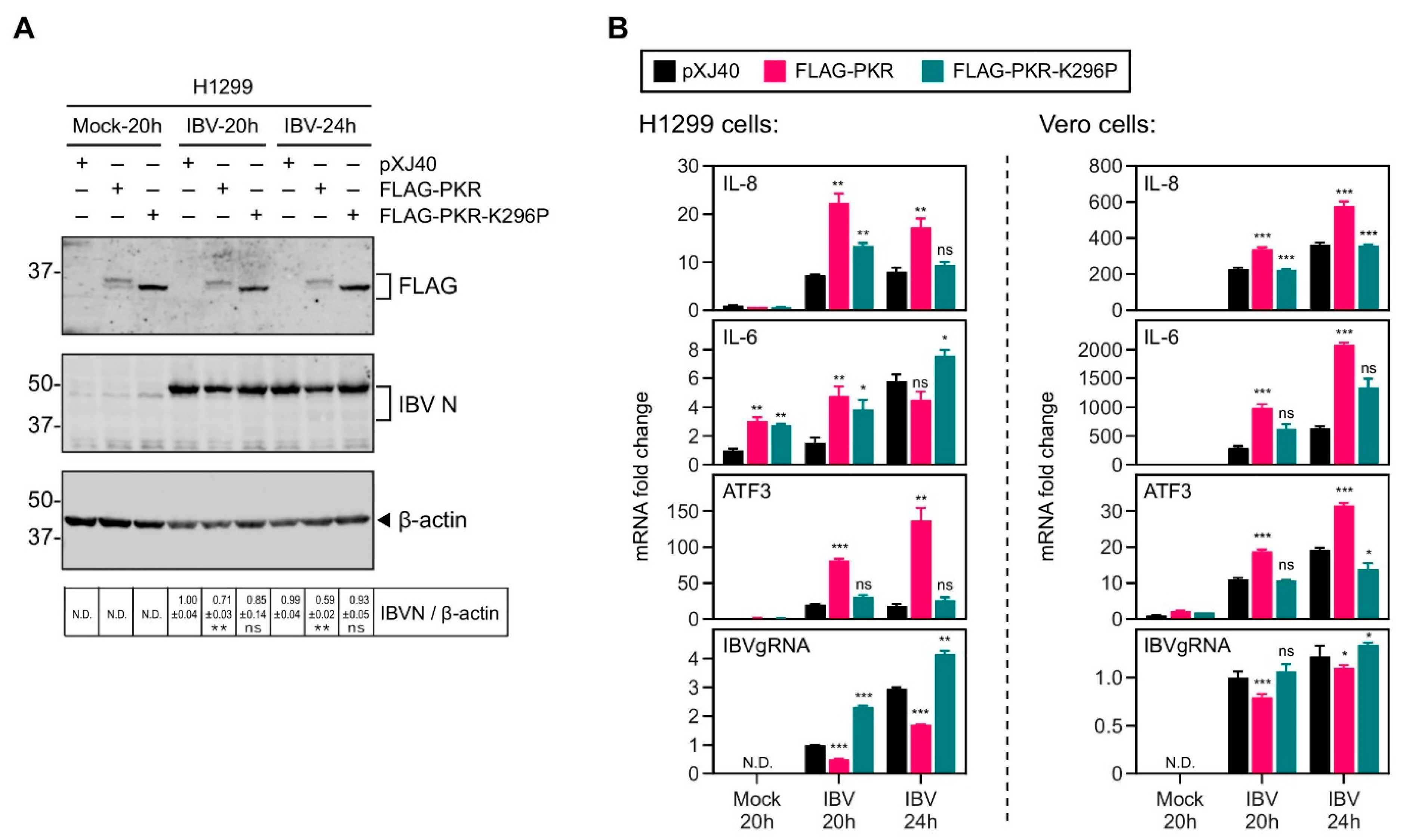
2.2. Overexpression of PKR UpRegulated the IBV-Induced IL-8 Expression
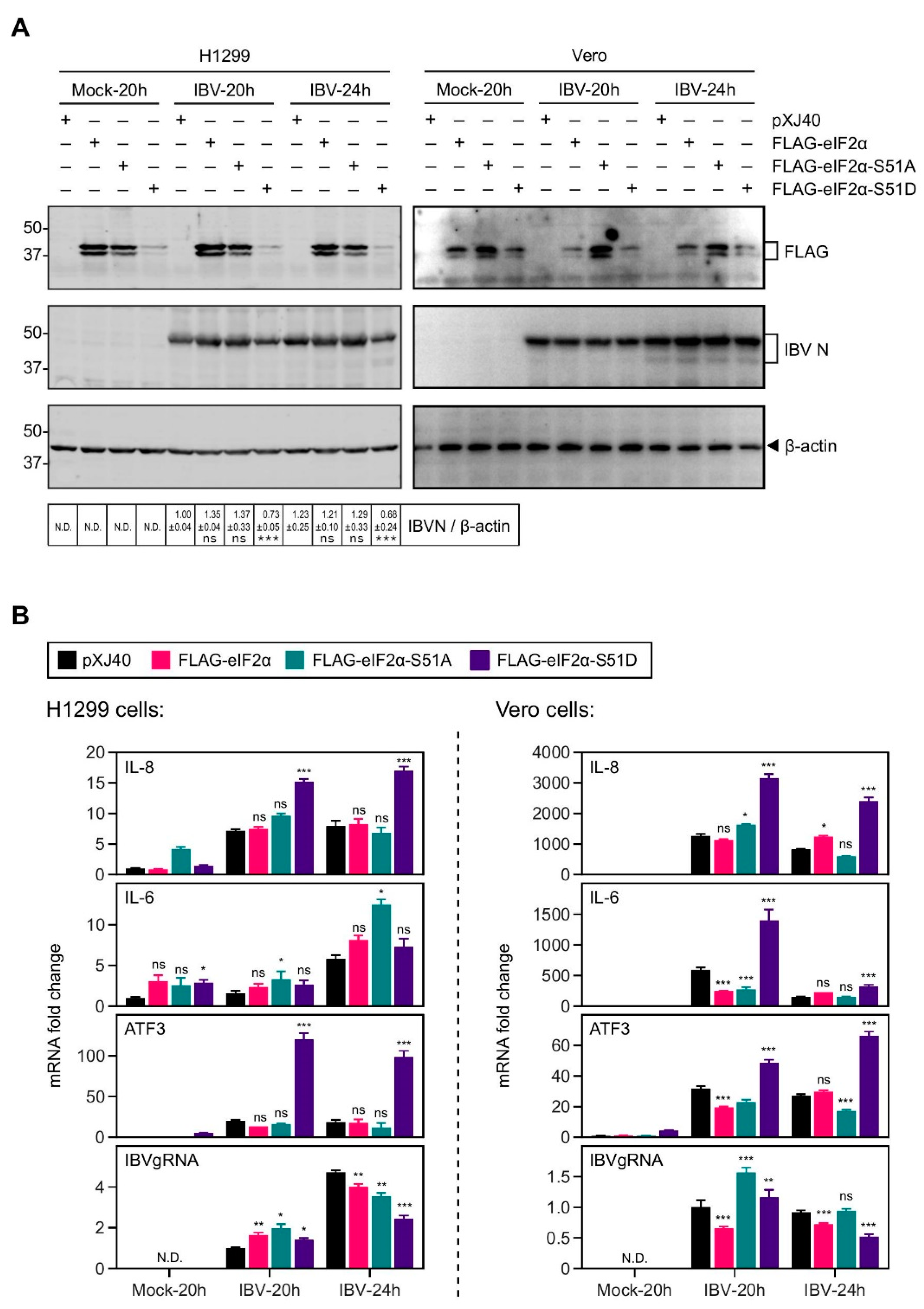
2.3. Overexpression of the Constitutively Active eIF2α Increased the IL-8 Induction by IBV Infection
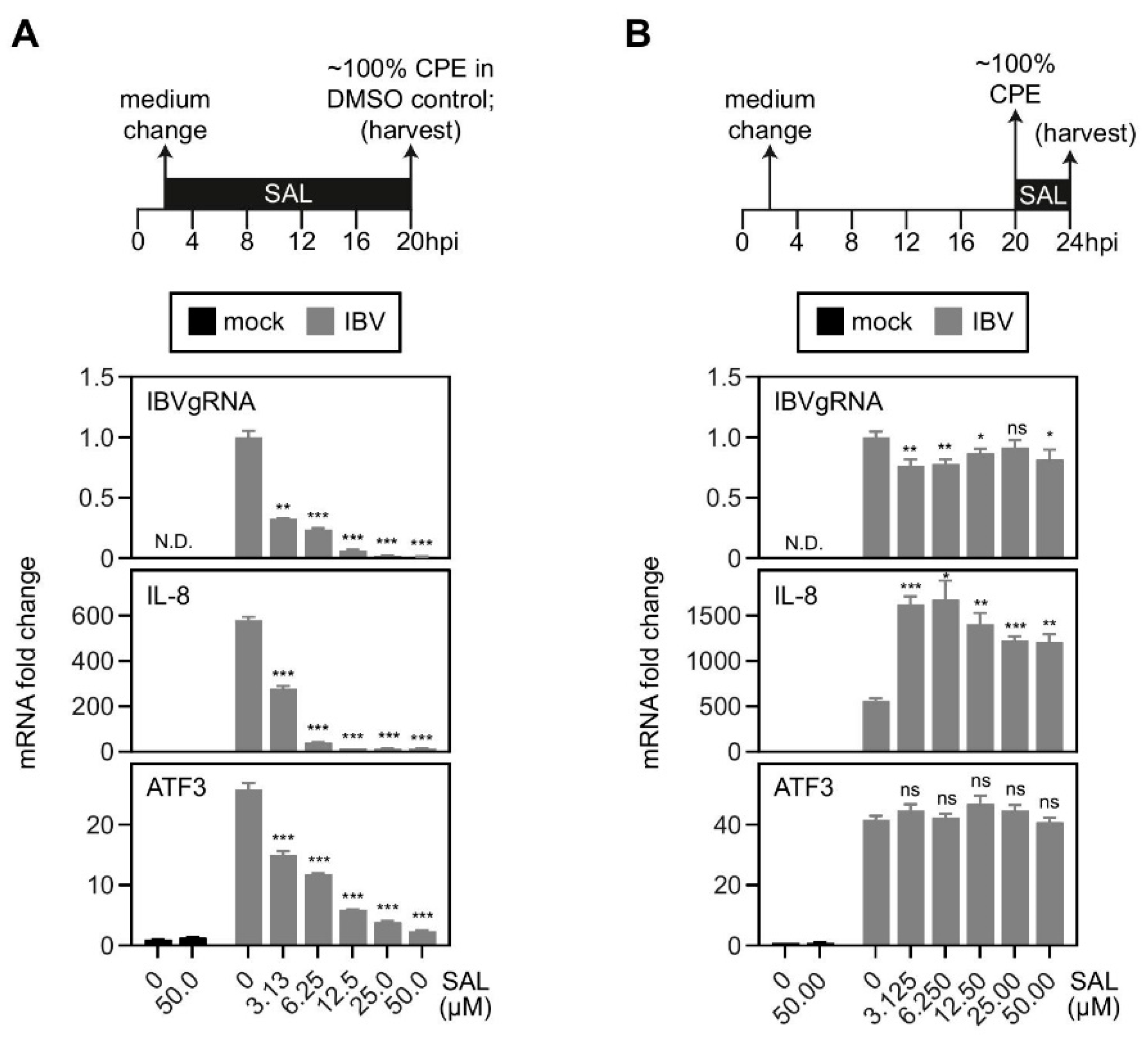
2.4. Inhibition of eIF2α Dephosphorylation Increased the Induction of IL-8 mRNA by IBV Infection
2.5. cJUN and cFOS Play a Synergistic Role in the IL-8 Induction during IBV Infection
2.6. Activation of the MKK7–JNK–cJUN Pathway Promoted the IBV-Induced IL-8 mRNA Expression
3. Discussions
4. Materials and Methods
4.1. Cell Culture and Virus Infection
4.2. Antibodies, Chemicals, and Reagents
4.3. Plasmid Construction and Transfection
4.4. SDS-PAGE and Western Blot Analysis
4.5. RNA Extraction and RT-qPCR Analysis
4.6. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.; Van Boheemen, S.; Bestebroer, T.; Osterhaus, A.; Fouchier, R. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Azhar, E.I.; El-Kafrawy, S.A.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Hashem, A.M.; Madani, T.A. Evidence for Camel-to-Human Transmission of MERS Coronavirus. N. Engl. J. Med. 2014, 370, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of Novel Coronavirus—Infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Masters, P.S. The Molecular Biology of Coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef] [PubMed]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Cavanagh, D. Coronaviruses in poultry and other birds. Avian Pathol. 2005, 34, 439–448. [Google Scholar] [CrossRef]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Didangelos, A. COVID-19 Hyperinflammation: What about Neutrophils? mSphere 2020, 5, 00367. [Google Scholar] [CrossRef]
- Sallenave, J.-M.; Guillot, L. Innate Immune Signaling and Proteolytic Pathways in the Resolution or Exacerbation of SARS-CoV-2 in Covid-19: Key Therapeutic Targets? Front. Immunol. 2020, 11, 1229. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tan, Y.; Ling, Y. Viral and host factors related to the clinical outcome of COVID-19. Nature 2020, 583, 437–440. [Google Scholar] [CrossRef]
- Yu, S.-Y.; Hu, Y.-W.; Liu, X.-Y.; Xiong, W.; Zhou, Z.-T.; Yuan, Z.-H. Gene expression profiles in peripheral blood mononuclear cells of SARS patients. World J. Gastroenterol. 2005, 11, 5037–5043. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Zhan, Y.; Wu, L.; Yu, X.; Zhang, W.; Ye, L.; Xu, S.; Sun, R.; Wang, Y.; et al. Analysis of Serum Cytokines in Patients with Severe Acute Respiratory Syndrome. Infect. Immun. 2004, 72, 4410–4415. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-J.; Su, I.-J.; Theron, M.; Wu, Y.-C.; Lai, S.-K.; Liu, C.-C.; Lei, H.-Y. An interferon-γ-related cytokine storm in SARS patients. J. Med. Virol. 2004, 75, 185–194. [Google Scholar] [CrossRef]
- Law, H.K.-W.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.M.; Lau, Y.L. Chemokine up-regulation in SARS-coronavirus–infected, monocyte-derived human dendritic cells. Blood 2005, 106, 2366–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, M.; Weber, F. Inhibition of cytokine gene expression and induction of chemokine genes in non-lymphatic cells infected with SARS coronavirus. Virol. J. 2006, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Huang, M.; Liu, D.X. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus–host interactions. Virus Res. 2014, 194, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Burwick, N.; Aktas, B.H. The eIF2-alpha kinase HRI: A potential target beyond the red blood cell. Expert Opin. Ther. Targets 2017, 21, 1171–1177. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Wang, Z.; Yu, L. GCN2 reduces inflammation by p-eIF2α/ATF4 pathway after intracerebral hemorrhage in mice. Exp. Neurol. 2019, 313, 16–25. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Sun, Z.; Zhang, Q. Intermittent-Hypoxia-Induced Autophagy Activation Through the ER-Stress-Related PERK/eIF2α/ATF4 Pathway is a Protective Response to Pancreatic β-Cell Apoptosis. Cell. Physiol. Biochem. 2018, 51, 2955–2971. [Google Scholar] [CrossRef]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during Coronavirus Infectious Bronchitis Virus Infection Modulates Apoptosis by Restricting Activation of the Extracellular Signal-Regulated Kinase Pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [Green Version]
- Kraähling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Mühlberger, E. Severe Acute Respiratory Syndrome Coronavirus Triggers Apoptosis via Protein Kinase R but Is Resistant to Its Antiviral Activity. J. Virol. 2008, 83, 2298–2309. [Google Scholar] [CrossRef] [Green Version]
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.M.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; De Groot, R.J.; et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016, 12, e1005982. [Google Scholar] [CrossRef]
- Wang, X.; Liao, Y.; Yap, P.L.; Png, K.J.; Tam, J.P.; Liu, D.X. Inhibition of Protein Kinase R Activation and Upregulation of GADD34 Expression Play a Synergistic Role in Facilitating Coronavirus Replication by Maintaining De Novo Protein Synthesis in Virus-Infected Cells. J. Virol. 2009, 83, 12462–12472. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Fu, F.; Ma, Y.; Zhang, X.; Li, L.; Feng, L.; Liu, P. The PERK Arm of the Unfolded Protein Response Negatively Regulates Transmissible Gastroenteritis Virus Replication by Suppressing Protein Translation and Promoting Type I Interferon Production. J. Virol. 2018, 92, 00431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Xia, Y. c-Jun, at the crossroad of the signaling network. Protein Cell 2011, 2, 889–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acquaviva, C.; Bossis, G.; Ferrara, P.; Brockly, F.; Jariel-Encontre, I.; Piechaczyk, M. Multiple Degradation Pathways for Fos Family Proteins. Ann. N. Y. Acad. Sci. 2002, 973, 426–434. [Google Scholar] [CrossRef]
- Bai, F.; Liu, K.; Li, H.; Wang, J.; Zhu, J.; Hao, P.; Zhu, L.; Zhang, S.; Shan, L.; Ma, W.; et al. Veratramine modulates AP-1-dependent gene transcription by directly binding to programmable DNA. Nucleic Acids Res. 2017, 46, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Cinatl, J.; Hoever, G.; Morgenstern, B.; Preiser, W.; Vogel, J.-U.; Hofmann, W.-K.; Bauer, G.; Michaelis, M.; Rabenau, H.F.; Doerr, H.W. Infection of cultured intestinal epithelial cells with severe acute respiratory syndrome coronavirus. Cell Mol. Life Sci. 2004, 61, 2100–2112. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Activation of the c-Jun NH2-terminal kinase pathway by coronavirus infectious bronchitis virus promotes apoptosis independently of c-Jun. Cell Death Dis. 2017, 8, 3215. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.-Y.; Chang, S.C.; Wu, H.-Y.; Yu, T.-C.; Wei, W.-C.; Lin, S.; Chien, C.-L.; Chang, M.-F. Upregulation of the Chemokine (C-C Motif) Ligand 2 via a Severe Acute Respiratory Syndrome Coronavirus Spike-ACE2 Signaling Pathway. J. Virol. 2010, 84, 7703–7712. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Leeson, A.; Andonov, A.; Li, Y.; Bastien, N.; Cao, J.; Osiowy, C.; Dobie, F.; Cutts, T.; Ballantine, M.; et al. Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 2003, 311, 870–876. [Google Scholar] [CrossRef]
- Obitsu, S.; Ahmed, N.; Nishitsuji, H.; Hasegawa, A.; Nakahama, K.-I.; Morita, I.; Nishigaki, K.; Hayashi, T.; Masuda, T.; Kannagi, M. Potential enhancement of osteoclastogenesis by severe acute respiratory syndrome coronavirus 3a/X1 protein. Arch. Virol. 2009, 154, 1457–1464. [Google Scholar] [CrossRef] [Green Version]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef] [Green Version]
- Varshney, B.; Lal, S.K. SARS-CoV Accessory Protein 3b Induces AP-1 Transcriptional Activity through Activation of JNK and ERK Pathways. Biochemistry 2011, 50, 5419–5425. [Google Scholar] [CrossRef]
- Varshney, B.; Agnihotram, S.; Tan, Y.-J.; Baric, R.; Lal, S.K. SARS Coronavirus 3b Accessory Protein Modulates Transcriptional Activity of RUNX1b. PLoS ONE 2012, 7, e29542. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, X.; Huang, M.; Tam, J.P.; Liu, D.X. Regulation of the p38 mitogen-activated protein kinase and dual-specificity phosphatase 1 feedback loop modulates the induction of interleukin 6 and 8 in cells infected with coronavirus infectious bronchitis virus. Virology 2011, 420, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.X.; Liang, J.Q.; Zhu, Q.C. Gammacoronavirus Avian Infectious Bronchitis Virus and Alphacoronavirus Porcine Epidemic Diarrhea Virus Exploit a Cell-Survival Strategy via Upregulation of cFOS to Promote Viral Replication. J. Virol. 2020, 95, 02107–02120. [Google Scholar] [CrossRef] [PubMed]
- Thornbrough, J.M.; Jha, B.K.; Yount, B.; Goldstein, S.A.; Li, Y.; Elliott, R.; Sims, A.C.; Baric, R.S.; Silverman, R.H.; Weiss, S.R. Middle East Respiratory Syndrome Coronavirus NS4b Protein Inhibits Host RNase L Activation. mBio 2016, 7, e00258-16. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, K.; Narayanan, K.; Wada, M.; Makino, S. Inhibition of Stress Granule Formation by Middle East Respiratory Syndrome Coronavirus 4a Accessory Protein Facilitates Viral Translation, Leading to Efficient Virus Replication. J. Virol. 2018, 92, e00902-18. [Google Scholar] [CrossRef] [Green Version]
- Comar, C.E.; Goldstein, S.A.; Li, Y.; Yount, B.; Baric, R.S.; Weiss, S.R. Antagonism of dsRNA-Induced Innate Immune Pathways by NS4a and NS4b Accessory Proteins during MERS Coronavirus Infection. mBio 2019, 10, e00319-19. [Google Scholar] [CrossRef] [Green Version]
- Versteeg, G.A.; Van De Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J.M. The Coronavirus Spike Protein Induces Endoplasmic Reticulum Stress and Upregulation of Intracellular Chemokine mRNA Concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-J.; Liu, C.Y.-Y.; Chiang, B.-L.; Chao, Y.-C.; Chen, C.-C. Induction of IL-8 Release in Lung Cells via Activator Protein-1 by Recombinant Baculovirus Displaying Severe Acute Respiratory Syndrome-Coronavirus Spike Proteins: Identification of Two Functional Regions. J. Immunol. 2004, 173, 7602–7614. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liao, Y.; Liu, D.X. The endoplasmic reticulum stress sensor IRE1α protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Yu, C.-J.; Luh, K.-T.; Kuo, S.-H.; Lee, Y.-C.; Yang, P.-C. Aberrant p53 Expression Correlates with Expression of Vascular Endothelial Growth Factor mRNA and Interleukin-8 mRNA and Neoangiogenesis in Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2002, 20, 900–910. [Google Scholar] [CrossRef]
- Lowe, J.M.; Menendez, D.; Bushel, P.R. p53 and NF-κB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014, 74, 2182–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatz, M.; Menendez, D.; Resnick, M.A. The Human TLR Innate Immune Gene Family Is Differentially Influenced by DNA Stress and p53 Status in Cancer Cells. Cancer Res. 2012, 72, 3948–3957. [Google Scholar] [CrossRef] [Green Version]
- Bechill, J.; Chen, Z.; Brewer, J.W.; Baker, S.C. Coronavirus Infection Modulates the Unfolded Protein Response and Mediates Sustained Translational Repression. J. Virol. 2008, 82, 4492–4501. [Google Scholar] [CrossRef] [Green Version]
- Cruz, J.L.G.; Sola, I.; Becares, M.; Alberca, B.; Plana, J.; Enjuanes, L.; Zuñiga, S. Coronavirus Gene 7 Counteracts Host Defenses and Modulates Virus Virulence. PLoS Pathog. 2011, 7, e1002090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavarino, G.; Claudio, N.; Dalet, A.; Terawaki, S.; Couderc, T.; Chasson, L.; Ceppi, M.; Schmidt, E.K.; Wenger, T.; Lecuit, M.; et al. Protein phosphatase 1 subunit Ppp1r15a/GADD34 regulates cytokine production in polyinosinic:polycytidylic acid-stimulated dendritic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3006–3011. [Google Scholar] [CrossRef] [Green Version]
- Clavarino, G.; Cláudio, N.; Couderc, T. Induction of GADD34 is necessary for dsRNA-dependent interferon-β production and participates in the control of Chikungunya virus infection. PLoS Pathog. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]
- Dalet, A.; Argüello, R.J.; Combes, A.; Spinelli, L.; Jaeger, S.; Fallet, M.; Manh, T.V.; Mendes, A.; Perego, J.; Reverendo, M.; et al. Protein synthesis inhibition and GADD34 control IFN-β heterogeneous expression in response to dsRNA. EMBO J. 2017, 36, 761–782. [Google Scholar] [CrossRef] [Green Version]
- Marks, F.; Klingmüller, U.; Müller-Decker, K. Chapter 11: Mitogen-activated protein kinase and nuclear factor κB modules. In Cellular Signal Processing: An Introduction to the Molecular Mechanisms of Signal Transduction, 2nd ed.; Garland Science Boca Raton: New York, NY, USA, 2017; pp. 395–422. [Google Scholar]
- Alfonso Pecchio, A.R.; Cardozo Gizzi, A.M.; Renner, M.L.; Molina-Calavita, M.; Caputto, B.L. c-Fos activates and physically interacts with specific enzymes of the pathway of synthesis of polyphosphoinositides. Mol. Biol. Cell 2011, 22, 4716–4725. [Google Scholar] [CrossRef]
- Guardeno, J.M.J.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Enjuanes, L. The PDZ-Binding Motif of Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Is a Determinant of Viral Pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.G.; Shen, S.; Tay, F.P.; Liu, D. Selection of and recombination between minor variants lead to the adaptation of an avian coronavirus to primate cells. Biochem. Biophys. Res. Commun. 2005, 336, 417–423. [Google Scholar] [CrossRef]
- Lim, K.; Liu, D. Characterization of the Two Overlapping Papain-like Proteinase Domains Encoded in Gene 1 of the Coronavirus Infectious Bronchitis Virus and Determination of the C-Terminal Cleavage Site of an 87-kDa Protein. Virology 1998, 245, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Kim, H.K.; Song, D.S.; An, D.J.; Park, B.K. Complete Genome Sequences of a Korean Virulent Porcine Epidemic Diarrhea Virus and Its Attenuated Counterpart. J. Virol. 2012, 86, 5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.Q.; Xiao, H.; Tam, J.P.; Liu, D. Sumoylation of the nucleocapsid protein of severe acute respiratory syndrome coronavirus. FEBS Lett. 2005, 579, 2387–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.H.; Davidson, I.; Matthes, H.; Garnier, J.-M.; Chambon, P. Cloning, expression, and transcriptional properties of the human enhancer factor TEF-1. Cell 1991, 65, 551–568. [Google Scholar] [CrossRef]
- Lei, K.; Nimnual, A.; Zong, W.-X.; Kennedy, N.J.; Flavell, R.A.; Thompson, C.B.; Bar-Sagi, D.; Davis, R.J. The Bax Subfamily of Bcl2-Related Proteins Is Essential for Apoptotic Signal Transduction by c-Jun NH2-Terminal Kinase. Mol. Cell Biol. 2002, 22, 4929–4942. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Q.C.; Li, S.; Yuan, L.X.; Chen, R.A.; Liu, D.X.; Fung, T.S. Induction of the Proinflammatory Chemokine Interleukin-8 Is Regulated by Integrated Stress Response and AP-1 Family Proteins Activated during Coronavirus Infection. Int. J. Mol. Sci. 2021, 22, 5646. https://doi.org/10.3390/ijms22115646
Zhu QC, Li S, Yuan LX, Chen RA, Liu DX, Fung TS. Induction of the Proinflammatory Chemokine Interleukin-8 Is Regulated by Integrated Stress Response and AP-1 Family Proteins Activated during Coronavirus Infection. International Journal of Molecular Sciences. 2021; 22(11):5646. https://doi.org/10.3390/ijms22115646
Chicago/Turabian StyleZhu, Qing Chun, Shumin Li, Li Xia Yuan, Rui Ai Chen, Ding Xiang Liu, and To Sing Fung. 2021. "Induction of the Proinflammatory Chemokine Interleukin-8 Is Regulated by Integrated Stress Response and AP-1 Family Proteins Activated during Coronavirus Infection" International Journal of Molecular Sciences 22, no. 11: 5646. https://doi.org/10.3390/ijms22115646
APA StyleZhu, Q. C., Li, S., Yuan, L. X., Chen, R. A., Liu, D. X., & Fung, T. S. (2021). Induction of the Proinflammatory Chemokine Interleukin-8 Is Regulated by Integrated Stress Response and AP-1 Family Proteins Activated during Coronavirus Infection. International Journal of Molecular Sciences, 22(11), 5646. https://doi.org/10.3390/ijms22115646