
Genome Evolutionary Dynamics Meets Functional Genomics: A Case Story on the Identification of SLC25A44


Abstract
:
1. Introduction
2. Results
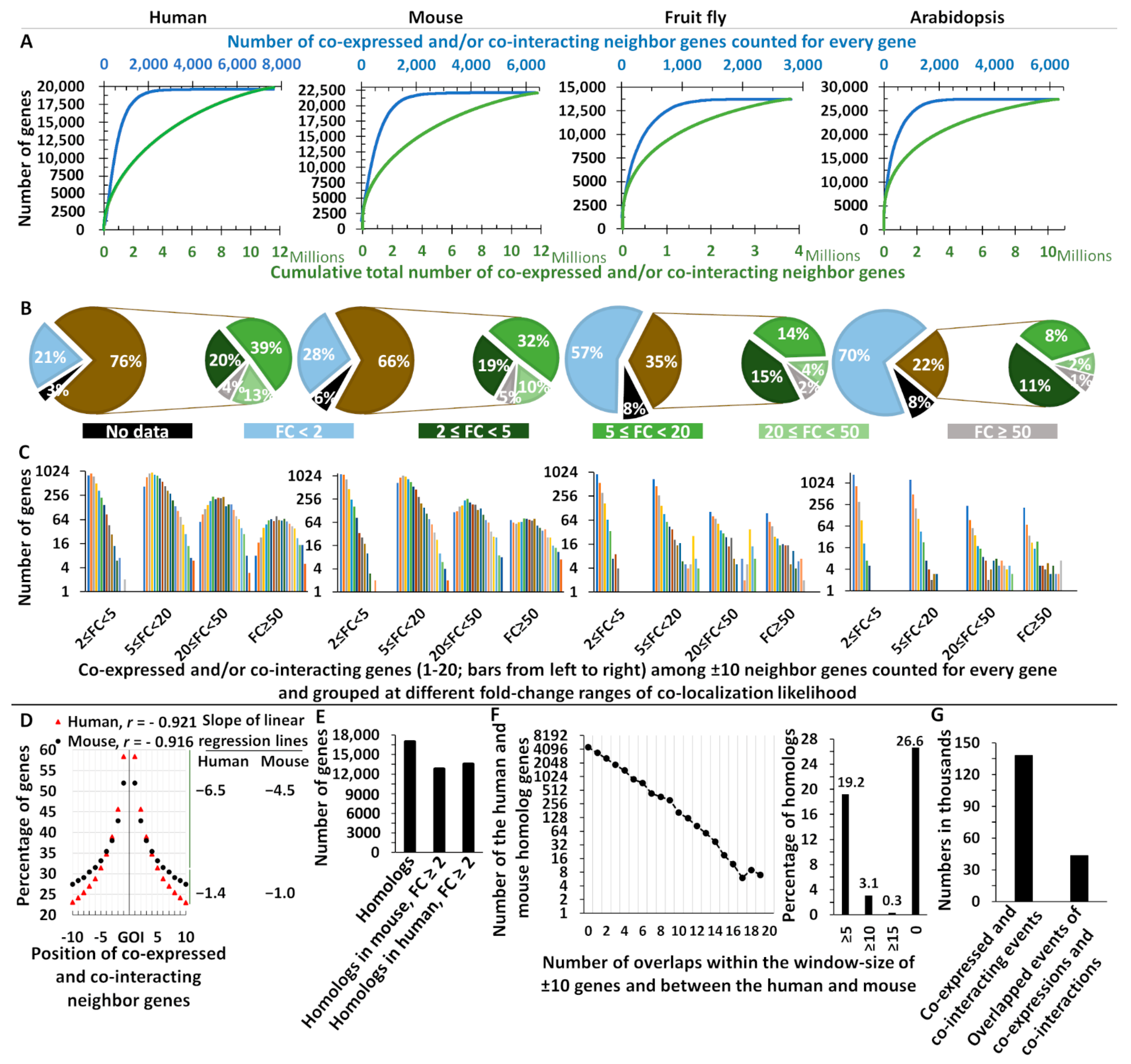
2.1. Genome-Wide Evidence for Co-Localization of Co-Interacting, Co-Expressed, and Functionally Related Genes
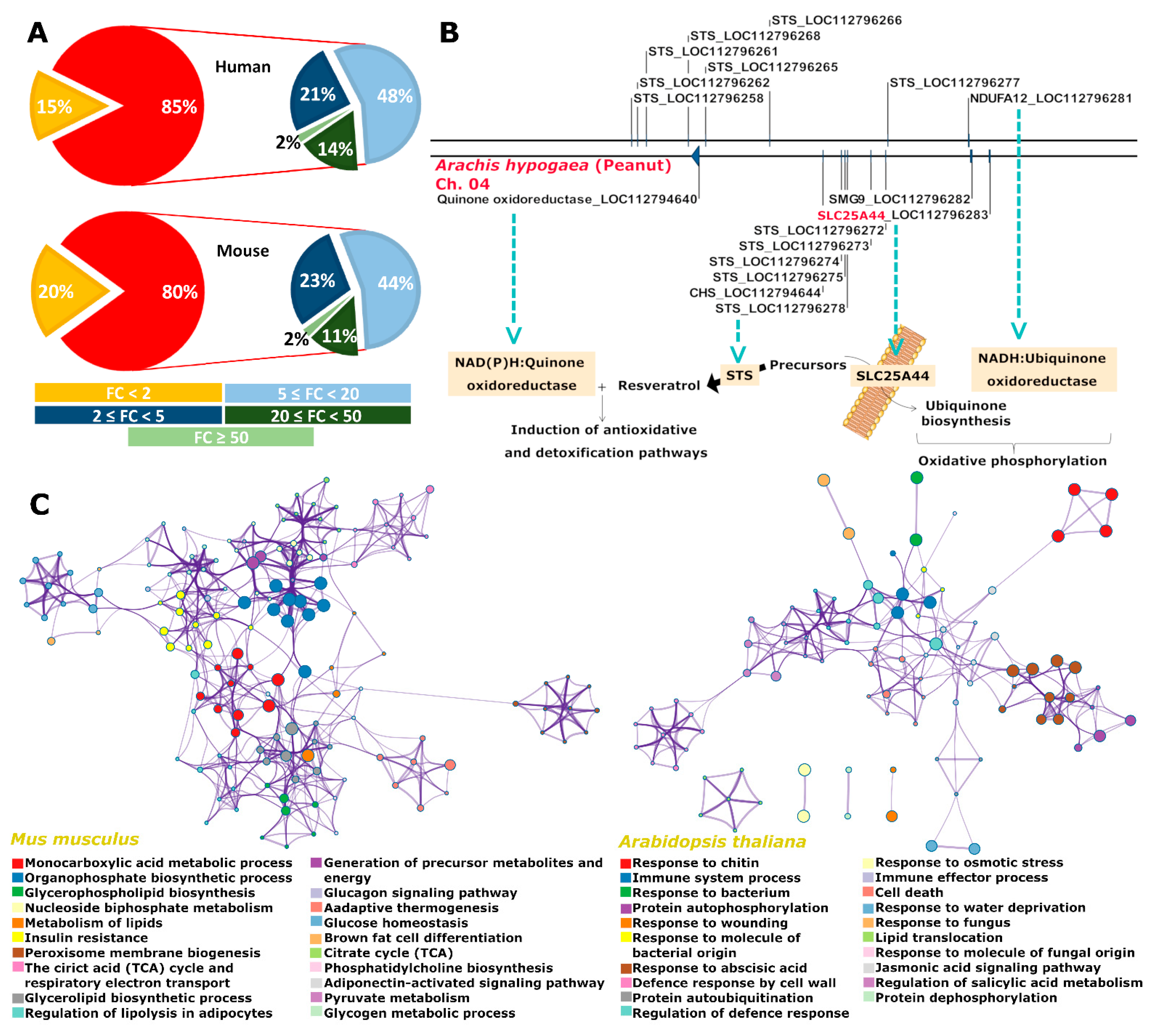
2.2. A Case Story on the Identification of Slc25A44
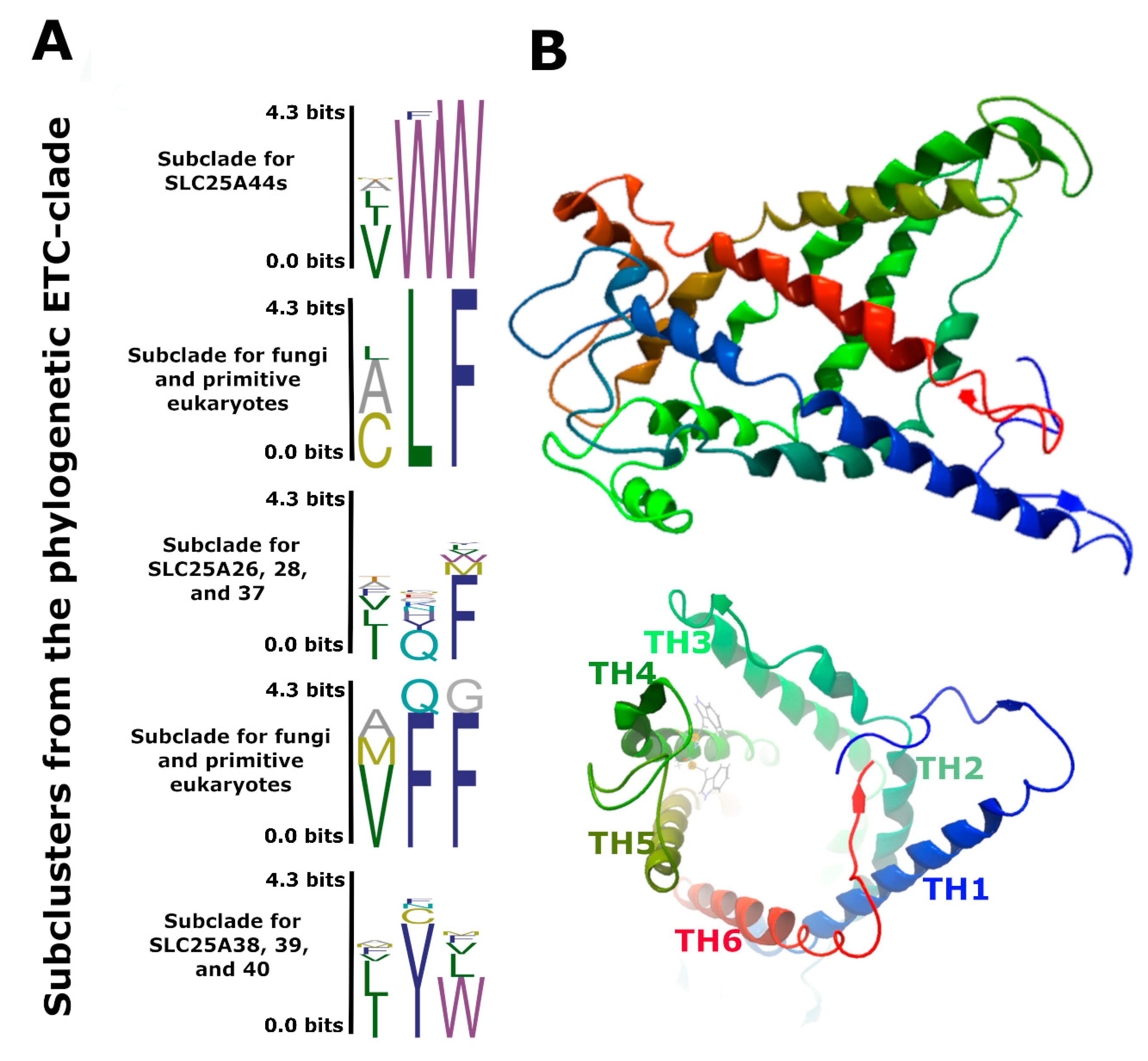
2.3. Slc25A44 Is a Highly Conserved Housekeeping Transporter Gene
2.4. Functional Characterization of AdSlc25A44 in Xenopus Oocytes
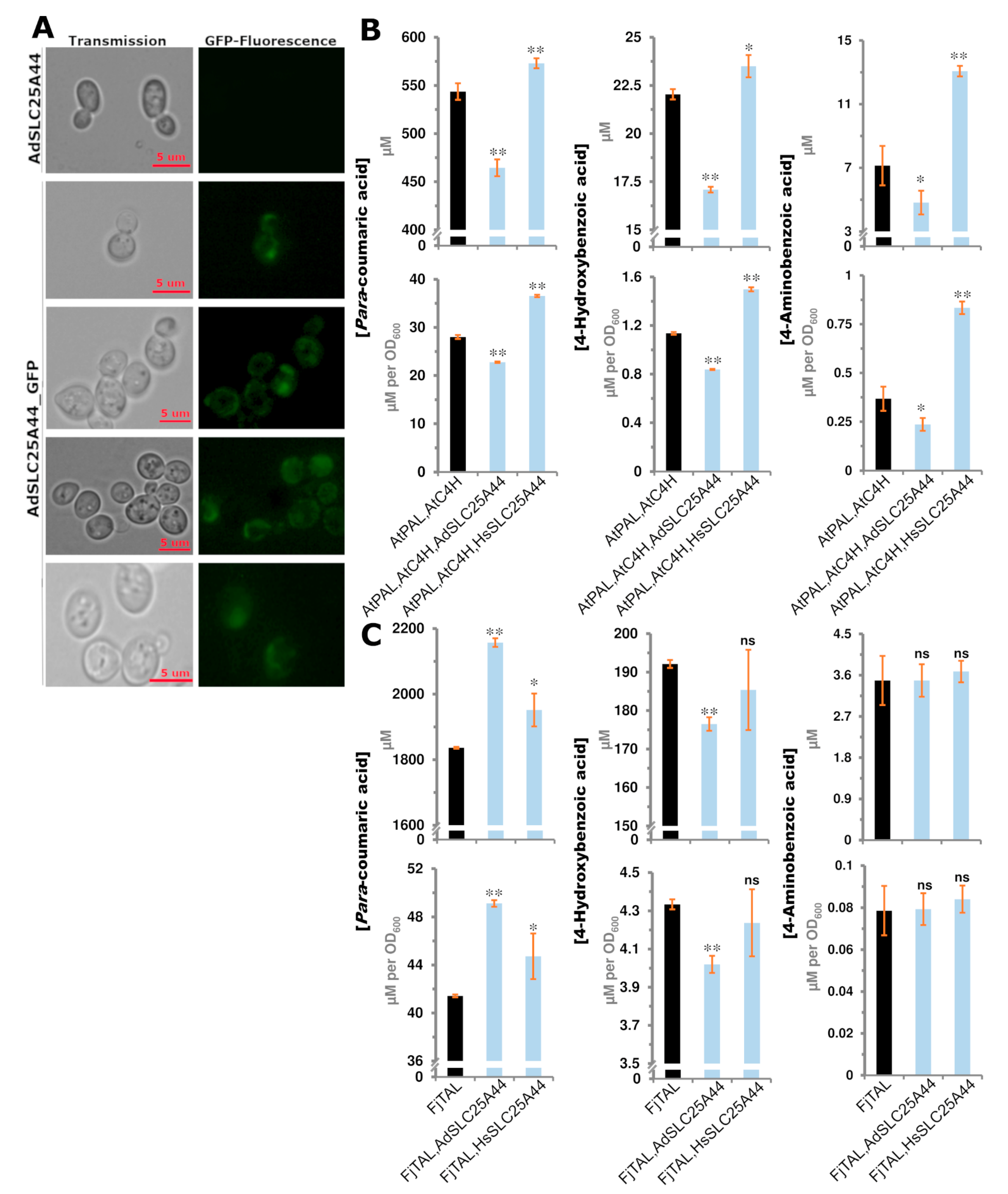
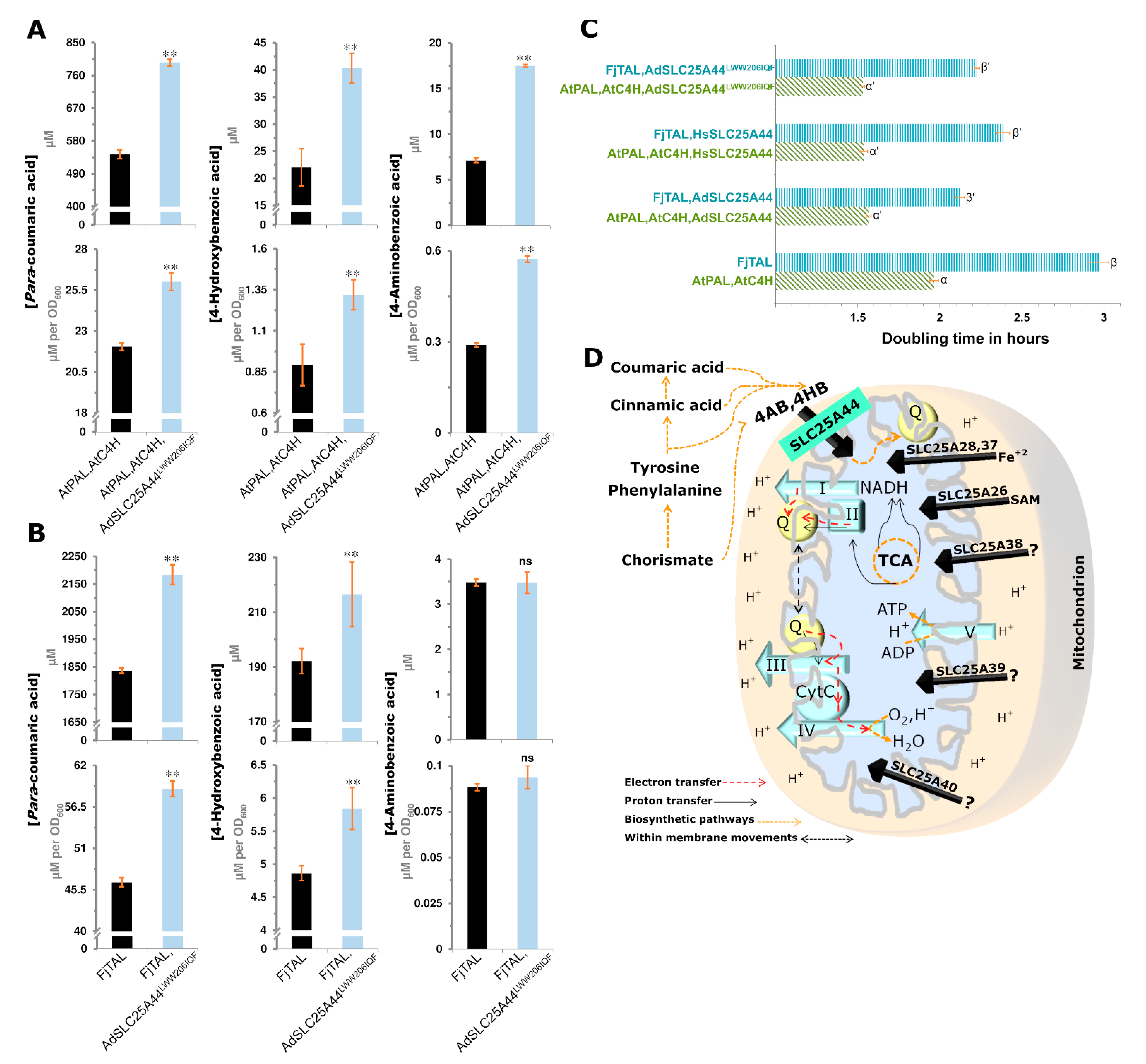
2.5. SLC25A44 Localized onto the Mitochondria, ER, and Nucleus of Yeast and Had Impact on the Production of Para-Coumaric, 4-Aminobenzoic, and 4-Hydroxybenzoic Acids
2.6. Rational Transporter Engineering toward Higher Production of Para-Courmaric Acid
3. Discussion
4. Materials and Methods
4.1. DNA Constructs
4.2. Yeast Strain Construction
4.3. Media and Yeast Cultivation Conditions
4.4. Transport Assays in Xenopus Oocytes
4.5. Chemicals and HPLC/LC-MS Analyses
4.6. Bioinformatics and Polymorphism Analysis
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duboule, D.; Dollé, P. The structural and functional organization of the murine HOX gene family resembles that of Drosophila homeotic genes. EMBO J. 1989, 8, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Darbani, B.; Motawia, M.S.; Olsen, C.E.; Nour-Eldin, H.H.; Møller, B.L.; Rook, F. The biosynthetic gene cluster for the cyanogenic glucoside dhurrin in Sorghum bicolor contains its co-expressed vacuolar MATE transporter. Sci. Rep. 2016, 6, 37079. [Google Scholar] [CrossRef] [Green Version]
- Darbani, B.; Stovicek, V.; van der Hoek, S.A.; Borodina, I. Engineering energetically efficient transport of dicarboxylic acids in yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2019, 116, 19415–19420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, D.; Maimann, A.B.; Macea, E.; Ocampo, C.H.; Cardona, G.; Pičmanová, M.; Darbani, B.; Olsen, C.E.; Debouck, D.; Raatz, B.; et al. Biosynthesis of cyanogenic glucosides in Phaseolus lunatus and the evolution of oxime-based defenses. Plant Direct 2020, 4, e00244. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, A.; Bhattacharya, N.; Olm, M.R.; Song, Y.S.; Banfield, J.F. Transporter genes in biosynthetic gene clusters predict metabolite characteristics and siderophore activity. Genome Res. 2020, 31, 239–250. [Google Scholar] [CrossRef]
- Darbani, B.; Kell, D.B.; Borodina, I. Energetic evolution of cellular Transportomes. BMC Genom. 2018, 19, 418. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Leroch, M.; Neuhaus, H.E.; Kirchberger, S.; Zimmermann, S.; Melzer, M.; Gerhold, J.; Tjaden, J. Identification of a Novel Adenine Nucleotide Transporter in the Endoplasmic Reticulum of Arabidopsis. Plant Cell 2008, 20, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, L.; Rottensteiner, H.; Girzalsky, W.; Scarcia, P.; Palmieri, F.; Erdmann, R. Identification and functional reconstitution of the yeast peroxisomal adenine nucleotide transporter. EMBO J. 2001, 20, 5049–5059. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.P.M.; Schwacke, R.; Flügge, U.-I. Solute transporters of the plastid envelope membrane. Annu. Rev. Plant Biol. 2005, 56, 133–164. [Google Scholar] [CrossRef]
- Takos, A.M.; Rook, F. Why biosynthetic genes for chemical defense compounds cluster. Trends Plant Sci. 2012, 17, 383–388. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, A.; Fisher, G.; Migliaccio, N.; Cammisa, G.; D’Aniello, E.; Spinelli, P. Amino acids and transaminases activity in ventricular CSF and in brain of normal and Alzheimer patients. Neurosci. Lett. 2005, 388, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-L.; Fan, Y.-G.; Yang, Z.-S.; Wang, Z.-Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium A global reference for human genetic variation. Nature 2015, 526, 68–74. [CrossRef] [Green Version]
- Wierckx, N.; Agrimi, G.; Lübeck, P.S.; Steiger, M.G.; Mira, N.P.; Punt, P.J. Metabolic specialization in itaconic acid production: A tale of two fungi. Curr. Opin. Biotechnol. 2019, 62, 153–159. [Google Scholar] [CrossRef]
- Sweetman, C.; Wong, D.C.; Ford, C.M.; Drew, D.P. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genom. 2012, 13, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrimi, G.; Di Noia, M.A.; Marobbio, C.M.T.; Fiermonte, G.; Lasorsa, F.M.; Palmieri, F. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 2004, 379, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nat. Cell Biol. 2006, 440, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef]
- Nilsson, R.; Schultz, I.J.; Pierce, E.L.; Soltis, K.A.; Naranuntarat, A.; Ward, D.M.; Baughman, J.M.; Paradkar, P.N.; Kingsley, P.D.; Culotta, V.C.; et al. Discovery of Genes Essential for Heme Biosynthesis through Large-Scale Gene Expression Analysis. Cell Metab. 2009, 10, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Kishita, Y.; Pajak, A.; Bolar, N.A.; Marobbio, C.M.T.; Maffezzini, C.; Miniero, D.V.; Monné, M.; Kohda, M.; Stranneheim, H.; Murayama, K.; et al. Intra-mitochondrial Methylation Deficiency Due to Mutations in SLC25A26. Am. J. Hum. Genet. 2015, 97, 761–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1823, 1604–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darbani, B.; Briat, J.-F.; Holm, P.B.; Husted, S.; Noeparvar, S.; Borg, S. Dissecting plant iron homeostasis under short and long-term iron fluctuations. Biotechnol. Adv. 2013, 31, 1292–1307. [Google Scholar] [CrossRef]
- Darbani, B.; Noeparvar, S.; Borg, S. Deciphering Mineral Homeostasis in Barley Seed Transfer Cells at Transcriptional Level. PLoS ONE 2015, 10, e0141398. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Baerenfaller, K.; Grossmann, J.; Grobei, M.A.; Hull, R.; Hirsch-Hoffmann, M.; Yalovsky, S.; Zimmermann, P.; Grossniklaus, U.; Gruissem, W.; Baginsky, S. Genome-Scale Proteomics Reveals Arabidopsis thaliana Gene Models and Proteome Dynamics. Science 2008, 320, 938–941. [Google Scholar] [CrossRef]
- Schmid, M.; Davison, T.S.; Henz, S.R.; Pape, U.J.; Demar, M.; Vingron, M.; Schölkopf, B.; Weigel, D.; Lohmann, J.U. A gene expression map of Arabidopsis thaliana development. Nat. Genet. 2005, 37, 501–506. [Google Scholar] [CrossRef]
- He, Y.-N.; Peng, J.-S.; Cai, Y.; Liu, D.-F.; Guan, Y.; Yi, H.-Y.; Gong, J.-M. Tonoplast-localized nitrate uptake transporters involved in vacuolar nitrate efflux and reallocation in Arabidopsis. Sci. Rep. 2017, 7, 6417. [Google Scholar] [CrossRef]
- Gutiérrez-Aguilar, M.; Baines, C.P. Physiological and pathological roles of mitochondrial SLC25 carriers. Biochem. J. 2013, 454, 371–386. [Google Scholar] [CrossRef] [Green Version]
- Huh, W.-K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nat. Cell Biol. 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Li, M.; Schneider, K.; Kristensen, M.; Borodina, I.; Nielsen, J. Engineering yeast for high-level production of stilbenoid antioxidants. Sci. Rep. 2016, 6, 36827. [Google Scholar] [CrossRef] [Green Version]
- Marbois, B.; Xie, L.X.; Choi, S.; Hirano, K.; Hyman, K.; Clarke, C.F. para-Aminobenzoic Acid Is a Precursor in Coenzyme Q6 Biosynthesis in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 27827–27838. [Google Scholar] [CrossRef] [Green Version]
- Payet, L.-A.; Leroux, M.; Willison, J.C.; Kihara, A.; Pelosi, L.; Pierrel, F. Mechanistic Details of Early Steps in Coenzyme Q Biosynthesis Pathway in Yeast. Cell Chem. Biol. 2016, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.; Kildegaard, K.R.; Li, M.; Borodina, I.; Nielsen, J. Establishment of a yeast platform strain for production of p-coumaric acid through metabolic engineering of aromatic amino acid biosynthesis. Metab. Eng. 2015, 31, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achnine, L.; Blancaflor, E.B.; Rasmussen, S.; Dixon, R.A. Colocalization of l-Phenylalanine Ammonia-Lyase and Cinnamate 4-Hydroxylase for Metabolic Channeling in Phenylpropanoid Biosynthesis. Plant Cell 2004, 16, 3098–3109. [Google Scholar] [CrossRef] [PubMed]
- Zaslaver, A.; Mayo, A.; Ronen, M.; Alon, U. Optimal gene partition into operons correlates with gene functional order. Phys. Biol. 2006, 3, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, J.N.; Bergendahl, L.T.; Marsh, J.A. Operon Gene Order Is Optimized for Ordered Protein Complex Assembly. Cell Rep. 2016, 14, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Hurst, L.D.; Pál, C.; Lercher, M.J. The evolutionary dynamics of eukaryotic gene order. Nat. Rev. Genet. 2004, 5, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.J.; Campbell, M.J.; Winzeler, E.A.; Steinmetz, L.; Conway, A.; Wodicka, L.; Wolfsberg, T.G.; Gabrielian, A.E.; Landsman, D.; Lockhart, D.J.; et al. A Genome-Wide Transcriptional Analysis of the Mitotic Cell Cycle. Mol. Cell 1998, 2, 65–73. [Google Scholar] [CrossRef]
- Cohen, B.A.; Mitra, R.D.; Hughes, J.D.; Church, G.M. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 2000, 26, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Boutanaev, A.M.; Kalmykova, A.I.; Shevelyov, Y.Y.; Nurminsky, D.I. Large clusters of co-expressed genes in the Drosophila genome. Nat. Cell Biol. 2002, 420, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Lercher, M.J.; Urrutia, A.O.; Hurst, L.D. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Genet. 2002, 31, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Sonnhammer, E.L.L. Genomic Gene Clustering Analysis of Pathways in Eukaryotes. Genome Res. 2003, 13, 875–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Threlfall, D.R.; Law, A.; Whistance, G.R. Cinnamic acid and p-coumaric acid, precursors of ubiquinone in higher plants, green algae and fungi. Biochem. J. 1970, 118, 55P. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, F.L.; Hatefi, Y.; Lester, R.L.; Widmer, C. Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta (BBA)-Bioenerg. 1957, 25, 220–221. [Google Scholar] [CrossRef]
- de Macêdo, J.P.; Burkard, G.S.; Niemann, M.; Barrett, M.P.; Vial, H.; Mäser, P.; Roditi, I.; Schneider, A.; Bütikofer, P. An Atypical Mitochondrial Carrier That Mediates Drug Action in Trypanosoma brucei. PLoS Pathog. 2015, 11, e1004875. [Google Scholar] [CrossRef] [Green Version]
- Yanos, M.E.; Bennett, C.F.; Kaeberlein, M. Genome-Wide RNAi Longevity Screens in Caenorhabditis elegans. Curr. Genom. 2012, 13, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Sukonina, V.; Ma, H.; Zhang, W.; Bartesaghi, S.; Subhash, S.; Heglind, M.; Foyn, H.; Betz, M.J.; Nilsson, D.; Lidell, M.E.; et al. FOXK1 and FOXK2 regulate aerobic glycolysis. Nat. Cell Biol. 2019, 566, 279–283. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial Diseases in Man and Mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.J.; Wang, Z.; Heymsfield, S.B.; Schautz, B.; Bosy-Westphal, A. Advances in the understanding of specific metabolic rates of major organs and tissues in humans. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 501–508. [Google Scholar] [CrossRef]
- Yoneshiro, T.; Wang, Q.; Tajima, K.; Matsushita, M.; Maki, H.; Igarashi, K.; Dai, Z.; White, P.J.; McGarrah, R.W.; Ilkayeva, O.R.; et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nat. Cell Biol. 2019, 572, 614–619. [Google Scholar] [CrossRef]
- Buryanovskyy, L.; Fu, Y.; Boyd, M.; Ma, Y.; Hsieh, T.-C.; Wu, J.M.; Zhang, Z. Crystal Structure of Quinone Reductase 2 in Complex with Resveratrol. Biochemistry 2004, 43, 11417–11426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gietz, R.D.; Woods, R.A. Yeast Transformation by the LiAc/SS Carrier DNA/PEG Method. Methods Mol. Biol. 2006, 313, 107–120. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants: Figure 1. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef] [PubMed]
- Toufighi, K.; Brady, S.M.; Austin, R.; Ly, E.; Provart, N.J. The Botany Array Resource: E-Northerns, Expression Angling, and promoter analyses. Plant J. 2005, 43, 153–163. [Google Scholar] [CrossRef]
- Yang, S.; Kim, C.Y.; Hwang, S.; Kim, E.; Kim, H.; Shim, H.; Lee, I. COEXPEDIA: Exploring biomedical hypotheses via co-expressions associated with medical subject headings (MeSH). Nucleic Acids Res. 2017, 45, D389–D396. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Akiyama, K.; Kurotani, A.; Iida, K.; Kuromori, T.; Shinozaki, K.; Sakurai, T. RARGE II: An Integrated Phenotype Database of Arabidopsis Mutant Traits Using a Controlled Vocabulary. Plant Cell Physiol. 2014, 55, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef] [PubMed]
- Youens-Clark, K.; Buckler, E.; Casstevens, T.; Chen, C.; DeClerck, G.; Derwent, P.; Dharmawardhana, P.; Jaiswal, P.; Kersey, P.; Karthikeyan, A.S.; et al. Gramene database in 2010: Updates and extensions. Nucleic Acids Res. 2011, 39, D1085–D1094. [Google Scholar] [CrossRef]
- The International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nat. Cell Biol. 2010, 467, 52–58. [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-Wide Identification of Episodic Selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernersson, R.; Pedersen, A.G. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003, 31, 3537–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S.L.K.; Frost, S.D.W. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Chr./Contig | Co-Localized Loci |
|---|---|---|
| Vitisvinifera | Chr. 14 | Slc25A44 (XP_010660575.1), NdufA4 (XP_002281305.1) |
| Chr. 16 | 4Cl2 (XP_002274994.1), NdufS6 (XP_002276949.1), Smg9 (XP_010662114.1), NdufA12 (XP_002276966.1), Cox11 (XP_002277478.1) | |
| Chr. 13 | Slc25A38 (XP_002277407.1), Atp5H/ATP synthase d (XP_002277452.1), Tim14-1 (XP_002277365.1) | |
| Cajanuscajan | Chr. 8 | Slc25A44 (XP_020221994.1), Lyrm (XP_020221999.1) |
| NW_017984070.1 | Chalcone synthase (XP_020230031.1), NdufA12 (XP_020230179.1), Smg9 (XP_020230144.1), 4Cl2 (XP_020230139.1 | |
| Chr. 2 | Slc25A38 (XP_020234579.1), Atp5H/ATP synthase d (XP_020202519.1) | |
| Glycine max | Chr. 9 | Slc25A44 (XP_025979445.1), Lyrm (XP_025979447.1) |
| Chr. 11 | 4Cl3 (NP_001237270.1), Smg9 (XP_014619235.1); NdufA12 (NP_001235085.1), Chalcone synthase (NP_001304585.2) | |
| Chr. 20 | Slc25A38 (XP_003556216.1), Atp5H/ATP synthase d (NP_001235072.1) | |
| Phaseolus vulgaris | Chr. 6 | Slc25A44 (XP_007147410.1), Lyrm (XP_007147403.1) |
| Chr. 2 | Chalcone synthase (XP_007157053.1), NdufA12 (XP_007157055.1), Smg9 (XP_007157056.1), 4Cl (XP_007157067.1) | |
| Chr. 4 | Slc25A38 (XP_007143676.1), Atp5H/ATP synthase d (XP_007143675.1) | |
| Vigna radiata | Chr. 10 | Slc25A44 (XP_022642859.1), Lyrm (XP_014517948.1) |
| Chr. 11 | 4Cl2 (XP_014521169.1), Smg9 (XP_014519466), NdufA12 (XP_014519966.1, XP_022643154.1), Chalcone synthase (XP_014520574.1) | |
| Chr. 8 | Slc25A38 (XP_014514167.1), Atp5H/ATP synthase d (XP_014511180.1) | |
| Cicer arietinum | Chr. 1 | Slc25A44 (XP_027189994.1), Lyrm (XP_004486516.1) |
| Chr. 8 | 4Cl2 (XP_004511423.1), Smg9 (XP_004511426.1), Chalcone synthase (XP_012574354.1) | |
| Chr. 4 | Slc25A38 (XP_004496329.1), Atp5H/ATP synthase d (XP_004496331.1) | |
| Medicago truncatula | Chr. 2 | Slc25A44 (XP_024632094.1), Lyrm (XP_003594632.1) |
| Chr. 5 | 4Cl2 (XP_003610843.1), Smg9 (XP_003610849.1), Chalcone synthase (XP_013453346.1) | |
| Chr. 1 | Slc25A38 (XP_003591971.1), Atp5H/ATP synthase d (XP_003591975.1) | |
| Lupinus angustifolius | Chr. 16 | Slc25A44 (XP_019418829.1), Lyrm (XP_019418830.1) |
| Chr. 17 | Smg9 (XP_019420313.1), Chalcone synthase (XP_019420991.1) | |
| Chr. 20 | Slc25A38 (XP_019427565.1), Atp5H/ATP synthase d (XP_019427572.1) | |
| Cucumis sativus | Chr. 3 | Slc25A44 (XP_004147664.2), Lyrm (XP_004147626.1) |
| Chr. 5 | Slc25A38 (XP_004142487.1), Atp5H/ATP synthase d (XP_004142418.1), Tim14-1 (XP_004142436.1) | |
| Prunus persica | Chr. 1 | Slc25A44 (XP_020410285.1), Lyrm (XP_020410759.1) |
| Chr. 2 | Slc25A38 (XP_007220258.1), Atp5H/ATP synthase d (XP_007218513.1), Tim14-1 (XP_007219829.1) | |
| Homo sapiens | Chr. 1 | Slc25A44 (NP_001273113.1), Smg5 (NP_001310543.1) |
| Chr. 10 | Slc25A28 (XP_011538541.1), Cox15 (NP_510870.1), CutC (NP_057044.2) | |
| Gorilla gorilla gorilla | Chr. 1 | Slc25A44 (XP_004026998.1), Smg5 (XP_018879956.1) |
| Chr. 10 | Slc25A28 (XP_018890384.2), Cox15 (XP_004049980.3), CutC (XP_030870896.1) | |
| Mus musculus | Chr. 3 | Slc25A44 (NP_001139348.1), Smg5 (NP_839977.2) |
| Chr. 19 | Slc25A28 (NP_660138.1), Cox15 (NP_659123.2), CutC (NP_001107034.1) | |
| Bos Taurus | Chr. 3 | Slc25A44 (NP_001071325.1), Smg5 (NP_001077148.1) |
| Chr. 26 | Slc25A28 (NP_001192481.1), Cox15 (NP_001070329.1), CutC (NP_001193825.1) | |
| Xenopus laevis | Chr. 8S | Slc25A44 (NP_001090232.1), Smg5 (XP_018089021.1) |
| Chr. 7S | Slc25A28 (XP_018083007.1, NP_001086329.1), ATP synthase 6v0e2 (NP_001165047.1), Cox15 (XP_018083005.1) | |
| Halyomorpha halys | NW_020110602.1 | Slc25A44 (XP_014285646.1), Uqcrq (Complex III Subunit, XP_024215595.1) |
| Saccharomyces cerevisiae | Chr. 16 | Agc1 (Aspartate-glutamate carrier, YPR021C), Atp20 (F1-F0 ATPase assembly protein, YPR020W) |
| Pichia membranifaciens | NW_017566982.1 | Agc1 like (XP_019020020.1), Sqr (Complex II subunit, XP_019020021.1) |
| Schizosaccharomyces pombe | Chr. 1 | Agc1 like (NP_593068.1), Atp10 (F1-F0 ATPase assembly protein, NP_593071.1) |
| Candida tenuis | NW_006281231.1 | Agc1 like (XP_006684445.1), NdufA8 (XP_006684447.1) |
| Scheffersomyces stipites | Chr. 1 | Agc1 like (XP_001387183.2), Cox19 (Complex IV chaperone, XP_001386938.2) |
| Model | Log Likelihood | cv(svr) 1 | ω1 2 | ω2 2 | ω3 2 |
|---|---|---|---|---|---|
| Unconstrained | −22,033 | 0.65 | 0.02 (83.38%) 3 | 0.20 (16.53%) 3 | 33.86 (0.08%) 3 |
| Constrained | −22,046 | 0.651 | 0.02 (80.89%) 3 | 0.12 (17.25%) 3 | 1.00 (1.86%) 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darbani, B. Genome Evolutionary Dynamics Meets Functional Genomics: A Case Story on the Identification of SLC25A44. Int. J. Mol. Sci. 2021, 22, 5669. https://doi.org/10.3390/ijms22115669
Darbani B. Genome Evolutionary Dynamics Meets Functional Genomics: A Case Story on the Identification of SLC25A44. International Journal of Molecular Sciences. 2021; 22(11):5669. https://doi.org/10.3390/ijms22115669
Chicago/Turabian StyleDarbani, Behrooz. 2021. "Genome Evolutionary Dynamics Meets Functional Genomics: A Case Story on the Identification of SLC25A44" International Journal of Molecular Sciences 22, no. 11: 5669. https://doi.org/10.3390/ijms22115669