
The E3 Ubiquitin-Protein Ligase RNF4 Promotes TNF-α-Induced Cell Death Triggered by RIPK1


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
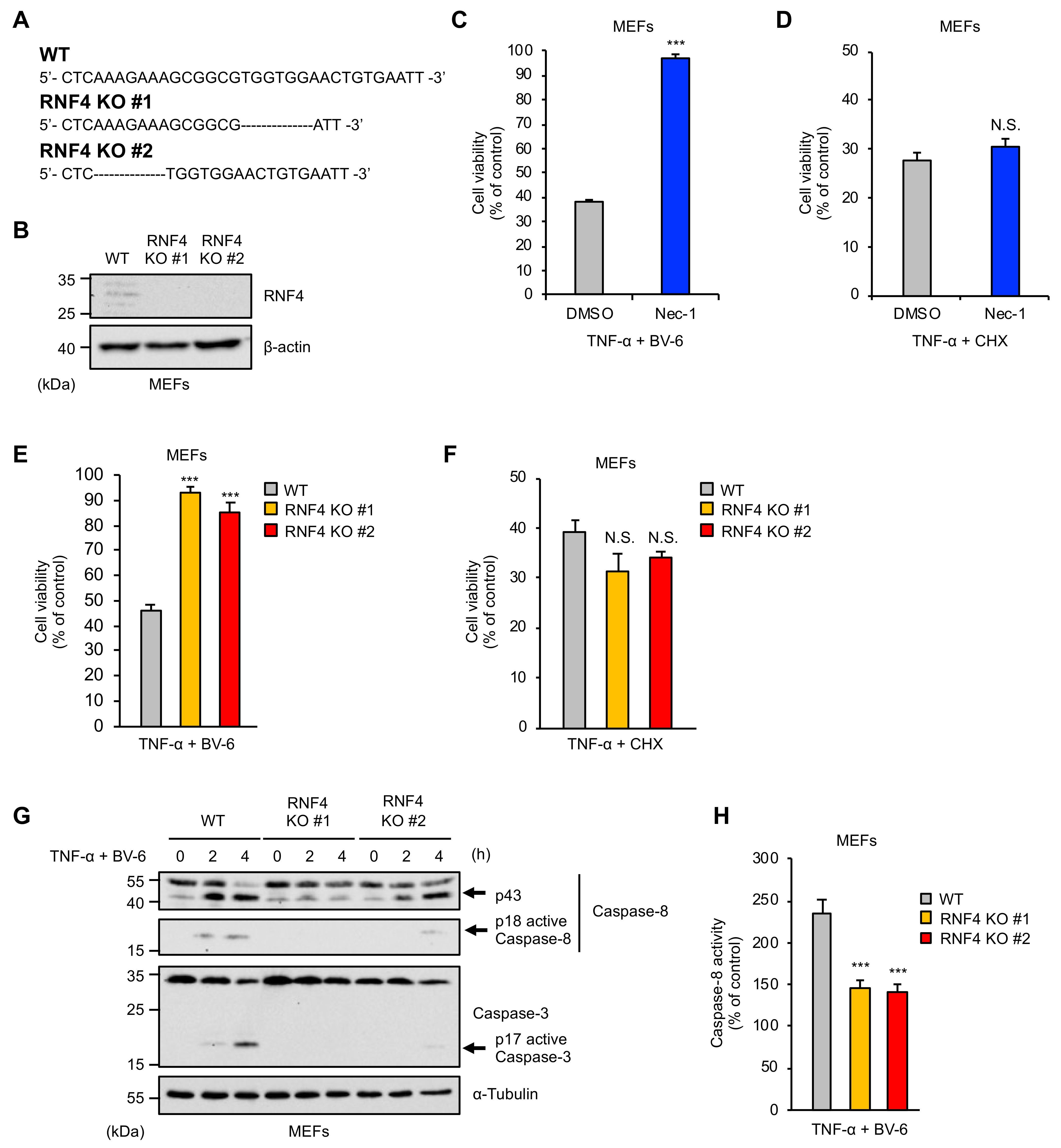
2.1. RNF4 Is Specifically Required for RIPK1-Mediated Cell Death Stimulated by TNF-α
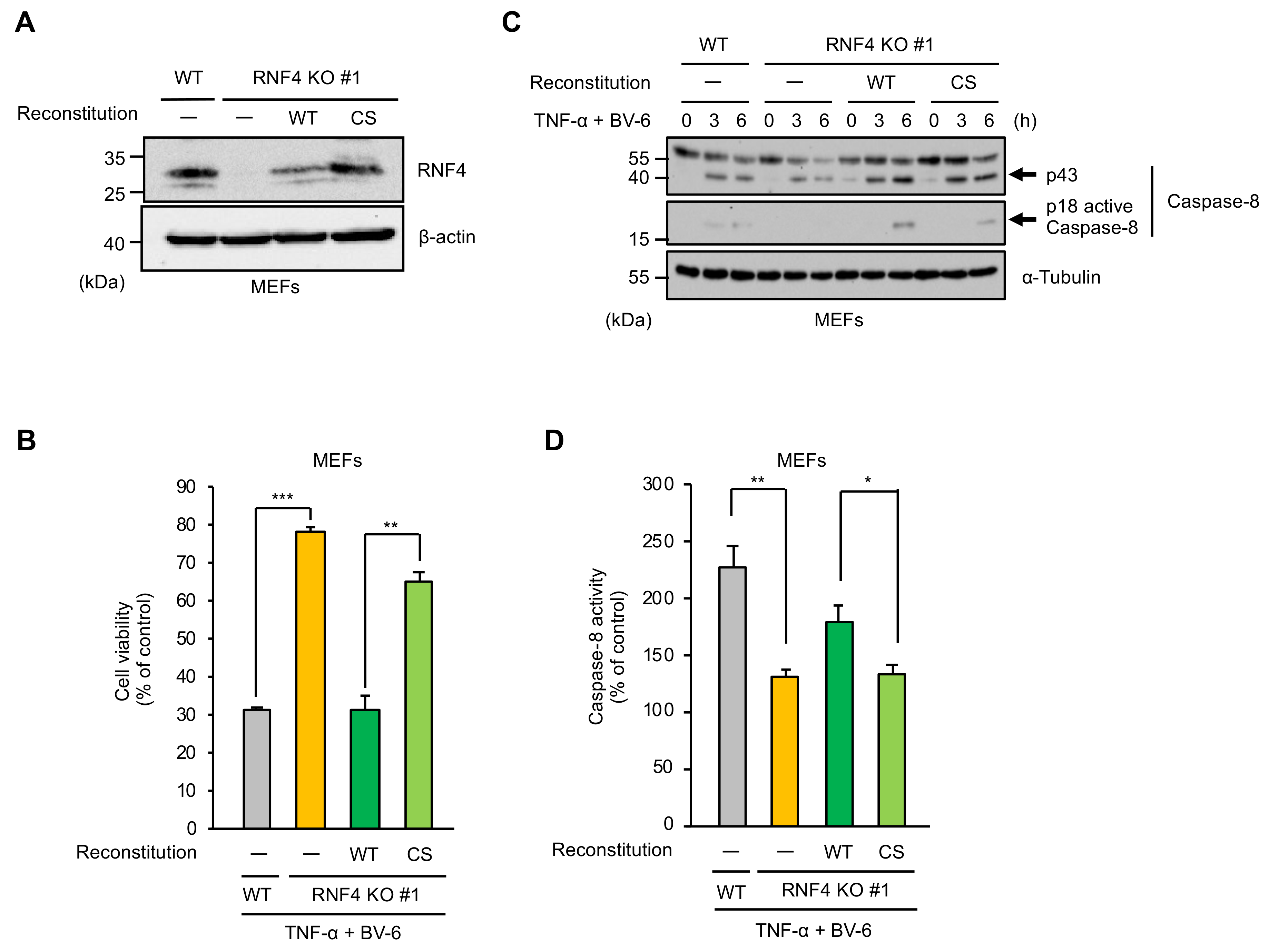
2.2. The E3 Ubiquitin Ligase Activity of RNF4 Is Required for TNF-α-Induced Apoptosis
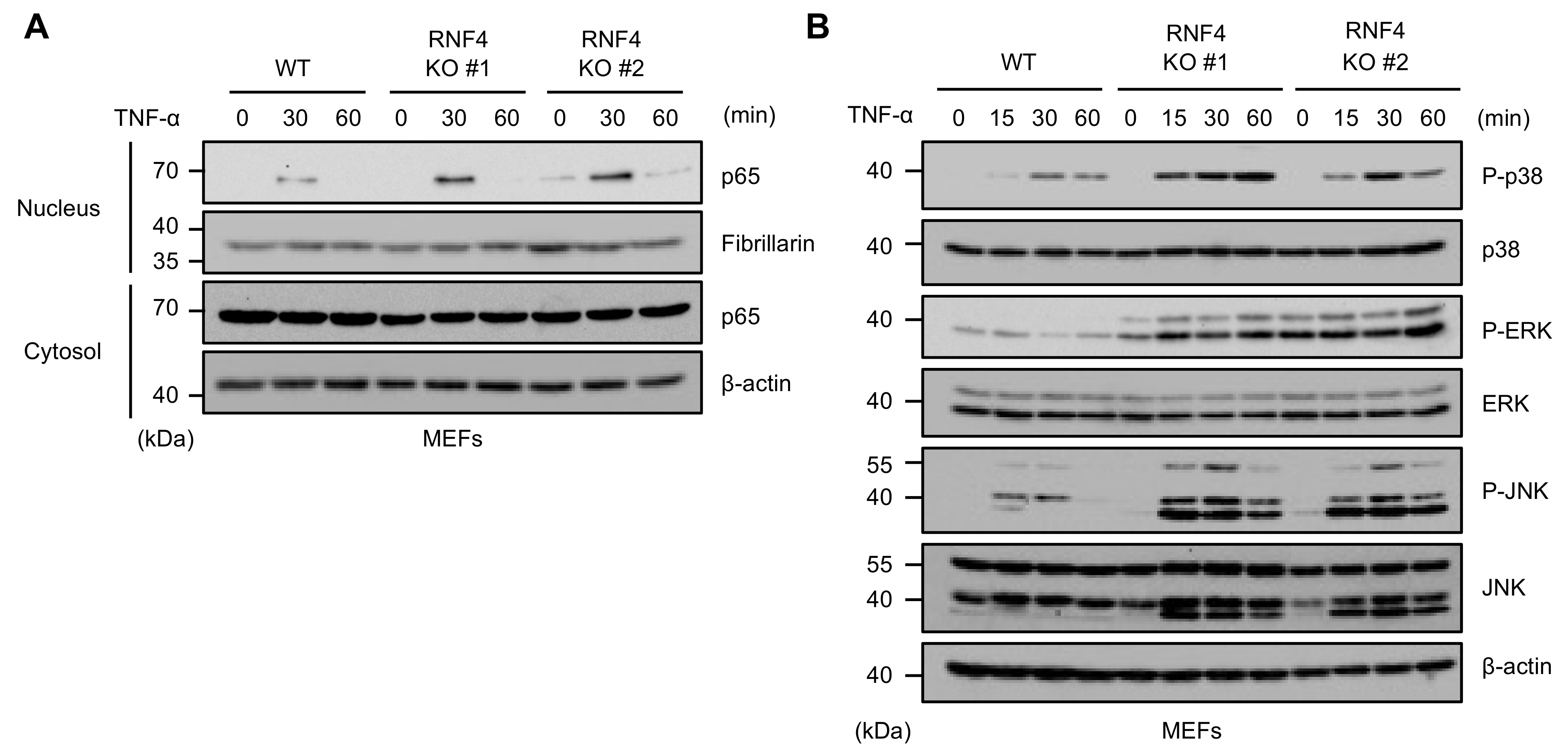
2.3. RNF4 Suppresses TNF-α-Induced Activation of the NF-κB and MAPK Signaling Pathways
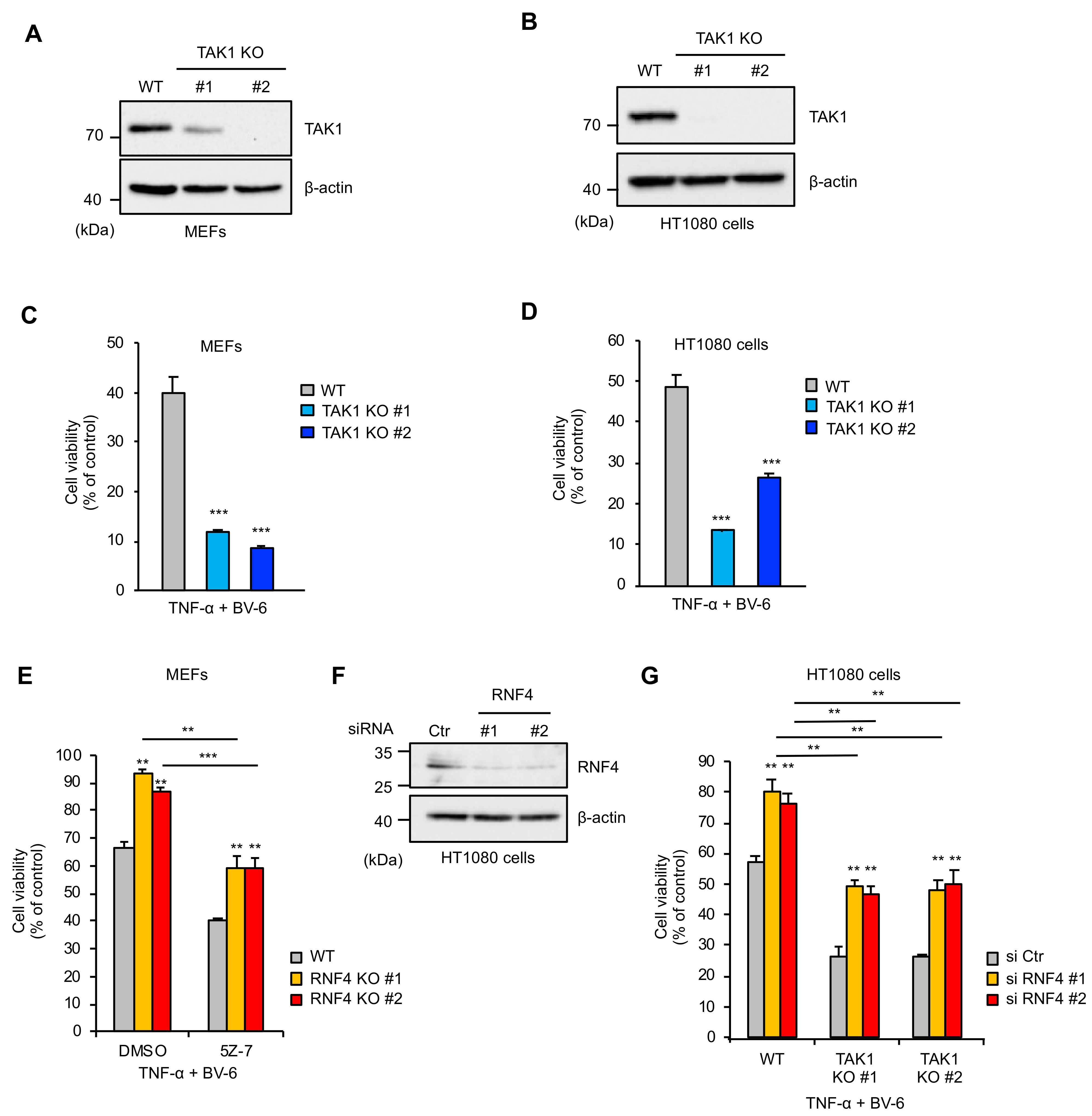
2.4. RNF4 Promotes TNF-α-Induced Cell Death Independently of Its Inhibitory Effects on the TAK1 Signaling
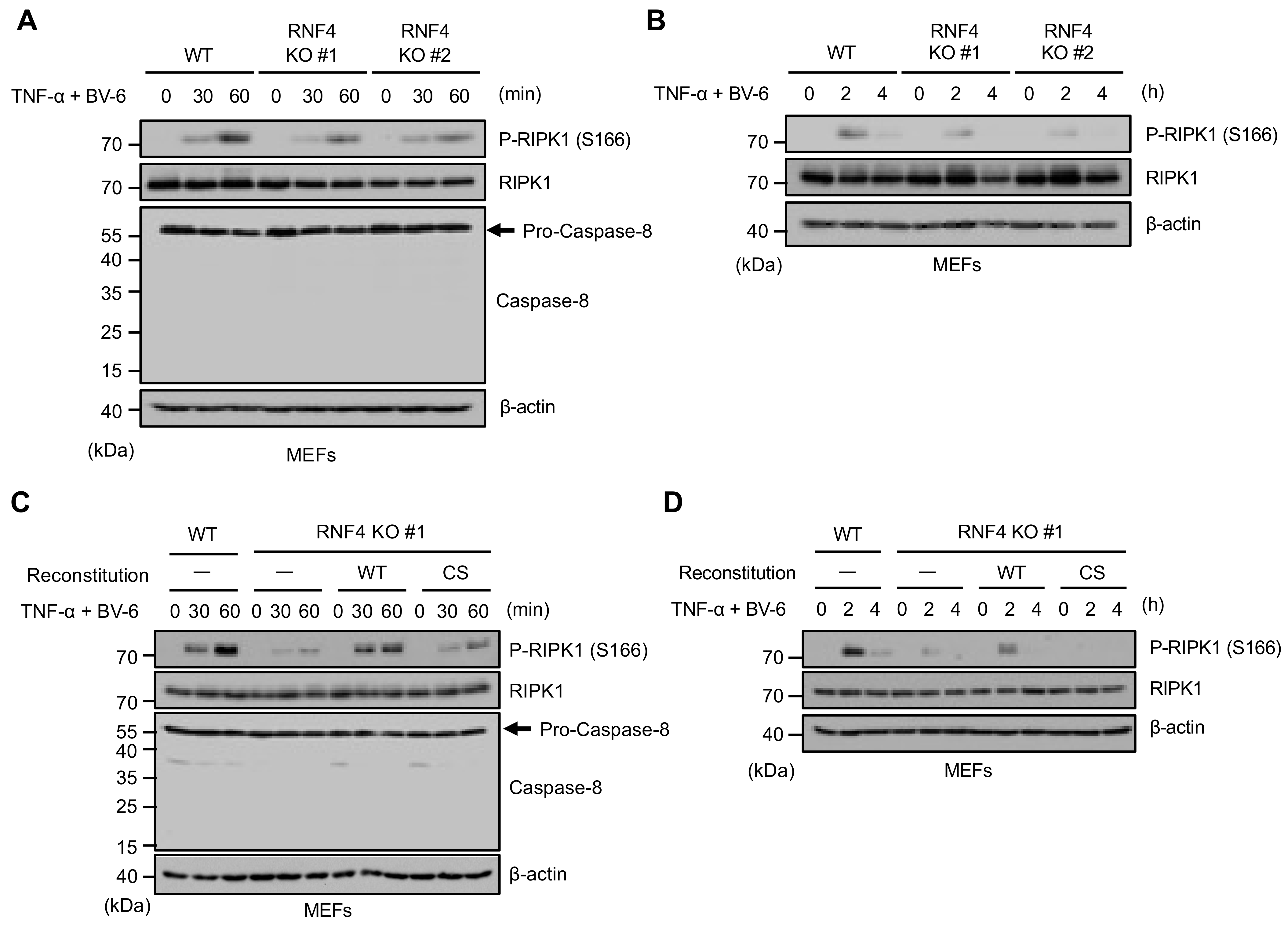
2.5. RNF4 Promotes Phosphorylation of RIPK1 at Ser166
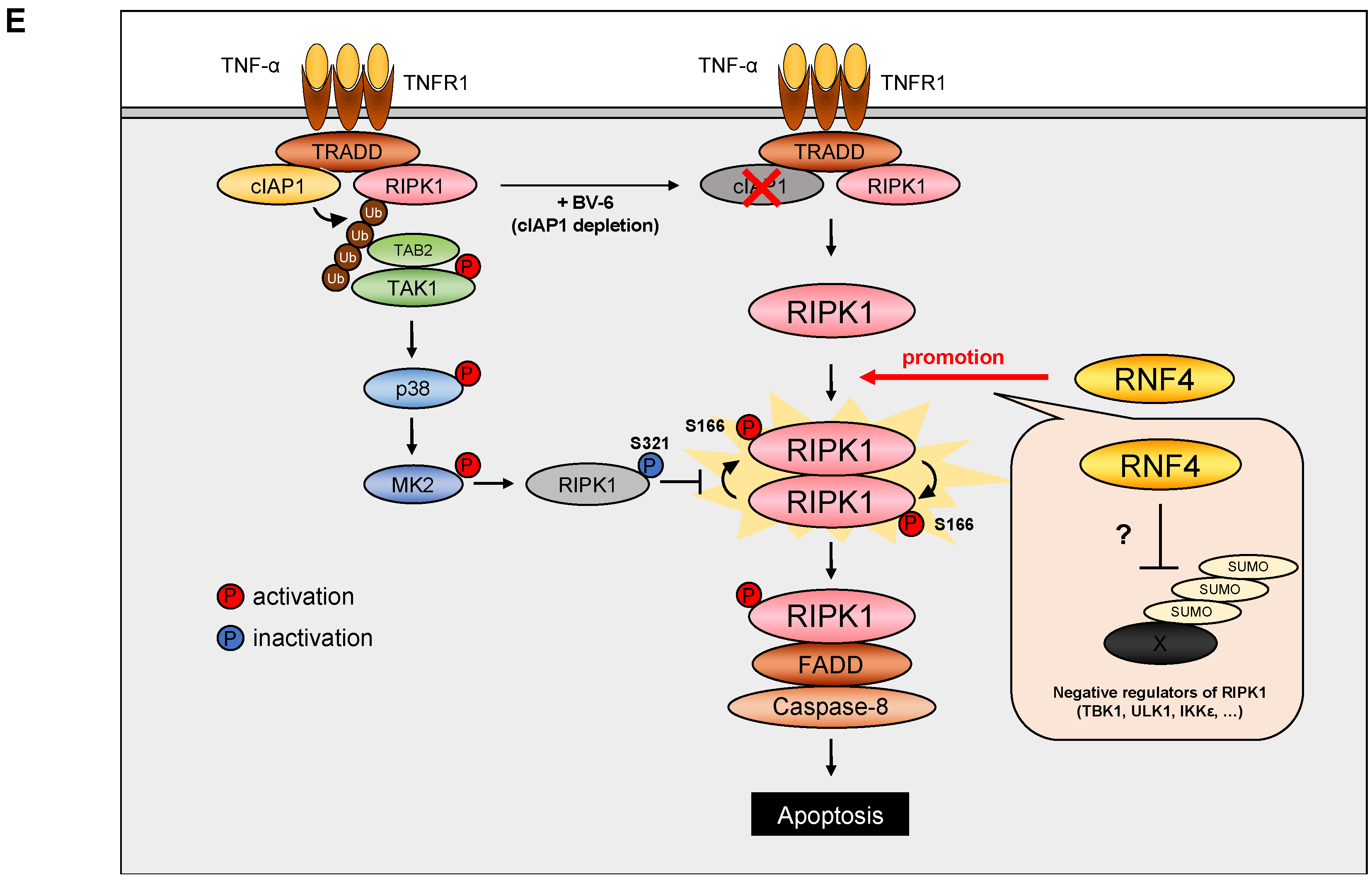
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. PMS/MTS Assay
4.3. Immunoblot Analysis
4.4. Generation of Knockout Cell Lines
4.5. Colorimetric Caspase-8 Assay
4.6. Generation of Reconstituted MEFs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood J. Am. Soc. Hematol. 2012, 119, 651–665. [Google Scholar] [CrossRef] [Green Version]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Chu, W.M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-Translational Modifications of the TAK1-TAB Complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Tsuchida, M.; Kogue, Y.; Spadini, C.; Hirata, Y.; Matsuzawa, A. Brefeldin A-Inhibited Guanine Nucleotide-Exchange Factor 1 (BIG1) Governs the Recruitment of Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) to Tumor Necrosis Factor Receptor 1 (TNFR1) Signaling Complexes. Int. J. Mol. Sci. 2016, 17, 1869. [Google Scholar] [CrossRef] [Green Version]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Du, F.; Wang, X. TNF-α induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Guo, X.; Yin, H.; Chen, Y.; Li, L.; Li, J.; Liu, Q. TAK1 regulates caspase 8 activation and necroptotic signaling via multiple cell death checkpoints. Cell Death Dis. 2016, 7, e2381. [Google Scholar] [CrossRef] [Green Version]
- Laurien, L.; Nagata, M.; Schünke, H.; Delanghe, T.; Wiederstein, J.L.; Kumari, S.; Schwarzer, R.; Corona, T.; Krüger, M.; Bertrand, M.J. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; John, S.W.; Liccardi, G. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 2017, 66, 698–710.e695. [Google Scholar] [CrossRef] [PubMed]
- Chiariotti, L.; Benvenuto, G.; Fedele, M.; Santoro, M.; Simeone, A.; Fusco, A.; Bruni, C.B. Identification and characterization of a novel RING-finger gene (RNF4) mapping at 4p16.3. Genomics 1998, 47, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, A.-M.; Poukka, H.; Karvonen, U.; Häkli, M.; Jänne, O.A.; Palvimo, J.J. Identification of a novel RING finger protein as a coregulator in steroid receptor-mediated gene transcription. Mol. Cell. Biol. 1998, 18, 5128–5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatham, M.H.; Geoffroy, M.-C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012, 26, 1179–1195. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Seifert, A.; Chua, J.S.; Maure, J.-F.; Golebiowski, F.; Hay, R.T. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 2012, 26, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; González-Prieto, R.; Xiao, Z.; Verlaan-de Vries, M.; Vertegaal, A.C. The STUbL RNF4 regulates protein group SUMOylation by targeting the SUMO conjugation machinery. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Luo, K.; Zhang, H.; Wang, L.; Yuan, J.; Lou, Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2012, 31, 3008–3019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xie, F.; Zhang, J.; Ten Dijke, P.; Zhou, F. SUMO-triggered ubiquitination of NR4A1 controls macrophage cell death. Cell Death Differ. 2017, 24, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Mu, R.; Chang, Y.; Wang, Y.-B.; Wu, M.; Tu, H.-Q.; Zhang, Y.-C.; Guo, S.-S.; Qin, X.-H.; Li, T. RNF4 negatively regulates NF-κB signaling by down-regulating TAB2. FEBS Lett. 2015, 589, 2850–2858. [Google Scholar] [CrossRef] [Green Version]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Häkli, M.; Lorick, K.L.; Weissman, A.M.; Jänne, O.A.; Palvimo, J.J. Transcriptional coregulator SNURF (RNF4) possesses ubiquitin E3 ligase activity. FEBS Lett. 2004, 560, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Wang, X.; Berleth, N.; Deitersen, J.; Wallot-Hieke, N.; Böhler, P.; Schlütermann, D.; Stuhldreier, F.; Cox, J.; Schmitz, K.; et al. The Autophagy-Initiating Kinase ULK1 Controls RIPK1-Mediated Cell Death. Cell Rep. 2020, 31, 107547. [Google Scholar] [CrossRef] [PubMed]
- Lafont, E.; Draber, P.; Rieser, E.; Reichert, M.; Kupka, S.; de Miguel, D.; Draberova, H.; von Mässenhausen, A.; Bhamra, A.; Henderson, S. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 2018, 20, 1389–1399. [Google Scholar] [CrossRef]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 2018, 174, 1477–1491.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.J.; Abed, M.; Heuberger, J.; Novak, R.; Zohar, Y.; Lopez, A.P.B.; Trausch-Azar, J.S.; Ilagan, M.X.G.; Benhamou, D.; Dittmar, G. RNF4-dependent oncogene activation by protein stabilization. Cell Rep. 2016, 16, 3388–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzzo, C.M.; Berndsen, C.E.; Zhu, J.; Gupta, V.; Datta, A.; Greenberg, R.A.; Wolberger, C.; Matunis, M.J. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci. Signal. 2012, 5, ra88. [Google Scholar] [CrossRef] [Green Version]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Müller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67.e57. [Google Scholar] [CrossRef] [PubMed]
- Avitan-Hersh, E.; Feng, Y.; Vaisman, A.O.; Ahmad, Y.A.; Zohar, Y.; Zhang, T.; Lee, J.S.; Lazar, I.; Khalil, S.S.; Feiler, Y. Regulation of eIF2α by RNF4 promotes melanoma tumorigenesis and therapy resistance. J. Investig. Dermatol. 2020, 140, 2466–2477. [Google Scholar] [CrossRef] [PubMed]
- Sriramachandran, A.M.; Dohmen, R.J. SUMO-targeted ubiquitin ligases. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Morrish, E.; Brumatti, G.; Silke, J. Future therapeutic directions for Smac-mimetics. Cells 2020, 9, 406. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, T.; Sekiguchi, Y.; Kudoh, Y.; Naganuma, R.; Kagi, T.; Nishidate, A.; Maeda, K.; Ishii, C.; Toyama, T.; Hirata, Y.; et al. Gefitinib initiates sterile inflammation by promoting IL-1β and HMGB1 release via two distinct mechanisms. Cell Death Dis. 2021, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, M.; Yokosawa, T.; Noguchi, T.; Shimada, T.; Yamada, M.; Sekiguchi, Y.; Hirata, Y.; Matsuzawa, A. Pro-apoptotic functions of TRAF2 in p53-mediated apoptosis induced by cisplatin. J. Toxicol. Sci. 2020, 45, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, T.; Suzuki, M.; Mutoh, N.; Hirata, Y.; Tsuchida, M.; Miyagawa, S.; Hwang, G.W.; Aoki, J.; Matsuzawa, A. Nuclear-accumulated SQSTM1/p62-based ALIS act as microdomains sensing cellular stresses and triggering oxidative stress-induced parthanatos. Cell Death Dis. 2018, 9, 1193. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Asai, Y.; Kagi, T.; Noguchi, T.; Yamada, M.; Hirata, Y.; Matsuzawa, A. TAK1 Mediates ROS Generation Triggered by the Specific Cephalosporins through Noncanonical Mechanisms. Int. J. Mol. Sci. 2020, 21, 9497. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, Y.; Yamada, M.; Noguchi, T.; Noomote, C.; Tsuchida, M.; Kudoh, Y.; Hirata, Y.; Matsuzawa, A. The anti-cancer drug gefitinib accelerates Fas-mediated apoptosis by enhancing caspase-8 activation in cancer cells. J. Toxicol. Sci. 2019, 44, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2015, 31, 1120–1123. [Google Scholar] [CrossRef]
- Kitamura, T.; Koshino, Y.; Shibata, F.; Oki, T.; Nakajima, H.; Nosaka, T.; Kumagai, H. Retrovirus-mediated gene transfer and expression cloning: Powerful tools in functional genomics. Exp. Hematol. 2003, 31, 1007–1014. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimada, T.; Kudoh, Y.; Noguchi, T.; Kagi, T.; Suzuki, M.; Tsuchida, M.; Komatsu, H.; Takahashi, M.; Hirata, Y.; Matsuzawa, A. The E3 Ubiquitin-Protein Ligase RNF4 Promotes TNF-α-Induced Cell Death Triggered by RIPK1. Int. J. Mol. Sci. 2021, 22, 5796. https://doi.org/10.3390/ijms22115796
Shimada T, Kudoh Y, Noguchi T, Kagi T, Suzuki M, Tsuchida M, Komatsu H, Takahashi M, Hirata Y, Matsuzawa A. The E3 Ubiquitin-Protein Ligase RNF4 Promotes TNF-α-Induced Cell Death Triggered by RIPK1. International Journal of Molecular Sciences. 2021; 22(11):5796. https://doi.org/10.3390/ijms22115796
Chicago/Turabian StyleShimada, Tatsuya, Yuki Kudoh, Takuya Noguchi, Tomohiro Kagi, Midori Suzuki, Mei Tsuchida, Hiromu Komatsu, Miki Takahashi, Yusuke Hirata, and Atsushi Matsuzawa. 2021. "The E3 Ubiquitin-Protein Ligase RNF4 Promotes TNF-α-Induced Cell Death Triggered by RIPK1" International Journal of Molecular Sciences 22, no. 11: 5796. https://doi.org/10.3390/ijms22115796
APA StyleShimada, T., Kudoh, Y., Noguchi, T., Kagi, T., Suzuki, M., Tsuchida, M., Komatsu, H., Takahashi, M., Hirata, Y., & Matsuzawa, A. (2021). The E3 Ubiquitin-Protein Ligase RNF4 Promotes TNF-α-Induced Cell Death Triggered by RIPK1. International Journal of Molecular Sciences, 22(11), 5796. https://doi.org/10.3390/ijms22115796