
Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
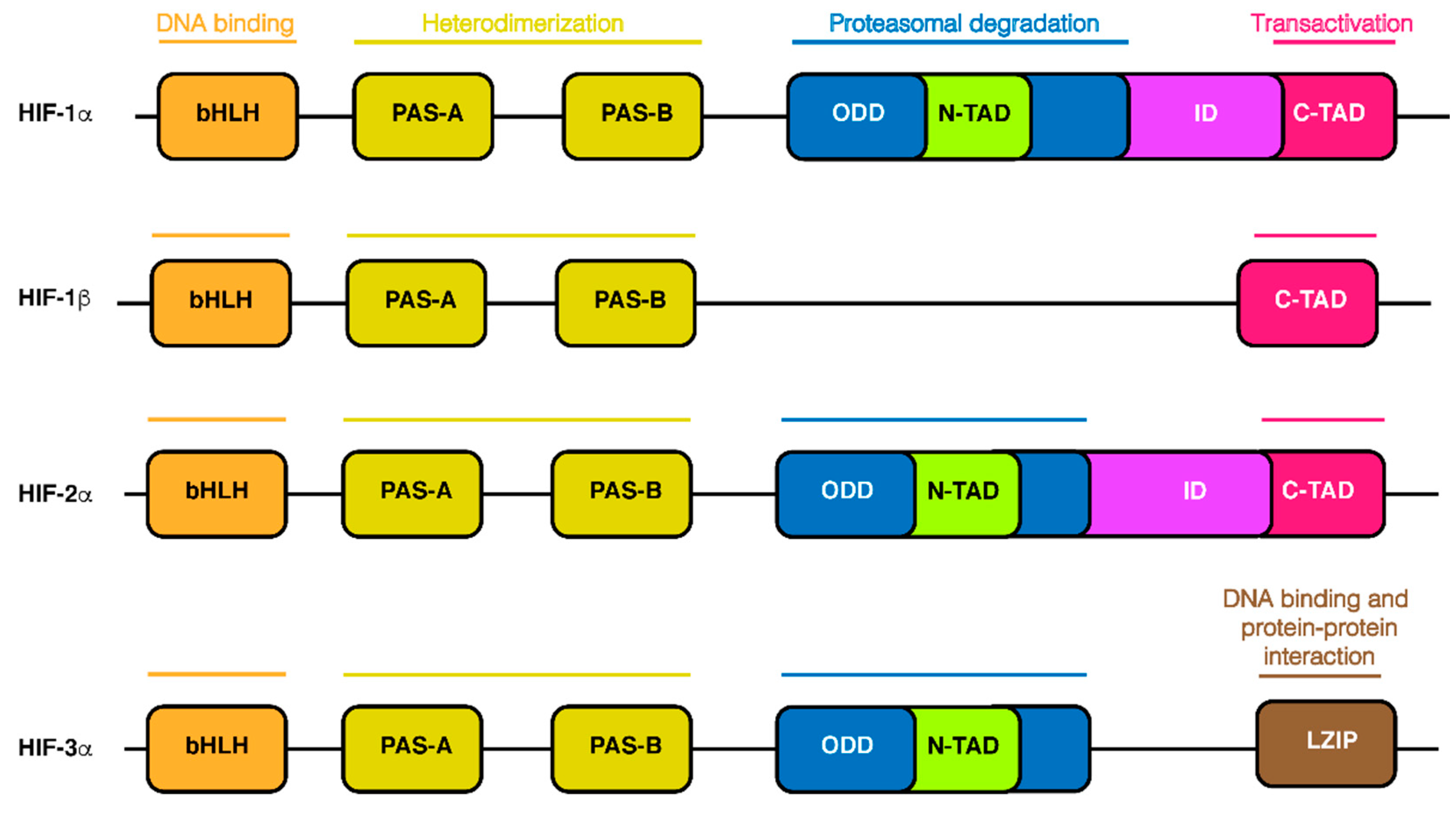
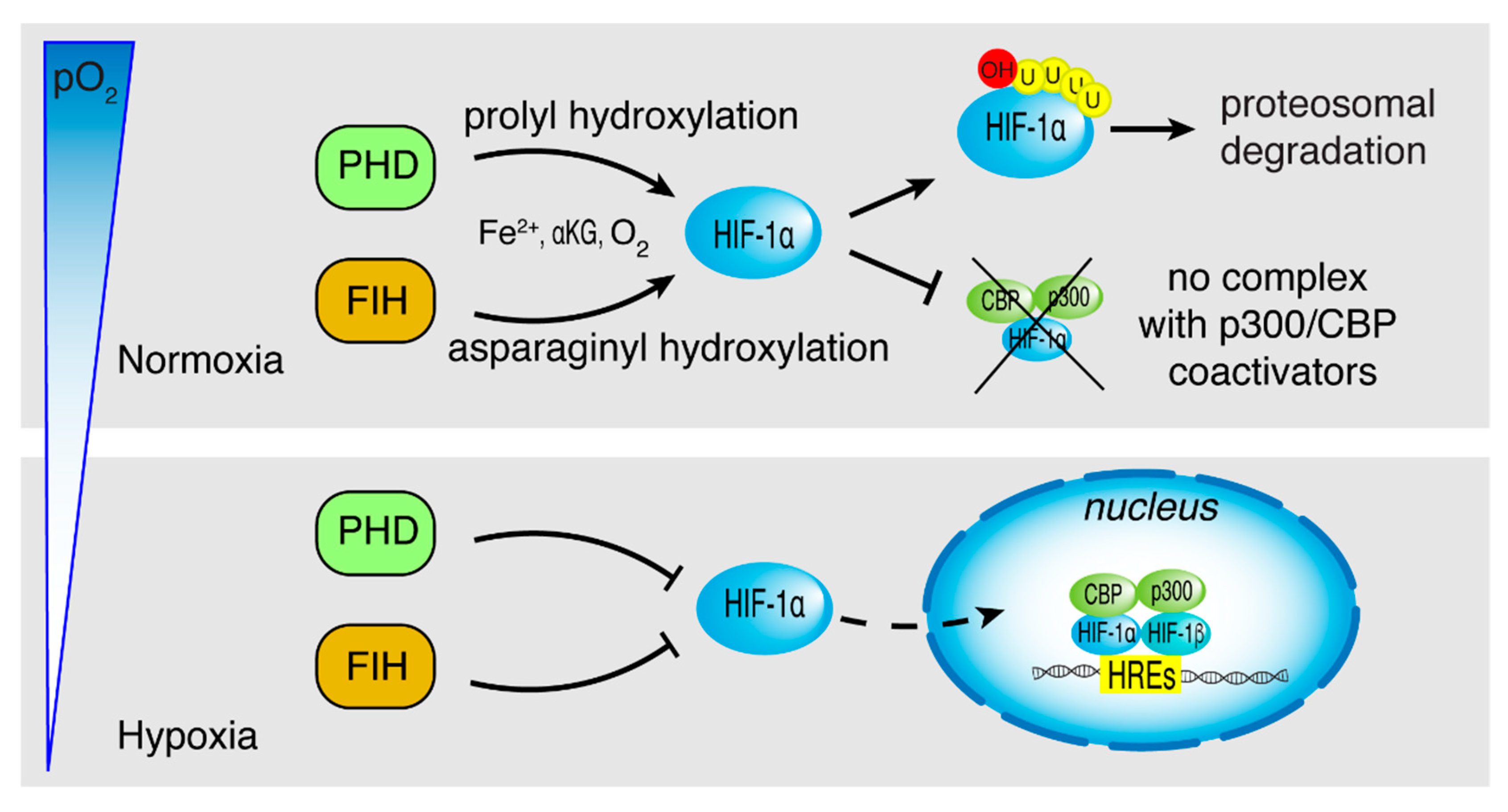
2. HIF-1α Structure
3. Metabolic Reprogramming in Hypoxia Induced by HIF-1
3.1. Glucose Metabolism
3.2. Lactate and Acidification
3.3. Lipid Metabolism
3.4. Amino Acids’ Metabolism
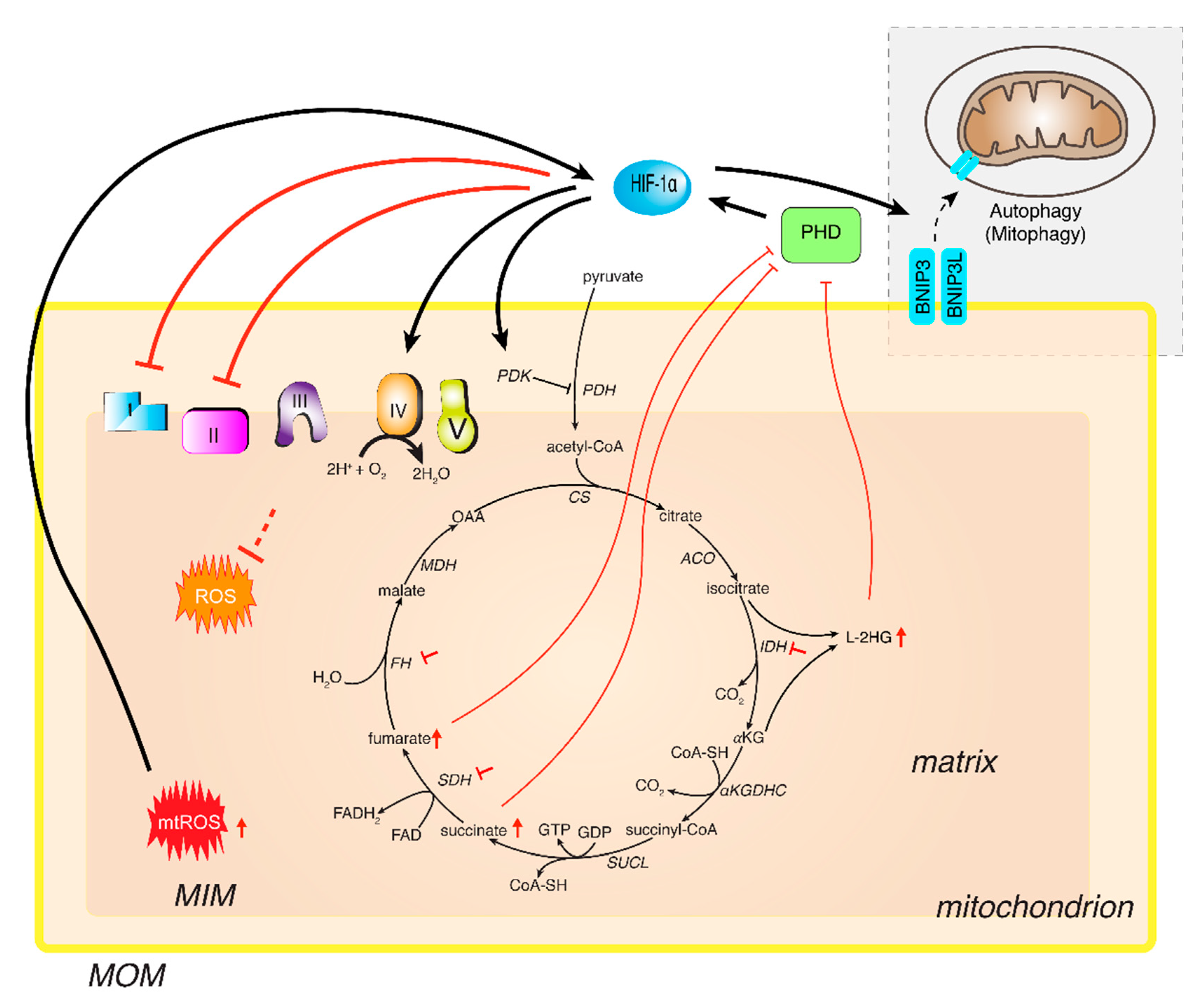
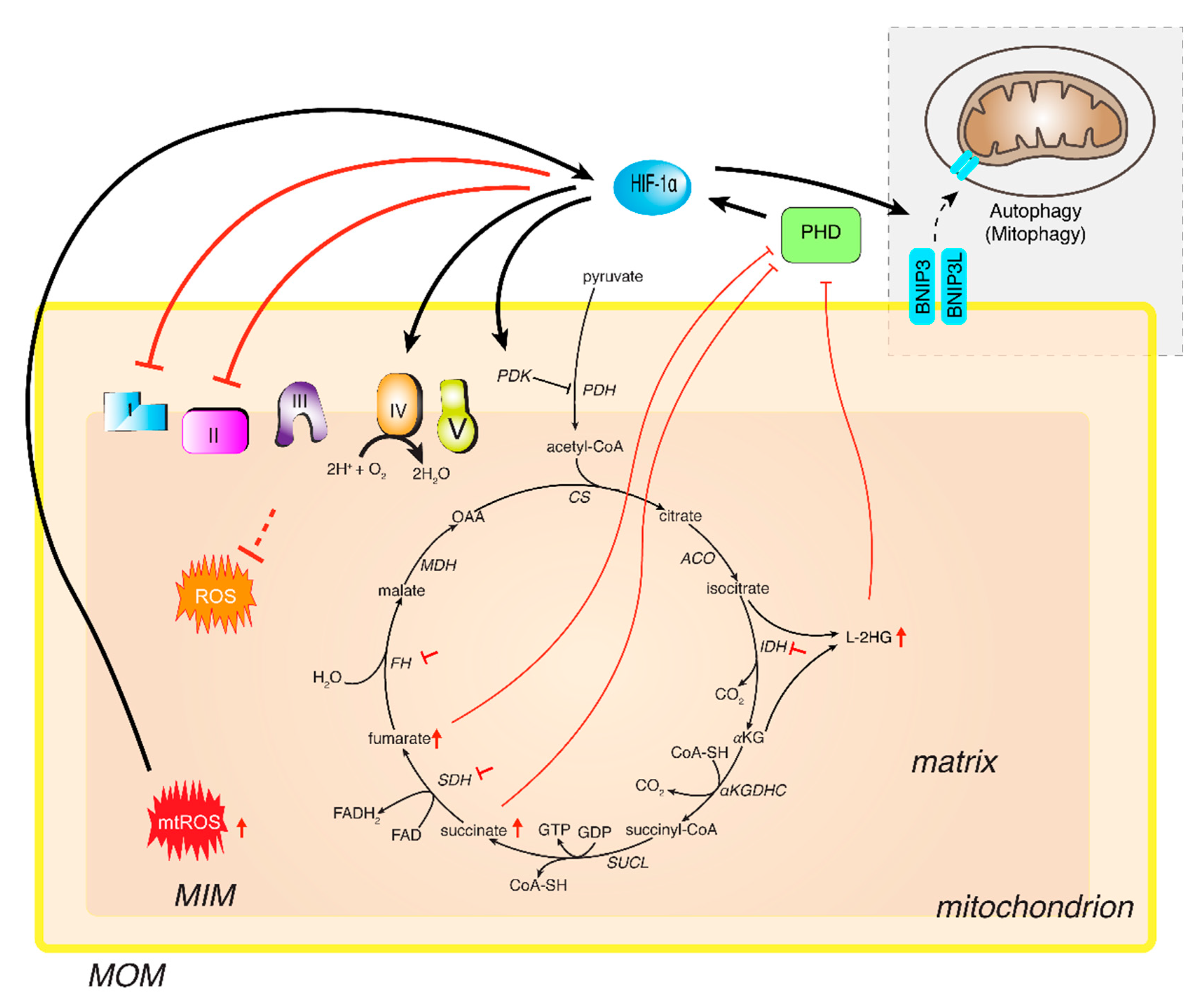
4. Impact of Hypoxia on Mitochondrial Function
4.1. TCA Cycle
4.2. Electron Transport Chain
4.3. Mitochondrial Biogenesis and Autophagy
4.4. ROS
5. Targeting HIF-1 in Cancer Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Ann. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickaert, A.; Saiselet, M.; Dom, G.; De Deken, X.; Dumont, J.E.; Feron, O.; Sonveaux, P.; Maenhaut, C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 2017, 36, 2637–2642. [Google Scholar] [CrossRef] [Green Version]
- Payen, V.L.; Porporato, P.E.; Baselet, B.; Sonveaux, P. Metabolic changes associated with tumor metastasis, Part 1: Tumor pH, glycolysis and the pentose phosphate pathway. Cell. Mol. Life Sci. 2016, 73, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016, 30, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.Y.; Yang, M.H. Metabolic reprogramming and epithelial-mesenchymal plasticity: Opportunities and challenges for cancer therapy. Front. Oncol. 2020, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Brune, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Wittig, I.; Heide, H.; Dehne, N.; Brune, B. Chronic hypoxia alters mitochondrial composition in human macrophages. Biochim. Biophys. Acta 2013, 1834, 2750–2760. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Wood, S.M.; Gleadle, J.M.; Pugh, C.W.; Hankinson, O.; Ratcliffe, P.J. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J. Biol. Chem. 1996, 271, 15117–15123. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, N.V.; Leung, S.W.; Semenza, G.L. The human hypoxia-inducible factor 1alpha gene: HIF1A structure and evolutionary conservation. Genomics 1998, 52, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiener, C.M.; Booth, G.; Semenza, G.L. In vivo expression of mRNAs encoding hypoxia-inducible factor 1. Biochem. Biophys. Res. Commun. 1996, 225, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jurgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Horstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [Green Version]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and characterization of hypoxia-inducible factor (HIF)-3alpha in human kidney: Suppression of HIF-mediated gene expression by HIF-3alpha. Biochem. Biophys. Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef]
- Maynard, M.A.; Qi, H.; Chung, J.; Lee, E.H.; Kondo, Y.; Hara, S.; Conaway, R.C.; Conaway, J.W.; Ohh, M. Multiple splice variants of the human HIF-3 alpha locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J. Biol. Chem. 2003, 278, 11032–11040. [Google Scholar] [CrossRef] [Green Version]
- Heidbreder, M.; Frohlich, F.; Johren, O.; Dendorfer, A.; Qadri, F.; Dominiak, P. Hypoxia rapidly activates HIF-3alpha mRNA expression. FASEB J. 2003, 17, 1541–1543. [Google Scholar] [CrossRef]
- Pasanen, A.; Heikkila, M.; Rautavuoma, K.; Hirsila, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3alpha is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int. J. Biochem. Cell Biol. 2010, 42, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Pappalardi, M.B.; McNulty, D.E.; Martin, J.D.; Fisher, K.E.; Jiang, Y.; Burns, M.C.; Zhao, H.; Ho, T.; Sweitzer, S.; Schwartz, B.; et al. Biochemical characterization of human HIF hydroxylases using HIF protein substrates that contain all three hydroxylation sites. Biochem. J. 2011, 436, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Zhang, H.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, P.; Weissmann, N.; Grimminger, F.; Hegel, C.; Bader, L.; Rose, F.; Fink, L.; Ghofrani, H.A.; Schermuly, R.T.; Schmidt, H.H.; et al. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free Radic. Biol. Med. 2004, 36, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.K.; Han, Y.M.; Kim, J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int. J. Biochem. Cell Biol. 2008, 40, 1043–1054. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal 2009, 2, ra73. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.; Snell, C.; Steers, G.; Turley, H.; Li, J.L.; Gunther, U.L.; et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.N.; Guo, P.; Lim, S.; Bassilian, S.; Lee, S.T.; Boren, J.; Cascante, M.; Go, V.L.; Boros, L.G. Metabolic sensitivity of pancreatic tumour cell apoptosis to glycogen phosphorylase inhibitor treatment. Br. J. Cancer 2004, 91, 2094–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brizel, D.M.; Schroeder, T.; Scher, R.L.; Walenta, S.; Clough, R.W.; Dewhirst, M.W.; Mueller-Klieser, W. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 349–353. [Google Scholar] [CrossRef]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfor, K.; Rofstad, E.K.; Mueller-Klieser, W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000, 60, 916–921. [Google Scholar] [PubMed]
- Garcia, C.K.; Goldstein, J.L.; Pathak, R.K.; Anderson, R.G.; Brown, M.S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: Implications for the Cori cycle. Cell 1994, 76, 865–873. [Google Scholar] [CrossRef]
- Jones, R.S.; Morris, M.E. Monocarboxylate Transporters: Therapeutic Targets and Prognostic Factors in Disease. Clin. Pharmacol. Ther. 2016, 100, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Leite, T.C.; Coelho, R.G.; Da Silva, D.; Coelho, W.S.; Marinho-Carvalho, M.M.; Sola-Penna, M. Lactate downregulates the glycolytic enzymes hexokinase and phosphofructokinase in diverse tissues from mice. FEBS Lett. 2011, 585, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; McIntyre, D.; Honess, D.; Hulikova, A.; Pacheco-Torres, J.; Cerdan, S.; Swietach, P.; Harris, A.L.; Griffiths, J.R. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. Br. J. Cancer 2018, 119, 622–630. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Brahimi-Horn, M.C.; Pouyssegur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Rathmell, J.C. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015, 36, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma. Cancers 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Chen, R.; Xu, M.; Nagati, J.; Garcia, J.A. Coordinate regulation of stress signaling and epigenetic events by Acss2 and HIF-2 in cancer cells. PLoS ONE 2017, 12, e0190241. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [Green Version]
- Peck, B.; Schulze, A. Lipid desaturation—the next step in targeting lipogenesis in cancer? FEBS J. 2016, 283, 2767–2778. [Google Scholar] [CrossRef]
- Gottgens, E.L.; van den Heuvel, C.N.; de Jong, M.C.; Kaanders, J.H.; Leenders, W.P.; Ansems, M.; Bussink, J.; Span, P.N. ACLY (ATP Citrate Lyase) Mediates Radioresistance in Head and Neck Squamous Cell Carcinomas and is a Novel Predictive Radiotherapy Biomarker. Cancers 2019, 11, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infantino, V.; Iacobazzi, V.; Palmieri, F.; Menga, A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 2013, 440, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Pierri, C.L.; Iacobazzi, V. Metabolic routes in inflammation: The citrate pathway and its potential as therapeutic target. Curr. Med. Chem. 2019, 26, 7104–7116. [Google Scholar] [CrossRef]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [Green Version]
- Triantafyllou, E.A.; Georgatsou, E.; Mylonis, I.; Simos, G.; Paraskeva, E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Kourti, M.; Ikonomou, G.; Giakoumakis, N.N.; Rapsomaniki, M.A.; Landegren, U.; Siniossoglou, S.; Lygerou, Z.; Simos, G.; Mylonis, I. CK1delta restrains lipin-1 induction, lipid droplet formation and cell proliferation under hypoxia by reducing HIF-1alpha/ARNT complex formation. Cell Signal 2015, 27, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Siniossoglou, S. Phospholipid metabolism and nuclear function: Roles of the lipin family of phosphatidic acid phosphatases. Biochim. Biophys. Acta 2013, 1831, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2alpha-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [Green Version]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schodel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Wu, H.; Cordasic, N.; Oefner, P.; Dietel, B.; Thiele, C.; Weidemann, A.; Eckardt, K.U.; Warnecke, C. Hypoxia-inducible protein 2 Hig2/Hilpda mediates neutral lipid accumulation in macrophages and contributes to atherosclerosis in apolipoprotein E-deficient mice. FASEB J. 2017, 31, 4971–4984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Tumanov, S.; Qiu, B.; Michalopoulou, E.; Spata, M.; Azzam, A.; Xie, H.; Simon, M.C.; Kamphorst, J.J. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 2018, 24, 2596–2605.e5. [Google Scholar] [CrossRef] [Green Version]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morotti, M.; Bridges, E.; Valli, A.; Choudhry, H.; Sheldon, H.; Wigfield, S.; Gray, N.; Zois, C.E.; Grimm, F.; Jones, D.; et al. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 12452–12461. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathmell, W.K.; Chen, S. VHL inactivation in renal cell carcinoma: Implications for diagnosis, prognosis and treatment. Expert Rev. Anticancer Ther. 2008, 8, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, A.; Gameiro, P.A.; Christodoulou, D.; Laviollette, L.; Schneider, M.; Chaves, F.; Stemmer-Rachamimov, A.; Yazinski, S.A.; Lee, R.; Stephanopoulos, G.; et al. Glutaminase and poly (ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J. Clin. Investig. 2017, 127, 1631–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Convertini, P.; Todisco, S.; De Santis, F.; Pappalardo, I.; Iacobazzi, D.; Castiglione Morelli, M.A.; Fondufe-Mittendorf, Y.N.; Martelli, G.; Palmieri, F.; Infantino, V. Transcriptional Regulation Factors of the Human Mitochondrial Aspartate/Glutamate Carrier Gene, Isoform 2 (SLC25A13): USF1 as Basal Factor and FOXA2 as Activator in Liver Cells. Int. J. Mol. Sci. 2019, 20, 1888. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Finlay, D.K. Glucose, glycolysis and lymphocyte responses. Mol. Immunol. 2015, 68, 513–519. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.Q.; Zhang, S.; Zhu, H.J.; Wang, W.; Zhu, J.H.; Wang, X.D.; Qiang, J.F. Increased expression of PHGDH and prognostic significance in colorectal cancer. Transl. Oncol. 2016, 9, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Z.; Heng, W.; Aiping, D.; Yafei, Q.; Shulan, Z. Expression and clinical significance of phosphoglycerate dehydrogenase and squamous cell carcinoma antigen in cervical cancer. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2013, 23, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 2016, 76, 4430–4442. [Google Scholar] [CrossRef] [Green Version]
- Iacobazzi, V.; Infantino, V.; Castegna, A.; Andria, G. Hyperhomocysteinemia: Related genetic diseases and congenital defects, abnormal DNA methylation and newborn screening issues. Mol. Genet. Metab. 2014, 113, 27–33. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Infantino, V.; Dituri, F.; Convertini, P.; Santarsiero, A.; Palmieri, F.; Todisco, S.; Mancarella, S.; Giannelli, G.; Iacobazzi, V. Epigenetic upregulation and functional role of the mitochondrial aspartate/glutamate carrier isoform 1 in hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 38–47. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, A.; Letouze, E.; Gimenez-Roqueplo, A.P.; Favier, J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int. J. Cancer 2014, 135, 2237–2248. [Google Scholar] [CrossRef] [PubMed]
- Vatrinet, R.; Leone, G.; De Luise, M.; Girolimetti, G.; Vidone, M.; Gasparre, G.; Porcelli, A.M. The alpha-ketoglutarate dehydrogenase complex in cancer metabolic plasticity. Cancer Metab. 2017, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Asano, Y.; Shintani, Y.; Aoyama, H.; Kioka, H.; Tsukamoto, O.; Hikita, M.; Shinzawa-Itoh, K.; Takafuji, K.; Higo, S.; et al. Higd1a is a positive regulator of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 1553–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordonez, A.; Corral-Escariz, M.; Soro, I.; Lopez-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.K.; Xu, I.M.; Chiu, D.K.; Tse, A.P.; Wei, L.L.; Law, C.T.; Lee, D.; Wong, C.M.; Wong, M.P.; Ng, I.O.; et al. NDUFA4L2 Fine-tunes Oxidative Stress in Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3105–3117. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.L.; et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef] [Green Version]
- Babot, M.; Birch, A.; Labarbuta, P.; Galkin, A. Characterisation of the active/de-active transition of mitochondrial complex I. Biochim. Biophys. Acta 2014, 1837, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Ciano, M.; Fuszard, M.; Heide, H.; Botting, C.H.; Galkin, A. Conformation-specific crosslinking of mitochondrial complex I. FEBS Lett. 2013, 587, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Puissegur, M.P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef] [Green Version]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landazuri, M.O.; Enriquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtulus, S.; Tripathi, P.; Opferman, J.T.; Hildeman, D.A. Contracting the ’mus cells’—does down-sizing suit us for diving into the memory pool? Immunol. Rev. 2010, 236, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Drogat, B.; Bouchecareilh, M.; North, S.; Petibois, C.; Deleris, G.; Chevet, E.; Bikfalvi, A.; Moenner, M. Acute L-glutamine deprivation compromises VEGF-a upregulation in A549/8 human carcinoma cells. J. Cell. Physiol. 2007, 212, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, D.; Shen, L.; Dong, K.; Wu, M.; Ou, Z.; Shi, D. Redox homeostasis protects mitochondria through accelerating ROS conversion to enhance hypoxia resistance in cancer cells. Sci. Rep. 2016, 6, 22831. [Google Scholar] [CrossRef] [Green Version]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug. Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Mylonis, I.; Chachami, G.; Simos, G. Specific inhibition of HIF activity: Can peptides lead the way? Cancers 2021, 13, 410. [Google Scholar] [CrossRef] [PubMed]
- Schonberger, T.; Fandrey, J.; Prost-Fingerle, K. Ways into Understanding HIF Inhibition. Cancers 2021, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Puppo, M.; Rapisarda, A.; Uranchimeg, B.; Cao, L.; Burger, A.M.; Ziche, M.; Melillo, G. Aminoflavone, a ligand of the aryl hydrocarbon receptor, inhibits HIF-1alpha expression in an AhR-independent fashion. Cancer Res. 2010, 70, 6837–6848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Shyu, K.G.; Lee, C.C.; Tsai, S.C.; Wang, B.W.; Hsien Lee, Y.; Lin, S. GL331 inhibits HIF-1alpha expression in a lung cancer model. Biochem. Biophys. Res. Commun. 2003, 302, 95–100. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, C.; Schovanek, J.; Wang, H.; Bullova, P.; Caisova, V.; Gupta, G.; Wolf, K.I.; Semenza, G.L.; Zhuang, Z.; et al. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017, 8, 22313–22324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkotinakou, I.M.; Kechagia, E.; Pazaitou-Panayiotou, K.; Mylonis, I.; Liakos, P.; Tsakalof, A. Calcitriol Suppresses HIF-1 and HIF-2 Transcriptional activity by reducing HIF-1/2alpha protein levels via a VDR-independent mechanism. Cells 2020, 9, 11. [Google Scholar] [CrossRef]
- Rapisarda, A.; Zalek, J.; Hollingshead, M.; Braunschweig, T.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Hewitt, S.M.; Shoemaker, R.H.; et al. Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res. 2004, 64, 6845–6848. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.L.; Zhong, D.; Zhou, W.; Malik, S.; Liotta, D.; Snyder, J.P.; Hamel, E.; Giannakakou, P. EF24, a novel curcumin analog, disrupts the microtubule cytoskeleton and inhibits HIF-1. Cell Cycle 2008, 7, 2409–2417. [Google Scholar] [CrossRef] [Green Version]
- Strowitzki, M.J.; Ritter, A.S.; Kimmer, G.; Schneider, M. Hypoxia-adaptive pathways: A pharmacological target in fibrotic disease? Pharmacol. Res. 2019, 147, 104364. [Google Scholar] [CrossRef]
- Xiong, G.; Stewart, R.L.; Chen, J.; Gao, T.; Scott, T.L.; Samayoa, L.M.; O’Connor, K.; Lane, A.N.; Xu, R. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nat. Commun. 2018, 9, 4456. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Huang, Y.C.; Huang, F.I.; Mehndiratta, S.; Lai, S.C.; Liou, J.P.; Yang, C.R. Anticancer activity of MPT0G157, a derivative of indolylbenzenesulfonamide, inhibits tumor growth and angiogenesis. Oncotarget 2015, 6, 18590–18601. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; He, K.; Sridhara, R.; Abraham, S.; Booth, B.P.; Verbois, L.; Morse, D.E.; Jee, J.M.; Pope, S.; et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 2318–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, H.M.; Dickinson, M. Romidepsin for cutaneous T-cell lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3509–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Grasse, L.; Shah, J.; Chandra, J. Panobinostat, a pan-histone deacetylase inhibitor: Rationale for and application to treatment of multiple myeloma. Drugs Today 2015, 51, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.Q.; Li, Z.B.; Newman, M.J.; Shan, S.; Wang, X.H.; Pan, D.S.; Zhang, J.; Dong, M.; Du, X.; Lu, X.P. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Scheuermann, T.H.; Li, Q.; Ma, H.W.; Key, J.; Zhang, L.; Chen, R.; Garcia, J.A.; Naidoo, J.; Longgood, J.; Frantz, D.E.; et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat. Chem. Biol. 2013, 9, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J. Biol. Chem. 2006, 281, 33095–33106. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1alpha to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Qian, D.Z.; Rey, S.; Wei, H.; Liu, J.O.; Semenza, G.L. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2353–2358. [Google Scholar] [CrossRef] [Green Version]
- Kung, A.L.; Zabludoff, S.D.; France, D.S.; Freedman, S.J.; Tanner, E.A.; Vieira, A.; Cornell-Kennon, S.; Lee, J.; Wang, B.; Wang, J. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 2004, 6, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyberopoulou, A.; Venieris, E.; Mylonis, I.; Chachami, G.; Pappas, I.; Simos, G.; Bonanou, S.; Georgatsou, E. MgcRacGAP interacts with HIF-1alpha and regulates its transcriptional activity. Cell Physiol. Biochem. 2007, 20, 995–1006. [Google Scholar] [CrossRef]
- Imayoshi, N.; Yoshioka, M.; Chauhan, J.; Nakata, S.; Toda, Y.; Fletcher, S.; Strovel, J.W.; Takata, K.; Ashihara, E. CG13250, a novel bromodomain inhibitor, suppresses proliferation of multiple myeloma cells in an orthotopic mouse model. Biochem. Biophys. Res. Commun. 2017, 484, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Befani, C.D.; Vlachostergios, P.J.; Hatzidaki, E.; Patrikidou, A.; Bonanou, S.; Simos, G.; Papandreou, C.N.; Liakos, P. Bortezomib represses HIF-1alpha protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. 2012, 90, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Akiyama, M.; Hayashi, T.; Chauhan, D.; Richardson, P.; Schlossman, R.; Podar, K.; Munshi, N.C.; Mitsiades, N.; et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003, 101, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, D.; Park, J.Y.; Jung, H.J.; Cho, Y.H.; Kim, H.K.; Han, J.; Choi, K.Y.; Kwon, H.J. NNC 55-0396, a T-type Ca2+ channel inhibitor, inhibits angiogenesis via suppression of hypoxia-inducible factor-1alpha signal transduction. J. Mol. Med. 2015, 93, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Pham, E.; Birrer, M.J.; Eliasof, S.; Garmey, E.G.; Lazarus, D.; Lee, C.R.; Man, S.; Matulonis, U.A.; Peters, C.G.; Xu, P.; et al. Translational impact of nanoparticle-drug conjugate CRLX101 with or without bevacizumab in advanced ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Jee, J.G.; Bae, J.S.; Liu, K.H.; Lee, Y.M. A group of novel HIF-1alpha inhibitors, glyceollins, blocks HIF-1alpha synthesis and decreases its stability via inhibition of the PI3K/AKT/mTOR pathway and Hsp90 binding. J. Cell. Physiol. 2015, 230, 853–862. [Google Scholar] [CrossRef]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001, 414, 550–554. [Google Scholar] [CrossRef]
- Yamashita, T.; Ohneda, O.; Nagano, M.; Iemitsu, M.; Makino, Y.; Tanaka, H.; Miyauchi, T.; Goto, K.; Ohneda, K.; Fujii-Kuriyama, Y.; et al. Abnormal heart development and lung remodeling in mice lacking the hypoxia-inducible factor-related basic helix-loop-helix PAS protein NEPAS. Mol. Cell. Biol. 2008, 28, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhao, S.; Nakada, K.; Kuge, Y.; Tamaki, N.; Okada, F.; Wang, J.; Shindo, M.; Higashino, F.; Takeda, K.; et al. Dominant-negative hypoxia-inducible factor-1 alpha reduces tumorigenicity of pancreatic cancer cells through the suppression of glucose metabolism. Am. J. Pathol. 2003, 162, 1283–1291. [Google Scholar] [CrossRef]
- Wang, Y.; Thompson, J.D.; Chan, W.K. A cell-penetrating peptide suppresses the hypoxia inducible factor-1 function by binding to the helix-loop-helix domain of the aryl hydrocarbon receptor nuclear translocator. Chem. Biol. Interact. 2013, 203, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Miranda, E.; Nordgren, I.K.; Male, A.L.; Lawrence, C.E.; Hoakwie, F.; Cuda, F.; Court, W.; Fox, K.R.; Townsend, P.A.; Packham, G.K.; et al. A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. J. Am. Chem. Soc. 2013, 135, 10418–10425. [Google Scholar] [CrossRef]
- Hetherington, K.; Hegedus, Z.; Edwards, T.A.; Sessions, R.B.; Nelson, A.; Wilson, A.J. Stapled peptides as HIF-1alpha/p300 inhibitors: Helicity enhancement in the bound state increases inhibitory potency. Chemistry 2020, 26, 7638–7646. [Google Scholar] [CrossRef]
- Kyle, H.F.; Wickson, K.F.; Stott, J.; Burslem, G.M.; Breeze, A.L.; Tiede, C.; Tomlinson, D.C.; Warriner, S.L.; Nelson, A.; Wilson, A.J.; et al. Exploration of the HIF-1alpha/p300 interface using peptide and Adhiron phage display technologies. Mol. Biosyst. 2015, 11, 2738–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P.; Dennis, M.F.; Everett, S.A.; Patel, K.B.; Stratford, M.R.; Tracy, M. Radicals from one-electron reduction of nitro compounds, aromatic N-oxides and quinones: The kinetic basis for hypoxia-selective, bioreductive drugs. Biochem. Soc. Symp. 1995, 61, 171–194. [Google Scholar] [PubMed] [Green Version]
- Borad, M.J.; Reddy, S.G.; Bahary, N.; Uronis, H.E.; Sigal, D.; Cohn, A.L.; Schelman, W.R.; Stephenson, J., Jr.; Chiorean, E.G.; Rosen, P.J.; et al. Randomized phase II trial of gemcitabine plus TH-302 versus gemcitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Chawla, S.P.; Cranmer, L.D.; Van Tine, B.A.; Reed, D.R.; Okuno, S.H.; Butrynski, J.E.; Adkins, D.R.; Hendifar, A.E.; Kroll, S.; Ganjoo, K.N. Phase II study of the safety and antitumor activity of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 3299–3306. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 2017, 32, 71–87.e7. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. https://doi.org/10.3390/ijms22115703
Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. International Journal of Molecular Sciences. 2021; 22(11):5703. https://doi.org/10.3390/ijms22115703
Chicago/Turabian StyleInfantino, Vittoria, Anna Santarsiero, Paolo Convertini, Simona Todisco, and Vito Iacobazzi. 2021. "Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target" International Journal of Molecular Sciences 22, no. 11: 5703. https://doi.org/10.3390/ijms22115703
APA StyleInfantino, V., Santarsiero, A., Convertini, P., Todisco, S., & Iacobazzi, V. (2021). Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. International Journal of Molecular Sciences, 22(11), 5703. https://doi.org/10.3390/ijms22115703