
Dysregulation of Astrocyte Ion Homeostasis and Its Relevance for Stroke-Induced Brain Damage

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
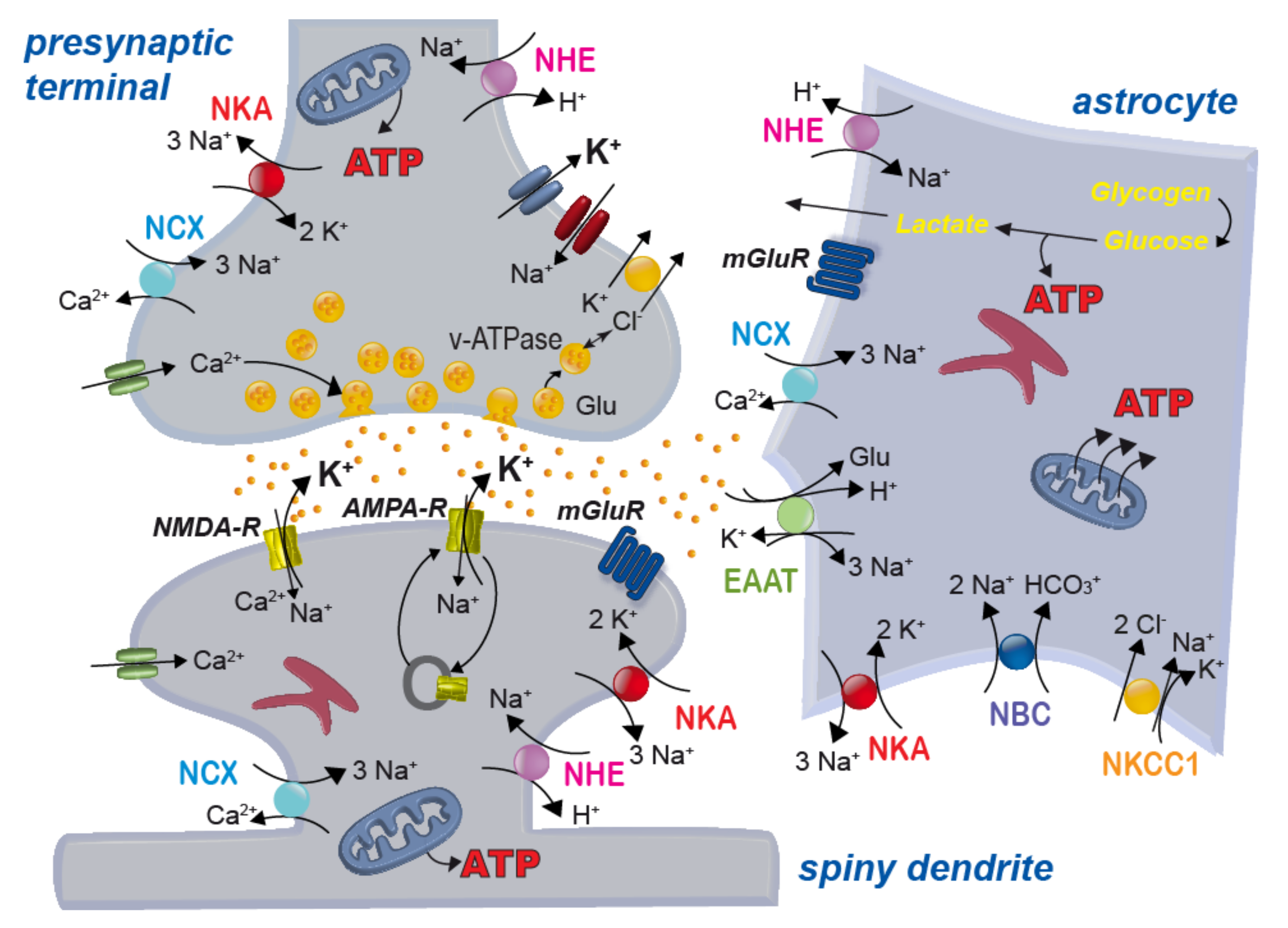
2. Energy Dependence of the Tripartite Synapse
2.1. Energy Needs of Neurons and Astrocytes
2.2. Neuro-Metabolic Coupling between Neurons and Astrocytes
2.3. Housekeeping by Astrocytes and the Tripartite Synapse
3. Relevance of Astrocytic Cation Homeostasis
3.1. Sodium Homeostasis
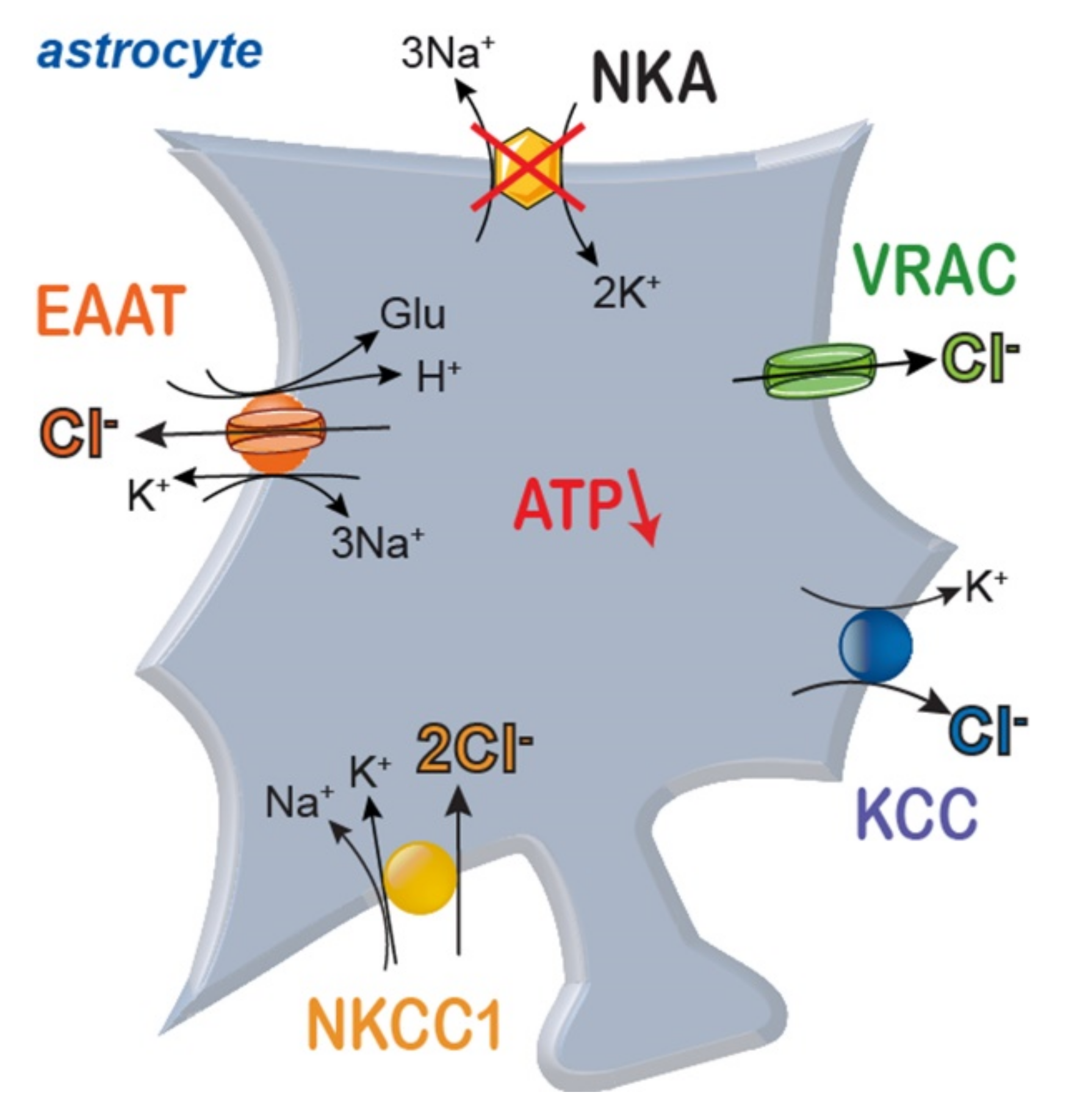
3.2. Sodium Dysregulation and Its Consequences under Ischemic Conditions
3.3. Potassium
3.4. pH
4. Relevance of Astrocytic Chloride Homeostasis
4.1. Regulation of Intracellular Chloride Levels
4.2. Chloride and Ischemia
5. Generation of Cell Swelling and Cerebral Edema
5.1. Volume and Water Transport in Animal Cells
5.2. Ion Concentrations and Osmolarity at the Gibbs-Donnan Equilibrium
5.3. Dynamics of Cell Volume Changes during Metabolic Stress
5.4. Cerebral Edema
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Astrup, J.; Siesjö, B.K.; Symon, L. Thresholds in cerebral ischemia—The ischemic penumbra. Stroke 1981, 12, 723–725. [Google Scholar] [CrossRef] [Green Version]
- Heiss, W.-D.; Graf, R.; Wienhard, K.; Löttgen, J.; Saito, R.; Fujita, T.; Rosner, G.; Wagner, R. Dynamic Penumbra Demonstrated by Sequential Multitracer PET after Middle Cerebral Artery Occlusion in Cats. Br. J. Pharmacol. 1994, 14, 892–902. [Google Scholar] [CrossRef] [Green Version]
- Berkhemer, O.A.; Fransen, P.S.S.; Beumer, D.; Berg, L.A.V.D.; Lingsma, H.F.; Yoo, A.J.; Schonewille, W.J.; Vos, J.A.; Nederkoorn, P.J.; Wermer, M.J.H.; et al. A Randomized Trial of Intraarterial Treatment for Acute Ischemic Stroke. New Engl. J. Med. 2015, 372, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Bandera, E.; Botteri, M.; Minelli, C.; Sutton, A.; Abrams, K.R.; Latronico, N. Cerebral blood flow threshold of ischemic penumbra and infarct core in acute ischemic stroke: A systematic review. Stroke 2006, 37, 1334–1339. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolay, H.; Güsoy-Özdemir, Y.; Sara, Y.; Onur, R.; Can, A.; Dalkara, T. Persistent Defect in Transmitter Release and Synapsin Phosphorylation in Cerebral Cortex After Transient Moderate Ischemic Injury. Stroke 2002, 33, 1369–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Feber, J.; Pavlidou, S.T.; Erkamp, N.; Van Putten, M.J.A.M.; Hofmeijer, J. Progression of Neuronal Damage in an In Vitro Model of the Ischemic Penumbra. PLoS ONE 2016, 11, e0147231. [Google Scholar] [CrossRef]
- Kim, B.J.; Menon, B.K.; Kim, J.Y.; Shin, D.-W.; Baik, S.H.; Jung, C.; Han, M.-K.; Demchuk, A.; Bae, H.-J. Endovascular Treatment After Stroke Due to Large Vessel Occlusion for Patients Presenting Very Late from Time Last Known Well. JAMA Neurol. 2021, 78, 21. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [Green Version]
- Kristián, T.; Siesjö, B.K. Calcium in Ischemic Cell Death. Stroke 1998, 29, 705–718. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef] [PubMed]
- Zandt, B.-J.; Haken, B.T.; Van Putten, M.J.A.M. Diffusing Substances during Spreading Depolarization: Analytical Expressions for Propagation Speed, Triggering, and Concentration Time Courses. J. Neurosci. 2013, 33, 5915–5923. [Google Scholar] [CrossRef] [Green Version]
- Ayata, C.; Lauritzen, M. Spreading Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol. Rev. 2015, 95, 953–993. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef]
- Hofmeijer, J.; van Putten, M.J.A.M. Ischemic cerebral damage: An appraisal of synaptic failure. Stroke 2012, 43, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Hartings, J.A.; Shuttleworth, C.W.; Kirov, S.A.; Ayata, C.; Hinzman, J.M.; Foreman, B.; Andrew, R.D.; Boutelle, M.G.; Brennan, K.C.; Carlson, A.P.; et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão’s legacy. Br. J. Pharmacol. 2016, 37, 1571–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Zoppo, G.J.; Sharp, F.R.; Heiss, W.-D.; Albers, G.W. Heterogeneity in the penumbra. Br. J. Pharmacol. 2011, 31, 1836–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 2000, 36, 291–300. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Zeng, X.-N.; Sun, X.-L.; Gao, L.; Fan, Y.; Ding, J.-H.; Hu, G. Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol. Cell. Neurosci. 2007, 34, 34–39. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into rather unexplored ter-rain-transcellular transport across the blood-brain barrier. Glia 2016, 64, 1097–1123. [Google Scholar] [CrossRef] [PubMed]
- Barreto, G.; White, R.E.; Ouyang, Y.; Xu, L.; Giffard, R.G. Astrocytes: Targets for Neuroprotection in Stroke. Central Nerv. Syst. Agents Med. Chem. 2011, 11, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziato, L.; Boscia, F.; Pignataro, G. Ionic Transporter Activity in Astrocytes, Microglia, and Oligodendrocytes During Brain Ischemia. Br. J. Pharmacol. 2013, 33, 969–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedergaard, M.; Dirnagl, U. Role of glial cells in cerebral ischemia. Glia 2005, 50, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Hirayama, Y.; Morizawa, Y.M. New roles of reactive astrocytes in the brain; an organizer of cerebral ischemia. Neurochem. Int. 2018, 119, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, G.R.; Ding, S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol. Dis. 2016, 85, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Take-bayashi, H.; et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 8, 28. [Google Scholar] [CrossRef]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol. Integr. Comp. Physiol. 1981, 241, R203–R212. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engl, E.; Attwell, D. Non-signalling energy use in the brain. J. Physiol. 2015, 593, 3417–3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erecińska, M.; Silver, I.A. Ions and energy in mammalian brain. Prog. Neurobiol. 1994, 43, 37–71. [Google Scholar] [CrossRef]
- Somjen, G.G. Ions in the Brain: Normal Function, Seizures, and Stroke; Oxford University Press: New York, NY, USA, 2004. [Google Scholar]
- Astrup, J.; Sørensen, P.M.; Sørensen, H.R. Oxygen and glucose consumption related to Na+-K+ transport in canine brain. Stroke 1981, 12, 726–730. [Google Scholar] [CrossRef] [Green Version]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated Energy Budgets for Neural Computation in the Neocortex and Cerebellum. Br. J. Pharmacol. 2012, 32, 1222–1232. [Google Scholar] [CrossRef]
- Lennie, P. The Cost of Cortical Computation. Curr. Biol. 2003, 13, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Sweadner, K.J. Isozymes of the Na+/K+-ATPase. Biochim. Biophys. Acta. 1989, 988, 185–220. [Google Scholar] [CrossRef]
- Kaplan, J.H. Biochemistry of Na, K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, K.; Stransky, L.; McGuire, C.; Forgac, M. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem. Sci. 2015, 40, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.; Nelson, J.C.; Bend, E.G.; Rodríguez-Laureano, L.; Tueros, F.G.; Cartagenova, L.; Underwood, K.; Jorgensen, E.M.; Colón-Ramos, D.A. Glycolytic Enzymes Localize to Synapses under Energy Stress to Support Synaptic Function. Neuron 2016, 90, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Sobieski, C.; Fitzpatrick, M.J.; Mennerick, S.J. Differential Presynaptic ATP Supply for Basal and High-Demand Transmission. J. Neurosci. 2017, 37, 1888–1899. [Google Scholar] [CrossRef] [Green Version]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na, K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef]
- Sweadner, K. Sodium, Potassium-Adenosine Triphosphatase and Its Isoforms; Neuroglia, R.B., Kettenmann, H., Eds.; Oxford University Press: New York, NY, USA, 1995; pp. 259–272. [Google Scholar]
- Verkhratsky, A.; Rose, C.R. Na+-dependent transporters: The backbone of astroglial homeostatic function. Cell Calcium 2020, 85, 102136. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nat. Cell Biol. 2000, 403, 316–321. [Google Scholar] [CrossRef]
- Rose, C.R.; Ziemens, D.; Verkhratsky, A. On the special role of NCX in astrocytes: Translating Na+-transients into intracellular Ca2+ signals. Cell Calcium 2020, 86, 102154. [Google Scholar] [CrossRef]
- Tønnesen, J.; Inavalli, V.K.; Nägerl, U.V. Super-Resolution Imaging of the Extracellular Space in Living Brain Tissue. Cell 2018, 172, 1108–1121e.15. [Google Scholar] [CrossRef] [Green Version]
- Grosche, J.; Kettenmann, H.; Reichenbach, A. Bergmann glial cells form distinct morphological structures to interact with cerebellar neurons. J. Neurosci. Res. 2002, 68, 138–149. [Google Scholar] [CrossRef]
- Khakh, B.S. Astrocyte–Neuron Interactions in the Striatum: Insights on Identity, Form, and Function. Trends Neurosci. 2019, 42, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Brown, A.M.; Ransom, B.R. Astrocyte glycogen and brain energy metabolism. Glia 2007, 55, 1263–1271. [Google Scholar] [CrossRef]
- Hertz, L.; Xu, J.; Song, D.; Du, T.; Li, B.; Yan, E.; Peng, L. Astrocytic glycogenolysis: Mechanisms and functions. Metab. Brain Dis. 2014, 30, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessières, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Waagepetersen, H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef]
- Chatton, J.-Y.; Magistretti, P.J.; Barros, L.F. Sodium signaling and astrocyte energy metabolism. Glia 2016, 64, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Lerchundi, R.; Huang, N.; Rose, C.R. Quantitative Imaging of Changes in Astrocytic and Neuronal Adenosine Tri-phosphate Using Two Different Variants of ATeam. Front Cell Neurosci. 2020, 14, 80. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.; Chatton, J.-Y. Astrocyte sodium signaling and neuro-metabolic coupling in the brain. Neuroscience 2016, 323, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Barros, L.F. Metabolic signaling by lactate in the brain. Trends Neurosci. 2013, 36, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Hirrlinger, J.; Nave, K.-A. Adapting brain metabolism to myelination and long-range signal transduction. Glia 2014, 62, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.F.; Weber, B. CrossTalk proposal: An important astrocyte-to-neuron lactate shuttle couples neuronal activity to glucose utilisation in the brain. J. Physiol. 2018, 596, 347–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sickmann, H.M.; Walls, A.B.; Schousboe, A.; Bouman, S.D.; Waagepetersen, H.S. Functional significance of brain gly-cogen in sustaining glutamatergic neurotransmission. J. Neurochem. 2009, 109, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Walls, A.B. CrossTalk opposing view: Lack of evidence supporting an astrocyte-to-neuron lactate shuttle coupling neuronal activity to glucose utilisation in the brain. J. Physiol. 2018, 596, 351–353. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Rothman, D.L.; Dienel, G.A. Development of a Model to Test Whether Glycogenolysis Can Support Astrocytic Energy Demands of Na+, K+-ATPase and Glutamate-Glutamine Cycling, Sparing an Equivalent Amount of Glucose for Neurons. Adv. Neurobiol. 2019, 23, 385–433. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Untiet, V.; Rose, C.R. Ionic signalling in astroglia beyond calcium. J. Physiol. 2020, 598, 1655–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, R.; Harris, K.M. Three-Dimensional Relationships between Hippocampal Synapses and Astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Grosche, J.; Matyash, V.; Möller, T.; Verkhratsky, A.; Reichenbach, A.; Kettenmann, H. Microdomains for neuron–glia interaction: Parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 1999, 2, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Newman, E. Regulation of potassium by glial cells in the central nervous system. In Astrocytes in (Patho)Physiology of the Nervous System; Parpura, V., Haydon, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 151–175. [Google Scholar]
- Heinemann, U.; Lux, H.D. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res. 1977, 120, 231–249. [Google Scholar] [CrossRef]
- Connors, B.; Ransom, B.; Kunis, D.; Gutnick, M. Activity-dependent K+ accumulation in the developing rat optic nerve. Sci. 1982, 216, 1341–1343. [Google Scholar] [CrossRef]
- Rasmussen, R.; O’Donnell, J.; Ding, F.; Nedergaard, M. Interstitial ions: A key regulator of state-dependent neural activity? Prog. Neurobiol. 2020, 193, 101802. [Google Scholar] [CrossRef]
- Hertz, L.; Song, D.; Xu, J.; Peng, L.; Gibbs, M.E. Role of the Astrocytic Na+, K+-ATPase in K+ Homeostasis in Brain: K+ Uptake, Signaling Pathways and Substrate Utilization. Neurochem. Res. 2015, 40, 2505–2516. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.R.; Stoica, A.; Macaulay, N. Managing Brain Extracellular K+ during Neuronal Activity: The Physiological Role of the Na+/K+-ATPase Subunit Isoforms. Front. Physiol. 2016, 7, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karus, C.; Mondragão, M.A.; Ziemens, D.; Rose, C.R. Astrocytes restrict discharge duration and neuronal sodium loads during recurrent network activity. Glia 2015, 63, 936–957. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Ransom, B.R. Intracellular sodium homeostasis in rat hippocampal astrocytes. J. Physiol. 1996, 491, 291–305. [Google Scholar] [CrossRef]
- Rose, C.R.; Ransom, B.R. Regulation of intracellular sodium in cultured rat hippocampal neurones. J. Physiol. 1997, 499, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Steinhäuser, C.; Seifert, G.; Bedner, P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 2012, 60, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Rothstein, J.D. Glutamate transporters: Animal models to neurologic disease. Neurobiol. Dis. 2004, 15, 461–473. [Google Scholar] [CrossRef]
- Hübel, N.; Hosseini-Zare, M.S.; Ziburkus, J.; Ullah, G. The role of glutamate in neuronal ion homeostasis: A case study of spreading depolarization. PLoS Comput. Biol. 2017, 13, e1005804. [Google Scholar] [CrossRef] [Green Version]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate Metabolism in the Brain Focusing on Astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Sonnewald, U. Glutamate synthesis has to be matched by its degradation—where do all the carbons go? J. Neurochem. 2014, 131, 399–406. [Google Scholar] [CrossRef]
- Panatier, A.; Robitaille, R. Astrocytic mGluR5 and the tripartite synapse. Neuroscience 2016, 323, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; McConnell, E.; Pare, J.F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Gluta-mate-dependent neuroglial calcium signaling differs between young and adult brain. Science 2013, 339, 197–200. [Google Scholar] [CrossRef] [Green Version]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca2+ signalling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Fiacco, T.A.; McCarthy, K.D. Multiple Lines of Evidence Indicate That Gliotransmission Does Not Occur under Phys-iological Conditions. J. Neurosci. 2018, 38, 3–13. [Google Scholar] [CrossRef]
- Agulhon, C.; Petravicz, J.; McMullen, A.B.; Sweger, E.J.; Minton, S.K.; Taves, S.R.; Casper, K.B.; Fiacco, T.A.; McCarthy, K.D. What Is the Role of Astrocyte Calcium in Neurophysiology? Neuron 2008, 59, 932–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Dunphy, J.; Tolman, M.; Foley, J.C.; Haydon, P.G. Astrocytic control of synaptic function. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160154. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.R.; Karus, C. Two sides of the same coin: Sodium homeostasis and signaling in astrocytes under physiological and pathophysiological conditions. Glia 2013, 61, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N. Molecular mechanisms of K+clearance and extracellular space shrinkage—Glia cells as the stars. Glia 2020, 68, 2192–2211. [Google Scholar] [CrossRef]
- Theparambil, S.M.; Naoshin, Z.; Thyssen, A.; Deitmer, J.W. Reversed electrogenic sodium bicarbonate cotransporter 1 is the major acid loader during recovery from cytosolic alkalosis in mouse cortical astrocytes. J. Physiol. 2015, 593, 3533–3547. [Google Scholar] [CrossRef] [Green Version]
- Deitmer, J.W.; Rose, C.R. REMOVED: Ion changes and signalling in perisynaptic glia. Brain Res. 2009, 63, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A. Structure, function, and plasticity of GABA transporters. Front. Cell. Neurosci. 2014, 8, 161. [Google Scholar] [CrossRef] [Green Version]
- Schousboe, A.; Wellendorph, P.; Frølund, B.; Clausen, R.P.; Krogsgaard-Larsen, P. Astrocytic GABA Transporters: Pharmacological Properties and Targets for Antiepileptic Drugs. Adv. Neurobiol. 2017, 16, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S.; Parpura, V.; Verkhratsky, A. Sodium dynamics: Another key to astroglial excitability? Trends Neurosci. 2012, 35, 497–506. [Google Scholar] [CrossRef]
- Felix, L.; Delekate, A.; Petzold, G.C.; Rose, C.R. Sodium Fluctuations in Astroglia and Their Potential Impact on As-trocyte Function. Front Physiol. 2020, 11, 871. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.; Gerkau, N.J.; Derouiche, A.; Kleinhans, C.; Moshrefi-Ravasdjani, B.; Fredrich, M.; Kafitz, K.W.; Seifert, G.; Steinhäuser, C.; Rose, C.R. Rapid sodium signaling couples glutamate uptake to breakdown of ATP in perivascular astrocyte endfeet. Glia 2017, 65, 293–308. [Google Scholar] [CrossRef]
- Bennay, M.; Langer, J.; Meier, S.D.; Kafitz, K.W.; Rose, C.R. Sodium signals in cerebellar Purkinje neurons and Bergmann glial cells evoked by glutamatergic synaptic transmission. Glia 2008, 56, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.; Rose, C.R. Synaptically induced sodium signals in hippocampal astrocytes in situ. J. Physiol. 2009, 587, 5859–5877. [Google Scholar] [CrossRef] [PubMed]
- Moshrefi-Ravasdjani, B.; Ziemens, D.; Pape, N.; Färfers, M.; Rose, C.R. Action Potential Firing Induces Sodium Tran-sients in Macroglial Cells of the Mouse Corpus Callosum. Neuroglia 2018, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Ziemens, D.; Oschmann, F.; Gerkau, N.J.; Rose, C.R. Heterogeneity of Activity-Induced Sodium Transients between Astrocytes of the Mouse Hippocampus and Neocortex: Mechanisms and Consequences. J. Neurosci. 2019, 39, 2620–2634. [Google Scholar] [CrossRef] [Green Version]
- Langer, J.; Stephan, J.; Theis, M.; Rose, C.R. Gap junctions mediate intercellular spread of sodium between hippocampal astrocytes in situ. Glia 2011, 60, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Augustin, V.; Bold, C.; Wadle, S.L.; Langer, J.; Jabs, R.; Philippot, C.; Weingarten, D.J.; Rose, C.R.; Steinhäuser, C.; Stephan, J. Functional anisotropic panglial networks in the lateral superior olive. Glia 2016, 64, 1892–1911. [Google Scholar] [CrossRef] [PubMed]
- Moshrefi-Ravasdjani, B.; Hammel, E.L.; Kafitz, K.W.; Rose, C.R. Astrocyte Sodium Signalling and Panglial Spread of Sodium Signals in Brain White Matter. Neurochem. Res. 2017, 42, 2505–2518. [Google Scholar] [CrossRef]
- Rose, C.R.; Waxman, S.G.; Ransom, B.R. Effects of Glucose Deprivation, Chemical Hypoxia, and Simulated Ischemia on Na+Homeostasis in Rat Spinal Cord Astrocytes. J. Neurosci. 1998, 18, 3554–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerkau, N.J.; Rakers, C.; Petzold, G.C.; Rose, C.R. Differential effects of energy deprivation on intracellular sodium homeostasis in neurons and astrocytes. J. Neurosci. Res. 2017, 95, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.J. Effect of anoxia on ion distribution in the brain. Physiol. Rev. 1985, 65, 101–148. [Google Scholar] [CrossRef]
- Longuemare, M.C.; Rose, C.R.; Farrell, K.; Ransom, B.R.; Waxman, S.G.; Swanson, R.A. K(+)-induced reversal of as-trocyte glutamate uptake is limited by compensatory changes in intracellular Na+. Neuroscience 1999, 93, 285–292. [Google Scholar] [CrossRef]
- Silver, I.A.; Deas, J.; Erecińska, M. Ion homeostasis in brain cells: Differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience 1997, 78, 589–601. [Google Scholar] [CrossRef]
- Kintner, D.B.; Look, A.; Shull, G.E.; Sun, D. Stimulation of astrocyte Na+/H+ exchange activity in response to in vitro ischemia depends in part on activation of ERK1/2. Am. J. Physiol. Cell Physiol. 2005, 289, C934–C945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerkau, N.J.; Rakers, C.; Durry, S.; Petzold, G.C.; Rose, C.R. Reverse NCX Attenuates Cellular Sodium Loading in Metabolically Compromised Cortex. Cereb. Cortex 2017, 28, 4264–4280. [Google Scholar] [CrossRef]
- Rossi, D.J.; Brady, J.D.; Mohr, C. Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci. 2007, 10, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Fern, R. Ischemic Tolerance in Pre-Myelinated White Matter: The Role of Astrocyte Glycogen in Brain Pathology. Br. J. Pharmacol. 2015, 35, 951–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, R.A.; Choi, D.W. Glial Glycogen Stores Affect Neuronal Survival during Glucose Deprivation in vitro. Br. J. Pharmacol. 1993, 13, 162–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, L.; Dienel, G.A. Lactate transport and transporters: General principles and functional roles in brain cells. J. Neurosci. Res. 2005, 79, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Somjen, G.G. Na+ and K+ Concentrations, Extra- and Intracellular Voltages, and the Effect of TTX in Hypoxic Rat Hippocampal Slices. J. Neurophysiol. 2000, 83, 735–745. [Google Scholar] [CrossRef]
- Xie, M.; Wang, W.; Kimelberg, H.K.; Zhou, M. Oxygen and Glucose Deprivation-Induced Changes in Astrocyte Membrane Potential and Their Underlying Mechanisms in Acute Rat Hippocampal Slices. Br. J. Pharmacol. 2007, 28, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wang, W.; Lutton, A.D.; Kiyoshi, C.M.; Ma, B.; Taylor, A.T.; Olesik, J.W.; McTigue, D.M.; Askwith, C.C.; Zhou, M. Dissipation of transmembrane potassium gradient is the main cause of cerebral ischemia-induced depolarization in astrocytes and neurons. Exp. Neurol. 2018, 303, 1–11. [Google Scholar] [CrossRef]
- Fonnum, F.; Johnsen, A.; Hassel, B. Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia 1997, 21, 106–113. [Google Scholar] [CrossRef]
- Szatkowski, M.; Attwell, D. Triggering and execution of neuronal death in brain ischaemia: Two phases of glutamate release by different mechanisms. Trends Neurosci. 1994, 17, 359–365. [Google Scholar] [CrossRef]
- Pietrobon, D.; Moskowitz, M.A. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 2014, 15, 379–393. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Rothstein, J.D. Glutamate Transporters in Neurologic Disease. Arch. Neurol. 2001, 58, 365–370. [Google Scholar] [CrossRef]
- Aizawa, H.; Sun, W.; Sugiyama, K.; Itou, Y.; Aida, T.; Cui, W.; Toyoda, S.; Terai, H.; Yanagisawa, M.; Tanaka, K. Glial glutamate transporter GLT -1 determines susceptibility to spreading depression in the mouse cerebral cortex. Glia 2020, 68, 2631–2642. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Domercq, M.; Gomez, M.V.S. Glutamate-mediated glial injury: Mechanisms and clinical importance. Glia 2006, 53, 212–224. [Google Scholar] [CrossRef]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef]
- Vardjan, N.; Parpura, V.; Zorec, R. Loose excitation-secretion coupling in astrocytes. Glia 2016, 64, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakers, C.; Petzold, G.C. Astrocytic calcium release mediates peri-infarct depolarizations in a rodent stroke model. J. Clin. Investig. 2016, 127, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Boscia, F.; Begum, G.; Pignataro, G.; Sirabella, R.; Cuomo, O.; Casamassa, A.; Sun, D.; Annunziato, L. Glial Na+-dependent ion transporters in pathophysiological conditions. Glia 2016, 64, 1677–1697. [Google Scholar] [CrossRef] [Green Version]
- Fern, R.F.; Matute, C.; Stys, P.K. White matter injury: Ischemic and nonischemic. Glia 2014, 62, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Luo, L.; Sun, B.; Sun, D. Roles of glial ion transporters in brain diseases. Glia 2020, 68, 472–494. [Google Scholar] [CrossRef]
- Coles, J.A.; Schneider-Picard, G. Increase in glial intracellular K+ in drone retina caused by photostimulation but not mediated by an increase in extracellular K+. Glia 1989, 2, 213–222. [Google Scholar] [CrossRef]
- Ballanyi, K.; Grafe, P.; Bruggencate, G.T. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J. Physiol. 1987, 382, 159–174. [Google Scholar] [CrossRef]
- Kofuji, P.; Newman, E. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Rimmele, T.S.; Chatton, J.-Y. A Novel Optical Intracellular Imaging Approach for Potassium Dynamics in Astrocytes. PLoS ONE 2014, 9, e109243. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Dempsey, R.J.; Sun, D. Expression of Na+-K+-Cl− cotransporter in rat brain during development and its localization in mature astrocytes. Brain Res. 2001, 911, 43–55. [Google Scholar] [CrossRef]
- Olsen, M.L.; Sontheimer, H. Functional implications for Kir4.1 channels in glial biology: From K+buffering to cell differentiation. J. Neurochem. 2008, 107, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Milton, M.; Smith, P.D. It’s All about Timing: The Involvement of Kir4.1 Channel Regulation in Acute Ischemic Stroke Pathology. Front. Cell. Neurosci. 2018, 12, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leis, J.A.; Bekar, L.K.; Walz, W. Potassium homeostasis in the ischemic brain. Glia 2005, 50, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Menyhárt, Á.; Farkas, A.E.; Varga, D.P.; Frank, R.; Tóth, R.; Bálint, A.R.; Makra, P.; Dreier, J.P.; Bari, F.; Krizbai, I.A.; et al. Large-conductance Ca2+-activated potassium channels are potently involved in the inverse neurovascular response to spreading depolarization. Neurobiol. Dis. 2018, 119, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Varga, D.P.; Menyhárt, Á.; Puskás, T.; Bari, F.; Farkas, E.; Kis, Z.; Vécsei, L.; Toldi, J.; Gellért, L. Systemic administration of l -kynurenine sulfate induces cerebral hypoperfusion transients in adult C57Bl/6 mice. Microvasc. Res. 2017, 114, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zhong, C.J.; Chen, M.M.; Lu, M.; Ding, J.H.; Du, R.H.; Hu, G. Astrocyte-specific deletion of Kir6.1/K-ATP channel aggravates cerebral ischemia/reperfusion injury through endoplasmic reticulum stress in mice. Exp. Neurol. 2019, 311, 225–233. [Google Scholar] [CrossRef]
- Su, G.; Kintner, D.B.; Flagella, M.; Shull, G.E.; Sun, D. Astrocytes from Na+-K+-Cl−cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am. J. Physiol. Physiol. 2002, 282, C1147–C1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenart, B.; Kintner, D.B.; Shull, G.E.; Sun, D. Na-K-Cl Cotransporter-Mediated Intracellular Na+ Accumulation Affects Ca2+ Signaling in Astrocytes in an In Vitro Ischemic Model. J. Neurosci. 2004, 24, 9585–9597. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Wang, Y.; Chen, H.; Kintner, D.B.; Cramer, S.W.; Gerdts, J.K.; Chen, X.; Shull, G.E.; Philipson, K.D.; Sun, D. A concerted role of Na+ -K+ -Cl- cotransporter and Na+/Ca2+ exchanger in ischemic damage. J. Cereb. Blood Flow Metab. 2008, 28, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Deitmer, J.W.; Theparambil, S.M.; Ruminot, I.; Noor, S.I.; Becker, H.M. Energy Dynamics in the Brain: Contributions of Astrocytes to Metabolism and pH Homeostasis. Front. Neurosci. 2019, 13, 1301. [Google Scholar] [CrossRef]
- Chesler, M. Failure and function of intracellular pH regulation in acute hypoxic-ischemic injury of astrocytes. Glia 2005, 50, 398–406. [Google Scholar] [CrossRef]
- Deitmer, J.W.; Rose, C.R. pH regulation and proton signalling by glial cells. Prog. Neurobiol. 1996, 48, 73–103. [Google Scholar] [CrossRef]
- Theparambil, S.M.; Hosford, P.S.; Ruminot, I.; Kopach, O.; Reynolds, J.R.; Sandoval, P.Y.; Rusakov, D.A.; Barros, L.F.; Gourine, A.V. Astrocytes regulate brain extracellular pH via a neuronal activity-dependent bicarbonate shuttle. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kraig, R.P.; Chesler, M. Astrocytic Acidosis in Hyperglycemic and Complete Ischemia. Br. J. Pharmacol. 1990, 10, 104–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Azad, P.; Zhao, H.W.; Wang, J.; Poulsen, O.; Freitas, B.C.; Muotri, A.R.; Haddad, G.G. The Na+/HCO3- co-transporter is protective during ischemia in astrocytes. Neuroscience 2016, 339, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarenko, A.; Chesler, M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia 2001, 34, 134–142. [Google Scholar] [CrossRef]
- Menyhart, A.; Zolei-Szenasi, D.; Puskas, T.; Makra, P.; Orsolya, M.T.; Szepes, B.E.; Toth, R.; Ivankovits-Kiss, O.; Obrenovitch, T.P.; Bari, F.; et al. Spreading depolarization remarkably exacerbates ischemia-induced tissue aci-dosis in the young and aged rat brain. Sci. Rep. 2017, 7, 1154. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.A.; Farrell, K.; Simon, R.P. Acidosis Causes Failure of Astrocyte Glutamate Uptake during Hypoxia. Br. J. Pharmacol. 1995, 15, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, O.M.; Menyhárt, Á.; Frank, R.; Hantosi, D.; Farkas, E.; Bari, F. Tissue Acidosis Associated with Ischemic Stroke to Guide Neuroprotective Drug Delivery. Biology 2020, 9, 460. [Google Scholar] [CrossRef] [PubMed]
- Mellergård, P.E.; Siesjö, B.K. Astrocytes fail to regulate intracellular pH at moderately reduced extracellular pH. NeuroReport 1991, 2, 695–698. [Google Scholar] [CrossRef]
- Mutch, W.A.C.; Hansen, A.J. Extracellular pH Changes during Spreading Depression and Cerebral Ischemia: Mechanisms of Brain pH Regulation. Br. J. Pharmacol. 1984, 4, 17–27. [Google Scholar] [CrossRef]
- Kintner, U.B.; Su, G.; Lenart, B.; Ballard, A.J.; Meyer, J.W.; Ng, L.L.; Shull, G.E.; Sun, D. Increased tolerance to oxygen and glucose deprivation in astrocytes from Na+/H+ exchanger isoform 1 null mice. Am. J. Physiol. Physiol. 2004, 287, C12–C21. [Google Scholar] [CrossRef] [Green Version]
- Bondarenko, A.; Svichar, N.; Chesler, M. Role of Na+-H+ and Na+-Ca2+ exchange in hypoxia-related acute astrocyte death. Glia 2004, 49, 143–152. [Google Scholar] [CrossRef]
- Giffard, R.G.; Papadopoulos, M.C.; van Hooft, J.A.; Xu, L.; Giuffrida, R.; Monyer, H. The electrogenic sodium bicar-bonate cotransporter: Developmental expression in rat brain and possible role in acid vulnerability. J. Neurosci. 2000, 20, 1001–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, B.R.; MacAulay, N. Activity-dependent astrocyte swelling is mediated by pH-regulating mechanisms. Glia 2017, 65, 1668–1681. [Google Scholar] [CrossRef]
- Beppu, K.; Sasaki, T.; Tanaka, K.; Yamanaka, A.; Fukazawa, Y.; Shigemoto, R.; Matsui, K. Optogenetic Countering of Glial Acidosis Suppresses Glial Glutamate Release and Ischemic Brain Damage. Neuron 2014, 81, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, S.A.; Barres, B.A. The detrimental role of glial acidification during ischemia. Neuron 2014, 81, 221–223. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.S.; Mongin, A.A. Cell Volume Control in Healthy Brain and Neuropathologies. In Current Topics in Membranes; Academic Press: New York, NY, USA, 2018; Volume 81, pp. 385–455. [Google Scholar]
- Hübel, N.; Ullah, G. Anions Govern Cell Volume: A Case Study of Relative Astrocytic and Neuronal Swelling in Spreading Depolarization. PLoS ONE 2016, 11, e0147060. [Google Scholar] [CrossRef] [Green Version]
- MacAulay, N.; Zeuthen, T. Water transport between CNS compartments: Contributions of aquaporins and cotrans-porters. Neuroscience 2010, 168, 941–956. [Google Scholar] [CrossRef]
- Papadopoulos, M.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Andrew, R.D.; Labron, M.W.; Boehnke, S.E.; Carnduff, L.; Kirov, S.A. Physiological Evidence That Pyramidal Neurons Lack Functional Water Channels. Cereb. Cortex 2006, 17, 787–802. [Google Scholar] [CrossRef] [Green Version]
- Nagelhus, E.A.; Ottersen, O.P. Physiological Roles of Aquaporin-4 in Brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delpire, E.; Staley, K.J. Novel determinants of the neuronal Cl—concentration. J. Physiol. 2014, 592, 4099–4114. [Google Scholar] [CrossRef]
- Wilson, C.S.; Mongin, A.A. The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci. Lett. 2019, 689, 33–44. [Google Scholar] [CrossRef]
- Kettenmann, H.; Backus, K.H.; Schachner, M. gamma-Aminobutyric acid opens Cl-channels in cultured astrocytes. Brain Res. 1987, 404, 1–9. [Google Scholar] [CrossRef]
- Bevensee, M.O.; Apkon, M.; Boron, W.F. Intracellular pH regulation in cultured astrocytes from rat hippocampus. II. Electrogenic Na/HCO3 cotransport. J. Gen. Physiol. 1997, 110, 467–483. [Google Scholar] [CrossRef] [Green Version]
- Kimelberg, H. Active accumulation and exchange transport of chloride in astroglial cells in culture. Biochim. et Biophys. Acta (BBA) Biomembr. 1981, 646, 179–184. [Google Scholar] [CrossRef]
- Walz, W.; Mukerji, S. KCl movements during potassium-induced cytotoxic swelling of cultured astrocytes. Exp. Neurol. 1988, 99, 17–29. [Google Scholar] [CrossRef]
- Bekar, L.K.; Walz, W. Intracellular chloride modulates A-type potassium currents in astrocytes. Glia 2002, 39, 207–216. [Google Scholar] [CrossRef]
- Smith, Q.R.; Johanson, C.E.; Woodbury, D.M. Uptake of 36Cl and 22Na by the brain-cerebrospinal fluid system: Comparison of the permeability of the blood-brain and blood-cerebrospinal fluid barriers. J. Neurochem. 1981, 37, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Marandi, N.; Konnerth, A.; Garaschuk, O. Two-photon chloride imaging in neurons of brain slices. Pflügers Arch. Eur. J. Physiol. 2002, 445, 357–365. [Google Scholar] [CrossRef]
- Kovalchuk, Y.; Garaschuk, O. Two-Photon Chloride Imaging Using MQAE In Vitro and In Vivo. Cold Spring Harb. Protoc. 2012, 2012, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Gensch, T.; Untiet, V.; Franzen, A.; Kovermann, P.; Fahlke, C. Determination of Intracellular Chloride Concentrations by Fluorescence Lifetime Imaging. In Springer Series in Chemical Physics; Metzler, J.B., Ed.; Springer: Cham, Switzerland, 2015; pp. 189–211. [Google Scholar]
- Untiet, V.; Kovermann, P.; Gerkau, N.J.; Gensch, T.; Rose, C.R.; Fahlke, C. Glutamate transporter-associated anion channels adjust intracellular chloride concentrations during glial maturation. Glia 2017, 65, 388–400. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Tse, F.W.; Crichton, S.A.; Kettenmann, H. GABA-activated Cl- channels in astrocytes of hippocampal slices. J. Neurosci. 1989, 9, 3577–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, T.; Fritschy, J.M.; Grosche, J.; Pratt, G.D.; Mohler, H.; Kettenmann, H. Developmental regulation of voltage-gated K+ channel and GABAA receptor expression in Bergmann glial cells. J. Neurosci. 1994, 14, 2503–2514. [Google Scholar] [CrossRef] [Green Version]
- Egawa, K.; Yamada, J.; Furukawa, T.; Yanagawa, Y.; Fukuda, A. Cl—homeodynamics in gap junction-coupled astrocytic networks on activation of GABAergic synapses. J. Physiol. 2013, 591, 3901–3917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, S.D.; Kafitz, K.W.; Rose, C.R. Developmental profile and mechanisms of GABA-induced calcium signaling in hippocampal astrocytes. Glia 2008, 56, 1127–1137. [Google Scholar] [CrossRef]
- Kahle, K.T.; Khanna, A.R.; Alper, S.L.; Adragna, N.C.; Lauf, P.K.; Sun, D.; Delpire, E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 2015, 21, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnon, K.B.; Delpire, E. Physiology of SLC12 transporters: Lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am. J. Physiol. Physiol. 2013, 304, C693–C714. [Google Scholar] [CrossRef] [Green Version]
- Mount, D.B.; Mercado, A.; Song, L.; Xu, J.; George, A.L., Jr.; Delpire, E.; Gamba, G. Cloning and Characterization of KCC3 and KCC4, New Members of the Cation-Chloride Cotransporter Gene Family. J. Biol. Chem. 1999, 274, 16355–16362. [Google Scholar] [CrossRef] [Green Version]
- Su, G.; Kintner, U.B.; Sun, D. Contribution of Na+-K+-Cl−cotransporter to high-[K+]o- induced swelling and EAA release in astrocytes. Am. J. Physiol. Physiol. 2002, 282, C1136–C1146. [Google Scholar] [CrossRef] [Green Version]
- Fairman, W.A.; Vandenberg, R.J.; Arriza, J.L.; Kavanaught, M.P.; Amara, S. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nat. Cell Biol. 1995, 375, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Wadiche, J.; Amara, S.; Kavanaugh, M.P. Ion fluxes associated with excitatory amino acid transport. Neuron 1995, 15, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Larsson, H.; Picaud, S.; Werblin, F.; Lecar, H. Noise analysis of the glutamate-activated current in photoreceptors. Biophys. J. 1996, 70, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Machtens, J.-P.; Kortzak, D.; Lansche, C.; Leinenweber, A.; Kilian, P.; Begemann, B.; Zachariae, U.; Ewers, D.; De Groot, B.L.; Briones, R.; et al. Mechanisms of Anion Conduction by Coupled Glutamate Transporters. Cell 2015, 160, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.H.; Torres-Salazar, D.; Gonzalez-Suarez, A.D.; Amara, S.G.; Bahar, I. Substrate transport and anion permeation proceed through distinct pathways in glutamate transporters. eLife 2017, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Fahlke, C.; Kortzak, D.; Machtens, J.-P. Molecular physiology of EAAT anion channels. Pflügers Arch. Eur. J. Physiol. 2016, 468, 491–502. [Google Scholar] [CrossRef]
- Regan, M.R.; Huang, Y.H.; Kim, Y.S.; Dykes-Hoberg, M.I.; Jin, L.; Watkins, A.M.; Bergles, D.E.; Rothstein, J.D. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the devel-oping and mature CNS. J. Neurosci. 2007, 27, 6607–6619. [Google Scholar] [CrossRef] [Green Version]
- Jen, J.C.; Wan, J.; Palos, T.P.; Howard, B.D.; Baloh, R.W. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 2005, 65, 529–534. [Google Scholar] [CrossRef]
- Kovermann, P.; Untiet, V.; Kolobkova, Y.; Engels, M.; Baader, S.; Schilling, K.; Fahlke, C. Increased glutamate trans-porter-associated anion currents cause glial apoptosis in episodic ataxia 6. Brain Commun. 2020, 2, fcaa022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, N.; Kovermann, P.; Fahlke, C. A point mutation associated with episodic ataxia 6 increases glutamate trans-porter anion currents. Brain 2012, 135, 3416–3425. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, C.K.; Stein, V.; Keating, D.J.; Maier, H.; Rinke, I.; Rudhard, Y.; Hentschke, M.; Rune, G.M.; Jentsch, T.J.; Hubner, C.A. NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal de-velopment. J. Neurosci. 2009, 29, 3419–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delpire, E.; Lu, J.; England, R.; Dull, C.; Thorne, T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 1999, 22, 192–195. [Google Scholar] [CrossRef] [PubMed]
- MacNamara, E.F.; Koehler, A.E.; D’Souza, P.; Estwick, T.; Lee, P.; Vezina, G.; Fauni, H.; Braddock, S.R.; Torti, E.; Holt, J.M.; et al. Kilquist syndrome: A novel syndromic hearing loss disorder caused by homozygous deletion of SLC12A2. Hum. Mutat. 2019, 40, 532–538. [Google Scholar] [CrossRef]
- Delpire, E.; Wolfe, L.; Flores, B.; Koumangoye, R.; Schornak, C.C.; Omer, S.; Pusey, B.; Lau, C.; Markello, T.; Adams, D.R. A patient with multisystem dysfunction carries a truncation mutation in humanSLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Mol. Case Stud. 2016, 2, a001289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.; Matheson, C.; Sun, J.; Radeke, M.; Feinstein, S.; Miller, J. Distribution of Intracerebral Ventricularly Administered Neurotrophins in Rat Brain and Its Correlation with Trk Receptor Expression. Exp. Neurol. 1994, 127, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Dempsey, R.J.; Sun, D. Na+-K+-Cl− Cotransporter in Rat Focal Cerebral Ischemia. Br. J. Pharmacol. 2001, 21, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Sun, D. The role of Na-K-Cl co-transporter in cerebral ischemia. Neurol. Res. 2005, 27, 280–286. [Google Scholar] [CrossRef]
- Chen, H.; Luo, J.; Kintner, D.B.; Shull, G.E.; Sun, D. Na+-Dependent Chloride Transporter (NKCC1)-Null Mice Exhibit Less Gray and White Matter Damage after Focal Cerebral Ischemia. Br. J. Pharmacol. 2005, 25, 54–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Bhuiyan, M.I.H.; Jiang, T.; Song, S.; Shankar, S.; Taheri, T.; Li, E.; Schreppel, P.; Hintersteininger, M.; Yang, S.-S.; et al. A Novel Na+-K+-Cl- Cotransporter 1 Inhibitor STS66* Reduces Brain Damage in Mice After Ischemic Stroke. Stroke 2019, 50, 1021–1025. [Google Scholar] [CrossRef]
- Inoue, H.; Okada, Y. Roles of Volume-Sensitive Chloride Channel in Excitotoxic Neuronal Injury. J. Neurosci. 2007, 27, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Okada, Y.; Nilius, B. Biophysics and Physiology of the Volume-Regulated Anion Channel (VRAC)/Volume-Sensitive Outwardly Rectifying Anion Channel (VSOR). Pflügers Arch. Eur. J. Physiol. 2016, 468, 371–383. [Google Scholar] [CrossRef]
- Strange, K.; Yamada, T.; Denton, J.S. A 30-year journey from volume-regulated anion currents to molecular structure of the LRRC8 channel. J. Gen. Physiol. 2019, 151, 100–117. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.S.; Bach, M.D.; Ashkavand, Z.; Norman, K.R.; Martino, N.; Adam, A.P.; Mongin, A.A. Metabolic constraints of swelling-activated glutamate release in astrocytes and their implication for ischemic tissue damage. J. Neurochem. 2019, 151, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Sacher, A.; Nelson, N.; Ogi, J.T.; Wright, E.M.; Loo, D.D.; Eskandari, S. Presteady-state and steady-state kinetics and turnover rate of the mouse gamma-aminobutyric acid transporter (mGAT3). J. Membr. Biol. 2002, 190, 57–73. [Google Scholar] [CrossRef]
- Loo, D.D.; Eskandari, S.; Boorer, K.J.; Sarkar, H.K.; Wright, E.M. Role of Cl− in Electrogenic Na+-coupled Cotransporters GAT1 and SGLT1. J. Biol. Chem. 2000, 275, 37414–37422. [Google Scholar] [CrossRef] [Green Version]
- Roux, M.J.; Supplisson, S. Neuronal and Glial Glycine Transporters Have Different Stoichiometries. Neuron 2000, 25, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Zomot, E.; Bendahan, A.; Quick, M.; Zhao, Y.; Javitch, J.A.; Kanner, B.I. Mechanism of chloride interaction with neu-rotransmitter:sodium symporters. Nature 2007, 449, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, A.L.; Horowicz, P. The influence of potassium and chloride ions on the membrane potential of single muscle fibres. J. Physiol. 1959, 148, 127–160. [Google Scholar] [CrossRef] [PubMed]
- Dulhunty, A.F. The dependence of membrane potential on extracellular chloride concentration in mammalian skeletal muscle fibres. J. Physiol. 1978, 276, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Habela, C.W.; Ernest, N.J.; Swindall, A.F.; Sontheimer, H. Chloride Accumulation Drives Volume Dynamics Underlying Cell Proliferation and Migration. J. Neurophysiol. 2009, 101, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Sachs, F.; Sivaselvan, M.V. Cell volume control in three dimensions: Water movement without solute movement. J. Gen. Physiol. 2015, 145, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongin, A.A.; Orlov, S.N. Mechanisms of cell volume regulation and possible nature of the cell volume sensor. Pathophysiol. 2001, 8, 77–88. [Google Scholar] [CrossRef]
- Benfenati, V.; Ferroni, S. Water transport between CNS compartments: Functional and molecular interactions between aquaporins and ion channels. Neuroscience 2010, 168, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Rungta, R.L.; Choi, H.B.; Tyson, J.R.; Malik, A.; Dissing-Olesen, L.; Lin, P.J.; Cain, S.M.; Cullis, P.R.; Snutch, T.P.; MacVicar, B.A. The Cellular Mechanisms of Neuronal Swelling Underlying Cytotoxic Edema. Cell 2015, 161, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, N. The vacuolar H(+)-ATPase--one of the most fundamental ion pumps in nature. J. Exp. Biol. 1992, 172, 19–27. [Google Scholar] [CrossRef]
- Lloyd, A.C. The Regulation of Cell Size. Cell 2013, 154, 1194–1205. [Google Scholar] [CrossRef] [Green Version]
- Gurtovenko, A.A.; Vattulainen, I. Ion Leakage through Transient Water Pores in Protein-Free Lipid Membranes Driven by Transmembrane Ionic Charge Imbalance. Biophys. J. 2007, 92, 1878–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischbarg, J.; Kuang, K.Y.; Vera, J.C.; Arant, S.; Silverstein, S.C.; Loike, J.; Rosen, O.M. Glucose transporters serve as water channels. Proc. Natl. Acad. Sci. USA 1990, 87, 3244–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toft-Bertelsen, T.L.; Larsen, B.R.; Christensen, S.K.; Khandelia, H.; Waagepetersen, H.S.; MacAulay, N. Clearance of activity-evoked K(+) transients and associated glia cell swelling occur independently of AQP4: A study with an iso-form-selective AQP4 inhibitor. Glia 2021, 69, 28–41. [Google Scholar] [CrossRef]
- Iwamoto, M.; Oiki, S. Counting Ion and Water Molecules in a Streaming File through the Open-Filter Structure of the K Channel. J. Neurosci. 2011, 31, 12180–12188. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, M.B.; Bagnéris, C.; McCusker, E.C.; DeCaen, P.G.; Delling, M.; Clapham, D.E.; Ulmschneider, J.P.; Wallace, B.A. Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 2013, 110, 6364–6369. [Google Scholar] [CrossRef] [Green Version]
- Ando, H.; Kuno, M.; Shimizu, H.; Muramatsu, I.; Oiki, S. Coupled K+–Water Flux through the HERG Potassium Channel Measured by an Osmotic Pulse Method. J. Gen. Physiol. 2005, 126, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Vella, J.; Zammit, C.; Di Giovanni, G.; Muscat, R.; Valentino, M. The central role of aquaporins in the pathophysiology of ischemic stroke. Front. Cell. Neurosci. 2015, 9, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, R.D.; Hsieh, Y.-T.; Brisson, C.D. Spreading depolarization triggered by elevated potassium is weak or absent in the rodent lower brain. Br. J. Pharmacol. 2016, 37, 1735–1747. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of Cell Volume Regulation in Vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Zeuthen, T. Water-Transporting Proteins. J. Membr. Biol. 2009, 234, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. Br. J. Pharmacol. 2016, 36, 513–538. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Zhang, Y.; Liao, X.; Yang, R.; Lei, Y.; Luo, J. Potential Therapies for Cerebral Edema After Ischemic Stroke: A Mini Review. Front. Aging Neurosci. 2021, 12, 618819. [Google Scholar] [CrossRef]
- Hofmeijer, J.; Kappelle, L.J.; Algra, A.; Amelink, G.J.; van Gijn, J.; van der Worp, H.B.; Investigators, H. Surgical de-compression for space-occupying cerebral infarction (the Hemicraniectomy After Middle Cerebral Artery infarction with Life-threatening Edema Trial [HAMLET]): A multicentre, open, randomised trial. Lancet Neurol. 2009, 8, 326–333. [Google Scholar] [CrossRef]
- Nelson, P. Biological Physics. Energy, Information, Life; W.H. Freeman and Company: New York, NY, USA, 2008. [Google Scholar]
- Van Putten, M.J.A.M. Dynamics of Neural Networks: A Mathematical and Clinical Approach; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Plonsey, R.; Barr, R.C. Bioelecticity: A Qunantitarive Approach; Spinger: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Dijkstra, K.; Hofmeijer, J.; van Gils, S.A.; van Putten, M.J.A.M. A Biophysical Model for Cytotoxic Cell Swelling. J. Neurosci. 2016, 36, 11881–11890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, B.-J.; Zhang, H.; Binder, D.K.; Verkman, A. Aquaporin-4–dependent K+ and water transport modeled in brain extracellular space following neuroexcitation. J. Gen. Physiol. 2013, 141, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Halnes, G.; Østby, I.; Pettersen, K.H.; Omholt, S.W.; Einevoll, G.T. Electrodiffusive Model for Astrocytic and Neuronal Ion Concentration Dynamics. PLoS Comput. Biol. 2013, 9, e1003386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübel, N.; Andrew, R.D.; Ullah, G. Large extracellular space leads to neuronal susceptibility to ischemic injury in a Na+/K+ pumps–dependent manner. J. Comput. Neurosci. 2016, 40, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Kalia, M.; Meijer, H.G.E.; van Gils, S.A.; van Putten, M.J.A.M.; Rose, C.R. Ion dynamics at the energy-deprived tripartite synapse. bioRxiv 2021, arXiv:2021.03.19.436129. [Google Scholar]
- Glykys, J.; Dzhala, V.; Egawa, K.; Kahle, K.T.; Delpire, E.; Staley, A. Chloride dysregulation, seizures, and cerebral edema: A relationship with therapeutic potential Trends. Neurosciences 2017, 40, 276–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Suryanarayanan, A.; Chandra, D.; Homanics, G.E.; Olsen, R.W.; Spigelman, I. Functional Consequences of GABAA Receptor α4 Subunit Deletion on Synaptic and Extrasynaptic Currents in Mouse Dentate Granule Cells. Alcohol. Clin. Exp. Res. 2007, 32, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Briggs, M.M.; McIntosh, T.J. Water Permeability of Aquaporin-4 Channel Depends on Bilayer Composition, Thickness, and Elasticity. Biophys. J. 2012, 103, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Pangršič, T.; Potokar, M.; Haydon, P.G.; Zorec, R.; Kreft, M. Astrocyte swelling leads to membrane unfolding, not membrane insertion. J. Neurochem. 2006, 99, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Ito, U.; Ohno, K.; Nakamura, R.; Suganuma, F.; Inaba, Y. Brain edema during ischemia and after restoration of blood flow. Measurement of water, sodium, potassium content and plasma protein permeability. Stroke 1979, 10, 542–547. [Google Scholar] [CrossRef] [Green Version]
- Michinaga, S.; Koyama, Y. Pathogenesis of Brain Edema and Investigation into Anti-Edema Drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef] [Green Version]
- Hladky, S.B.; Barrand, M.A. Mechanisms of fluid movement into, through and out of the brain: Evaluation of the evidence. Fluids Barriers CNS 2014, 11, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nzou, G.; Wicks, R.T.; VanOstrand, N.R.; Mekky, G.A.; Seale, S.A.; El-Taibany, A.; Wicks, E.E.; Nechtman, C.M.; Marrotte, E.J.; Makani, V.S.; et al. Multicellular 3D Neurovascular Unit Model for Assessing Hypoxia and Neuroinflammation Induced Blood-Brain Barrier Dysfunction. Sci. Rep. 2020, 10, 9766. [Google Scholar] [CrossRef]
- Dostovic, Z.; Dostovic, E.; Smajlovic, D.; Avdic, L.; Ibrahimagic, O.C. Brain Edema After Ischaemic Stroke. Med. Arch. 2016, 70, 339–341. [Google Scholar] [CrossRef] [Green Version]
- Abbruscato, T.J.; Lopez, S.P.; Roder, K.; Paulson, J.R. Regulation of Blood-Brain Barrier Na, K, 2Cl-Cotransporter through Phosphorylation during in Vitro Stroke Conditions and Nicotine Exposure. J. Pharmacol. Exp. Ther. 2004, 310, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vemula, S.; Roder, K.E.; Yang, T.; Bhat, G.J.; Thekkumkara, T.J.; Abbruscato, T.J. A Functional Role for Sodium-Dependent Glucose Transport across the Blood-Brain Barrier during Oxygen Glucose Deprivation. J. Pharmacol. Exp. Ther. 2008, 328, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junc-tions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.E. Blood–Brain Barrier Na Transporters in Ischemic Stroke. Adv. Pharmacol. 2014, 71, 113–146. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Putten, M.J.A.M.; Fahlke, C.; Kafitz, K.W.; Hofmeijer, J.; Rose, C.R. Dysregulation of Astrocyte Ion Homeostasis and Its Relevance for Stroke-Induced Brain Damage. Int. J. Mol. Sci. 2021, 22, 5679. https://doi.org/10.3390/ijms22115679
van Putten MJAM, Fahlke C, Kafitz KW, Hofmeijer J, Rose CR. Dysregulation of Astrocyte Ion Homeostasis and Its Relevance for Stroke-Induced Brain Damage. International Journal of Molecular Sciences. 2021; 22(11):5679. https://doi.org/10.3390/ijms22115679
Chicago/Turabian Stylevan Putten, Michel J. A. M., Christoph Fahlke, Karl W. Kafitz, Jeannette Hofmeijer, and Christine R. Rose. 2021. "Dysregulation of Astrocyte Ion Homeostasis and Its Relevance for Stroke-Induced Brain Damage" International Journal of Molecular Sciences 22, no. 11: 5679. https://doi.org/10.3390/ijms22115679
APA Stylevan Putten, M. J. A. M., Fahlke, C., Kafitz, K. W., Hofmeijer, J., & Rose, C. R. (2021). Dysregulation of Astrocyte Ion Homeostasis and Its Relevance for Stroke-Induced Brain Damage. International Journal of Molecular Sciences, 22(11), 5679. https://doi.org/10.3390/ijms22115679