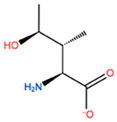
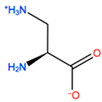
1. Introduction
Human phenotype development, evolution and physiopathological processes are regulated by several key actors. Among these,
HOX genes have been associated with the control of the final morphology [
1,
2]. Their increase or decrease in activity can often result in homeotic transformations provoking the formation of structures or organs in erroneous locations within the organism. Three different levels of
HOX genes evolutionary conservation have been identified: (1) at a molecular level, they all encode homeodomain transcription factors [
3]; (2) at a structural level,
HOX genes are usually organised in complexes, reflecting their phylogeny and regulatory aspects of their expression [
4,
5], and (3) at a functional level, they trigger similar effects in most animals and can work in substitution of an orthologue in other species [
6].
When becoming highly dysregulated and overexpressed,
HOX genes have been associated with a wide range of both solid and haematological cancers [
7].
For this reason, the processes modulated by
HOX genes have been extensively studied providing a substantial, although not exhaustive, analysis of them. Recent studies highlighted that
HOX genes also contribute to organogenesis [
8] by influencing a huge number of cellular functions such as differentiation, proliferation, migration or death [
9].
HOX proteins consist of two highly conserved portions: the hexapeptide (HX) motif and the homeodomain (HD). The HX motif establishes interactions with protein members of the PBC class, such as Pre-B-cell Leukemia Homeobox (PBX) proteins in humans [
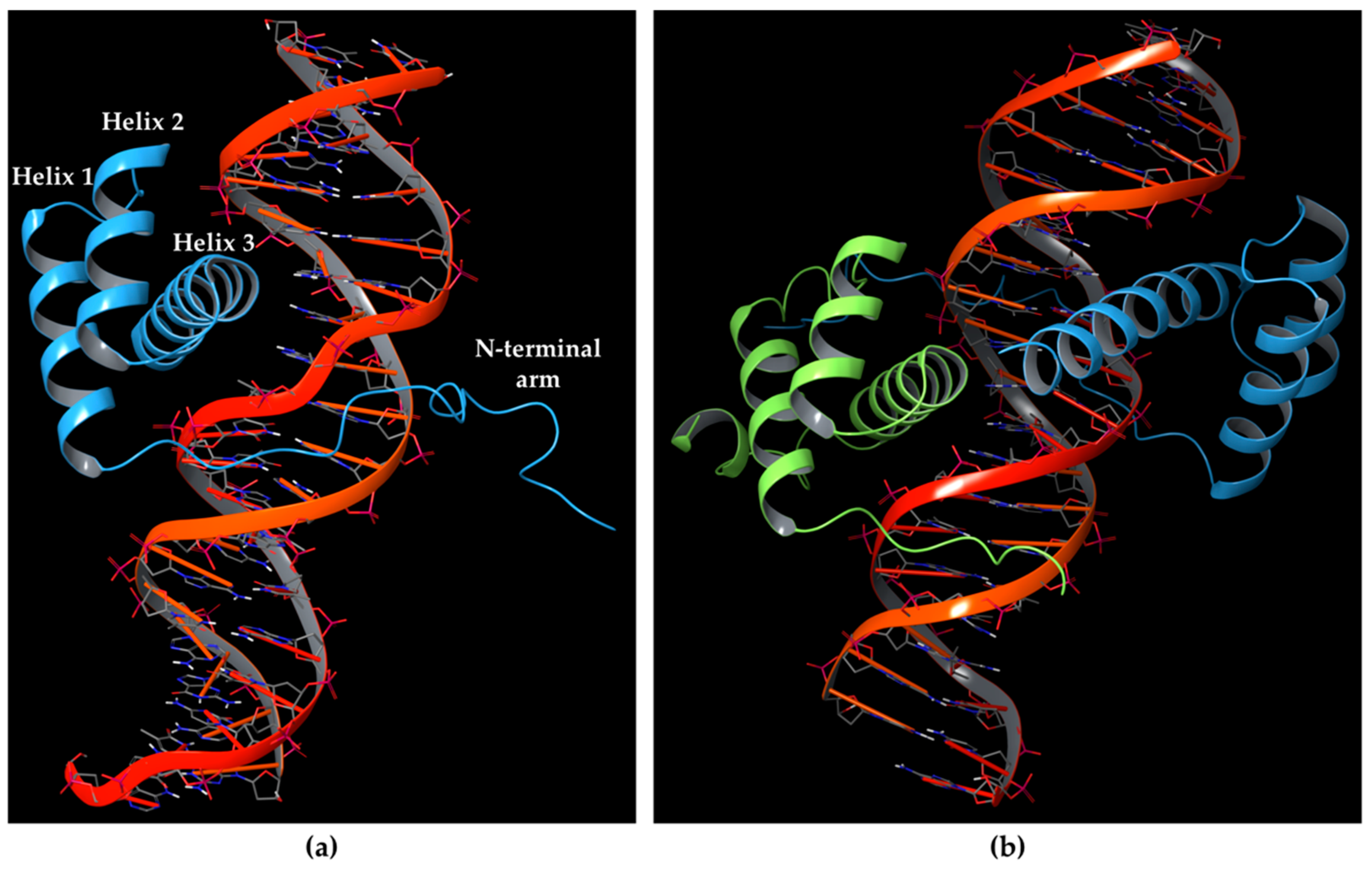
10]; while the HD motif corresponds to the DNA-binding domain. The HD folds into a triple-helix structure, including the N-terminal arm binding the minor groove of DNA, and helix 3 (also named the recognition helix) contacting the DNA in the major groove, as depicted in
Figure 1a. Residues involved in HOX HD helices 1 and 3 have shown to be the most conserved, and other amino acids of the N-terminal arm and loops between the helices have been reported well conserved among HOX proteins. Furthermore, the conservation of HD sequences is highly shared between HOX proteins, raising the issue of how they employ functional specificity [
11,
12,
13].
Indeed, the homeodomain of HOX proteins does not exhibit high specificity for DNA, by taking part in the molecular recognition through five amino acids [
14]. In this context, functional studies in the field of cancer and developmental biology highlighted the role of PBX proteins as HOX co-factors [
15,
16,
17], whereas PBX family members bind to HOX proteins 1-11 [
18,
19,
20]. These proteins may establish a cooperative binding to DNA [
21,
22] as depicted in
Figure 1b, indicating that the interaction of HOX proteins to PBX influences the DNA-binding of HOX by fostering a greater specificity [
23]. Furthermore, the HOX co-factors play other key roles influencing transcriptional events, by recruiting the RNA polymerase II and III or transcriptional inhibitors like HDAC, and post-translational events, by fostering the entry of HOX proteins into the nucleus.
Four different types of
PBX genes (PBX1-4) are encoded in the human genome. As for HOX proteins,
PBX genes also encode evolutionarily conserved homeodomains and other highly conserved regions [
16]. PBX proteins present two nuclear localization signals (NLSs) in the homeodomain and a nuclear export sequence (NES) [
24,
25,
26].
PBX proteins may participate in a DNA binding consensus through the formation of strong complexes with HOX1-11 proteins [
21,
28,
29]. The involved interactions have been shown to exhibit a highly conserved interaction mode between the HX motif of HOX and the three amino acid loop extension (TALE) or three-amino acid insertion peptide of PBX, which is located between helices 1 and 2 of the homeodomain [
18,
27,
29,
30,
31,
32,
33].
In 1995, Knoepfler and Kamps [
14] identified the minimal sequences that enable HOXB8 and HOXA5 proteins to bind cooperatively PBX1 protein, by performing deletion mutagenesis on the above-mentioned HOX proteins. This minimal sequence was the conserved pentapeptide motif
Y/F-P-W-M-R/K. In particular, mutations at tryptophan residue did not produce binding abrogation. Mutations of Trp135 to phenylalanine (W135F) or alanine (W135A) on HOXB8 did not alter the DNA binding but completely abolished the cooperativity of HOXB8 with PBX1. Met136 was also shown to be important but not essential for the DNA binding of PBX1, whereas its mutation to isoleucine (M136I) or alanine (M136A) strongly disrupted the cooperativity of HOXB8 with PBX1. Therefore, both residues, Trp135 and Met136, were considered crucial for the protein/DNA interaction, with particular attention for Trp135 [
32], since it was the only conserved amino acid among all HOX proteins pentapeptide sequences. Knoepfler and Kamps also assumed that the pentapeptide HOX motif stabilizes the trimeric HOX-PBX1-DNA complex by bringing a portion of the HOX protein surface into contact with PBX1 and enhancing DNA binding in the presence of PBX1 [
14]. However, the X-ray crystallographic structures of HOXB1-PBX1 and HOXA9-PBX1 in the presence of DNA revealed that the protein-protein-DNA contacts are stabilized by the interaction between HOX and PBX mediated not by a simple pentapeptide sequence, but by a conserved hexapeptide sequence in HOX proteins [
3,
18,
27,
31].
In 1999 Piper et al. [
18] found that a minimal portion of HOX containing hexapeptide and homeodomain was able to cooperatively stabilize DNA binding with PBX1. Hence, the identified consensus hexapeptide motif from HOX proteins was
φ-Y/F-P-W-M-K/R, where
φ stands for a hydrophobic residue. As reported above, tryptophan and methionine were conserved.
X-ray crystallographic structures of the ternary complex, HOX-PBX1-DNA (e.g., HOXA9 in PDB 1PUF, Resolution: 1.90 Å; and HOXB1 in PDB 1B72, Resolution: 2.35 Å) [
27] revealed that HOX protein and PBX1 establish contacts with opposite DNA faces, burying 2400 Å
2 of protein and DNA surface. The HOX hexapeptide mediates contacts with PBX1 within a hydrophobic pocket located between the TALE region and helix 3 of the PBX1 homeodomain.
PCR site-selection experiments performed by Piper et al. [
18] allowed us to identify the optimal HOXB1-PBX1 binding site on the 20 bp duplex DNA oligonucleotide, i.e., 5′-ATGATTGATCG-3′ [
34].
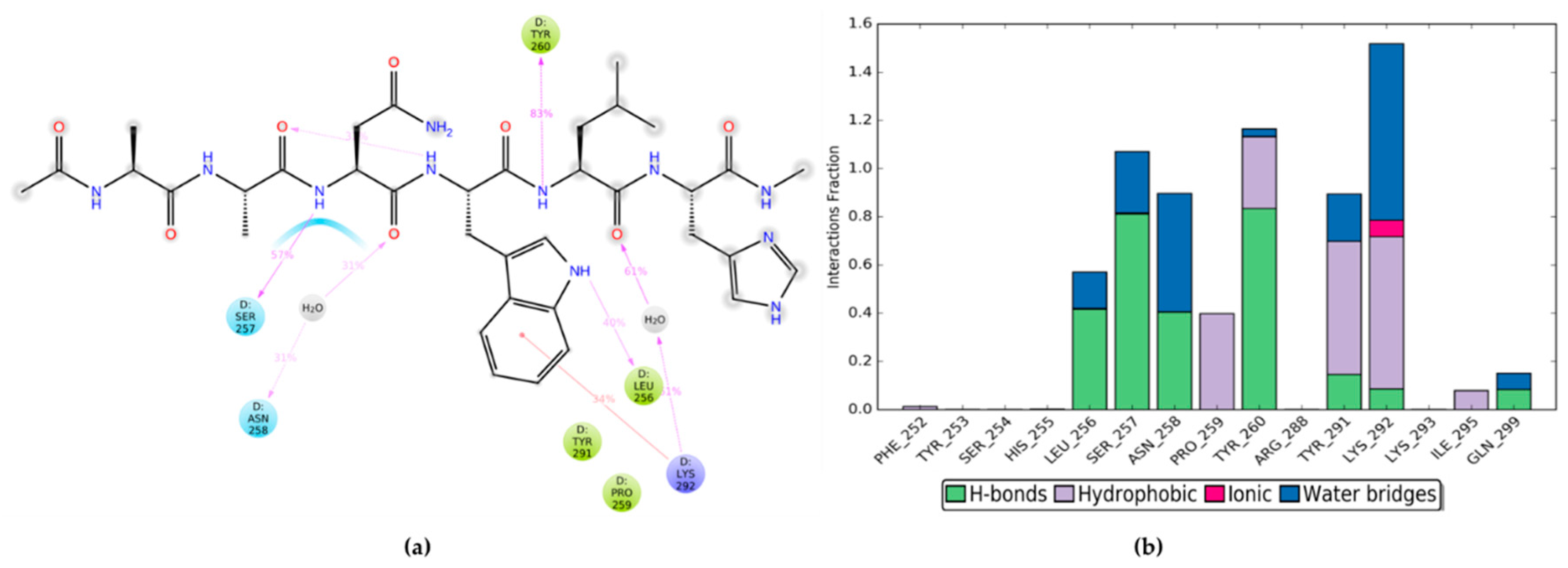
The PDB structure resolved by La Ronde-Le Blanc and Wolberger [
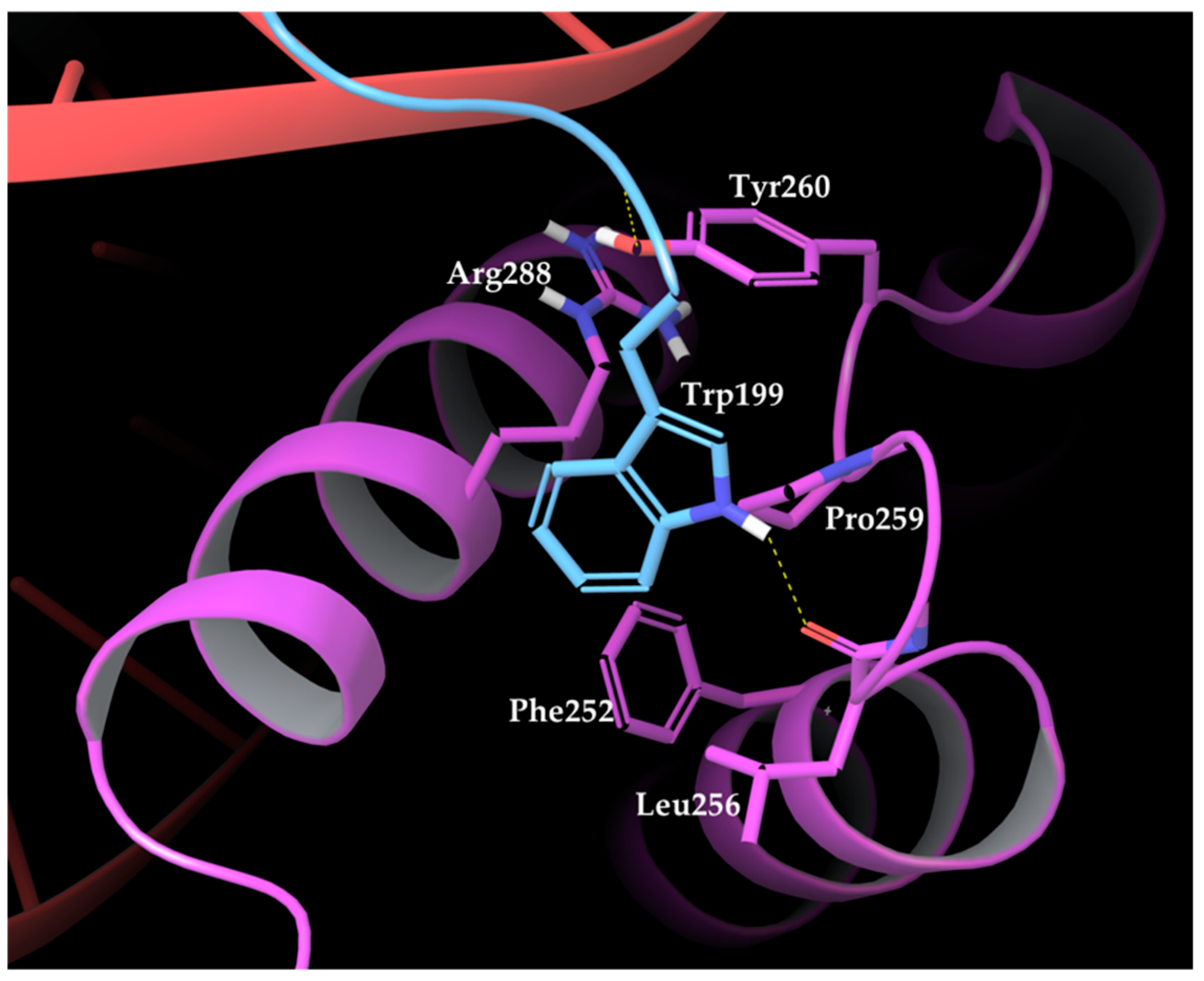
27] revealed that the interactions between HOXA9 and PBX1 are mediated by HOXA9 hexapeptide, consisting of residues 196 to 201 with the AANWLH sequence linked to the PBX1 homeodomain. As observable in the PBD structures, the hexapeptide residues mediate mainly hydrophobic contacts, whereas HOXA9 Trp199 side-chain inserts into a hydrophobic pocket of PBX1 formed by the C terminus of helix 3, a handle between helices 3 and 4, and the three–amino acid insertion. The key interactions involve HOXA9 Trp199 with its indole ring that forms van der Waals contacts with several PBX1 residues, such as the Phe252 side chain in helix 1, Leu256 within the TALE region, Pro259 and Tyr260 following the TALE peptide, and Arg288 in helix 3. Furthermore, Trp199 is highly buried into the PBX1 binding pocket by forming a hydrogen bond between the indole nitrogen and the backbone carbonyl of PBX1 Leu256 (
Figure 2).
The nitrogen atom at HOXA9 Leu200 backbone establishes van der Waals contacts with Ly292 and a hydrogen bond with Tyr260 hydroxyl group within the binding pocket of PBX1. Finally, His201 of HOXA9 hexapeptide forms a hydrogen bond with Lys292 of PBX1.
Moreover, Piper et al. [
18] on PBX1 conducted some mutational studies focused on the hexapeptide-contacting residues Leu252 and Pro259, which were substituted for alanine, triggering the disruption of the interactions with the hexapeptide in vitro and in a yeast two-hybrid assay [
35]. Furthermore, deletion assays at the three–amino acid insertion abolished the cooperative binding of PBX1 with HOX proteins [
36]. On the other hand, the deletion of the HOX hexapeptide caused the disappearance of cooperative interactions between PBX1 and HOX proteins [
14,
29,
37].
Although 3D structures of the ternary complex HOX-PBX-DNA have been experimentally solved, the drug discovery process for this protein-protein interaction (PPI) has met some issues typical of targeting a PPI through designing potential effective small molecule inhibitors [
38,
39]. However, an accepted strategy is to target the HOX-PBX binding interface at the highly conserved residues involving HOX hexapeptide and exploiting the hydrophobic nature of the PBX protein binding pocket. In the last decades, a small molecule inhibitor of this interaction was identified. However, its K
D was in the micromolar range (65 µM) and it was neglected for further experimental assays or clinical trials [
40]. On the other hand, in the last years, several peptides have been designed based on the hexapeptide consensus motif of HOX proteins, to work as a competitive antagonist of HOX-PBX binding [
41]. The most promising peptide among these was HXR9, an 18-amino acid peptide containing the hexapeptide sequence together with a polyarginine portion.
HXR9 was first shown to be cytotoxic to melanoma cell lines and primary melanoma cells and registered a reduction of B16F10 murine melanoma tumours growth in an orthotropic model [
42]. Other experimental studies reported that HXR9 was able to inhibit the growth of several tumour types in mouse xenograft models, including non-small cell lung [
43], breast [
44], ovarian [
44], and prostate cancer [
45], and mesothelioma [
46], melanoma [
47], and meningioma [
48].
Recently, modifications performed on the HXR9 sequence shed light on another peptide, i.e., the HTL001 peptide [
49] with the sequence WYPWMKKHHRRRRRRRRR, that was tested in cancer cells representative of 14 human and animal malignancies. HTL001 registered selective toxicity for cancer cells and safety for normal cells. To date, this peptide has reached the human clinical trials that are ongoing to assay the efficacy and safety. However, the mechanism associated with HOX-PBX inhibition and the resulting cell death through employing the HTL001 peptide is still to be fully elucidated, although generally in most solid tumours cell death is mediated by apoptosis [
42,
44,
45,
46,
50].
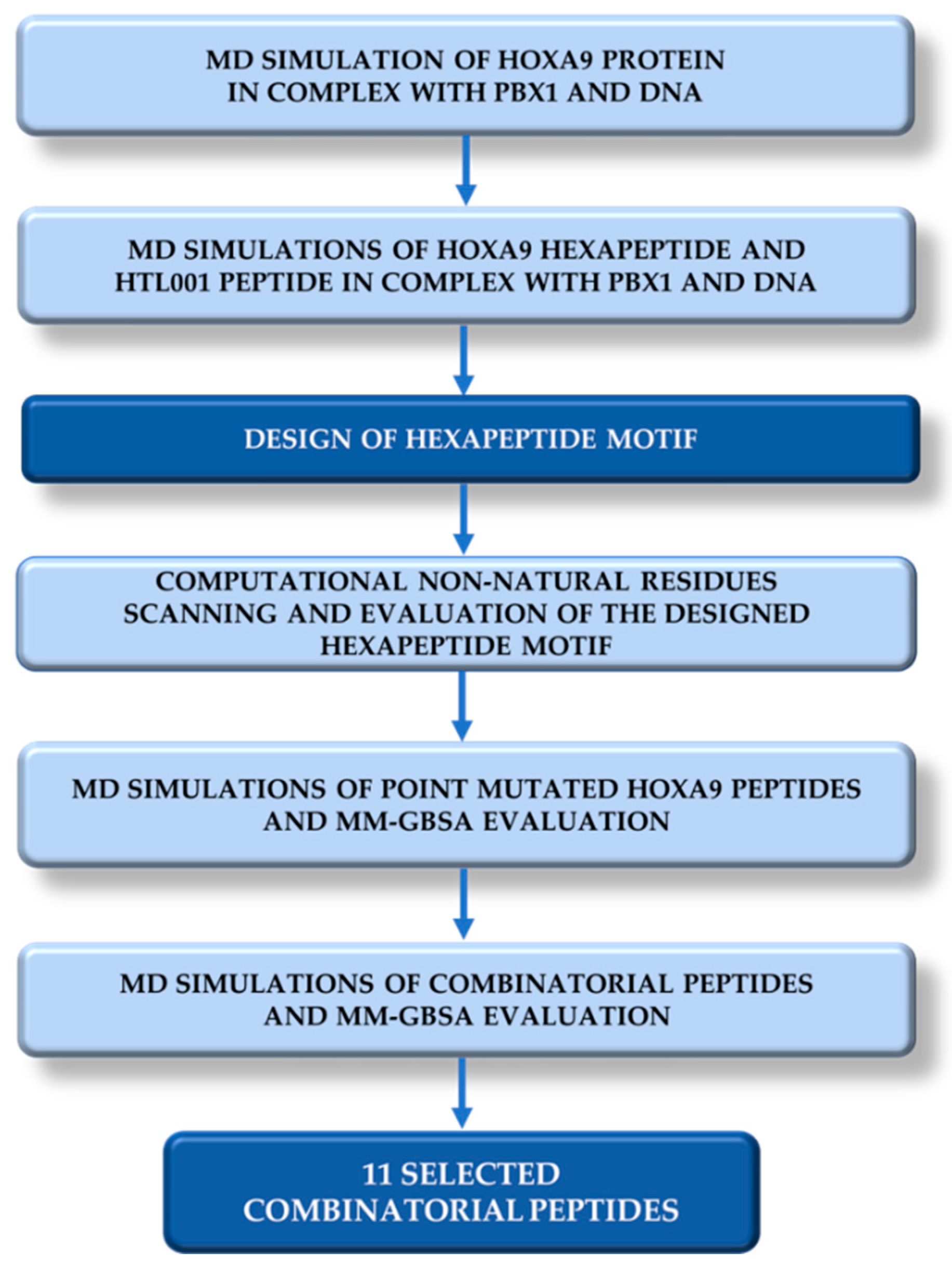
In this article, we describe a computer-based strategy to design novel peptides including non-standard amino acids to potentially bind PBX1 and inhibit HOX-PBX1 interaction (
Figure 3). For this purpose, a Molecular Dynamics (MD) simulation of 200 ns was performed on the ternary complex HOXA9-PBX1-DNA retrieved from the Protein Data Bank [
51] to identify key residues and interactions [
52]. Two other MD were run, one on HOXA9 hexapeptide (196-AANWLH-201) from PDB 1PUF and the other on the patented core peptide HTL001 without polyarginine coil in complex with PBX1-DNA. The resulting MD trajectories were then used to compute MM-GBSA (Molecular Mechanics–Generalised Born Surface Area) calculations to obtain ΔG
binding average values as references for the design of the new peptides. Subsequently, the HOXA9 hexapeptide sequence was submitted to a point mutational scanning exploiting a non-natural amino acid database populated by the Swiss Institute of Bioinformatics [
53,
54]. The mutants were selected according to ΔΔG
affinity and ΔΔG
stability values and, in complex with PBX1-DNA, were further explored by MD simulations and MM-GBSA calculations. All those complexes point-mutated peptide-PBX1-DNA reporting ΔG
binding average values lower compared to the reference ΔG
binding average values (involving HOXA9 hexapeptide and HTL001 core peptide) were chosen for the next steps of the work. Thus, the most promising mutated peptides were used as starting point to generate twelve combinatorial peptides. The newly generated peptides were reprocessed in MD and MM-GBSA calculations as peptide-PBX1-DNA complex. Finally, eleven peptides showed promising ΔG
binding values compared to the HOXA9 hexapeptide and HTL001 peptide.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}