
Prostaglandin E2 Enhances Gap Junctional Intercellular Communication in Clonal Epithelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
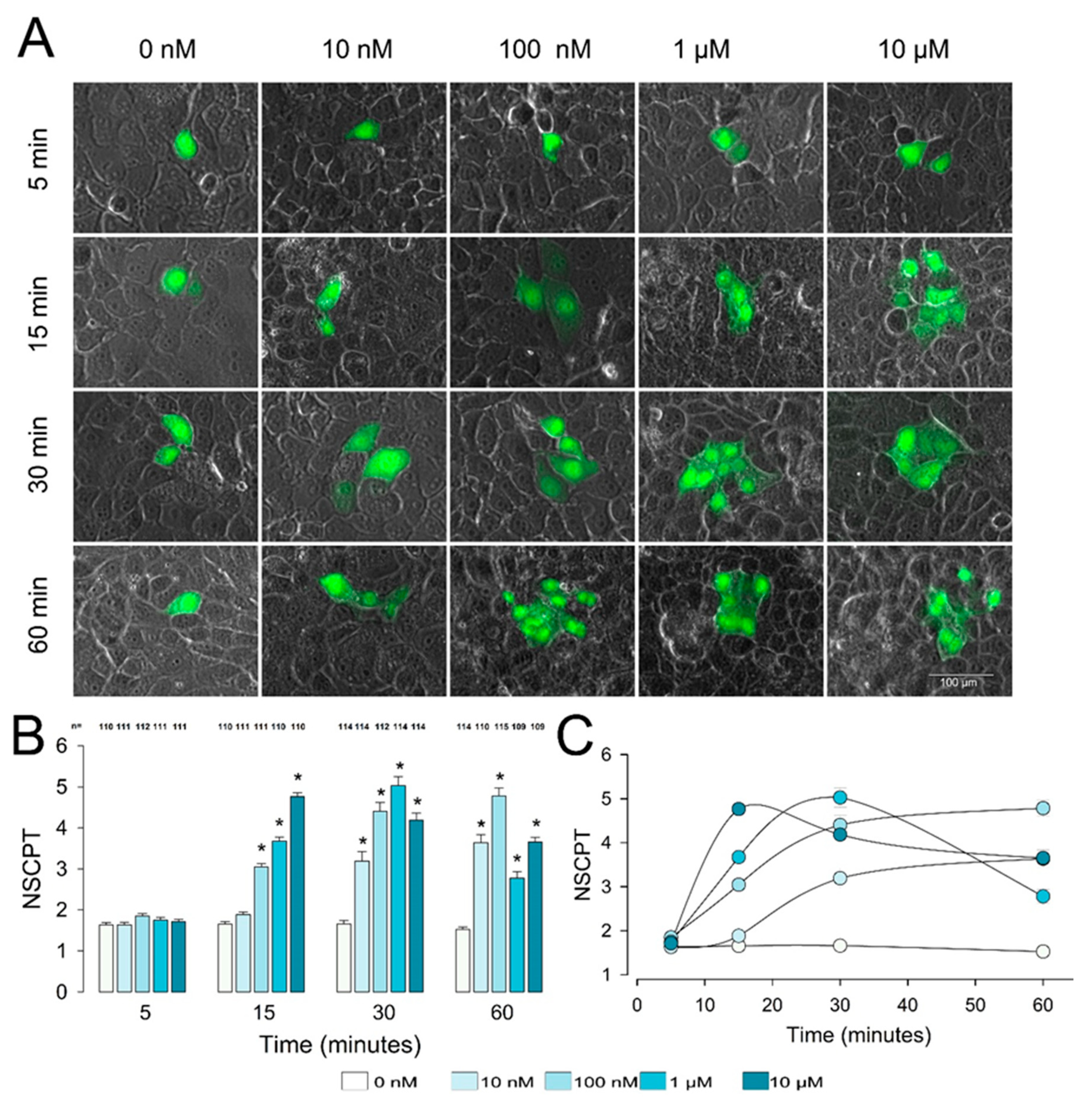
2.1. PGE2 Induces Enhancement of Gap Junctional Intercellular Communication in Epithelial Cells
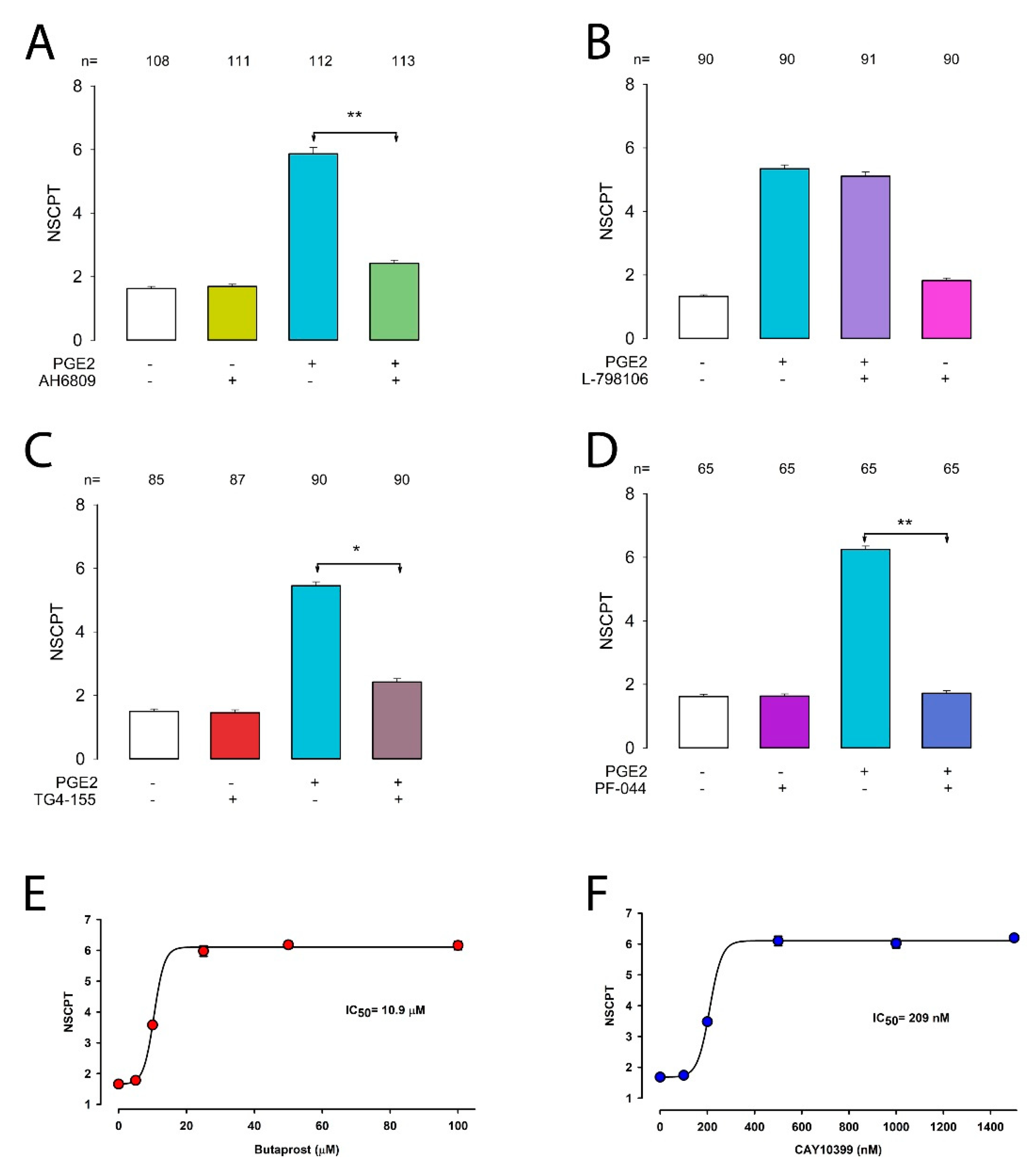
2.2. Effect of Gap Junction Blockers on PGE2-Induced GJIC Enhancement
2.3. PGE2-Induced Enhancement of GJIC Does Not Involve Synthesis of New Subunits
2.4. PGE2 Promotes Transport and Relocation of Gap Junction Components
2.5. Prostaglandin E2 Enhances GJIC via the EP2 Receptor
2.6. Inhibition of cAMP and PKA Diminishes the Effect of PGE2 on CIGJ
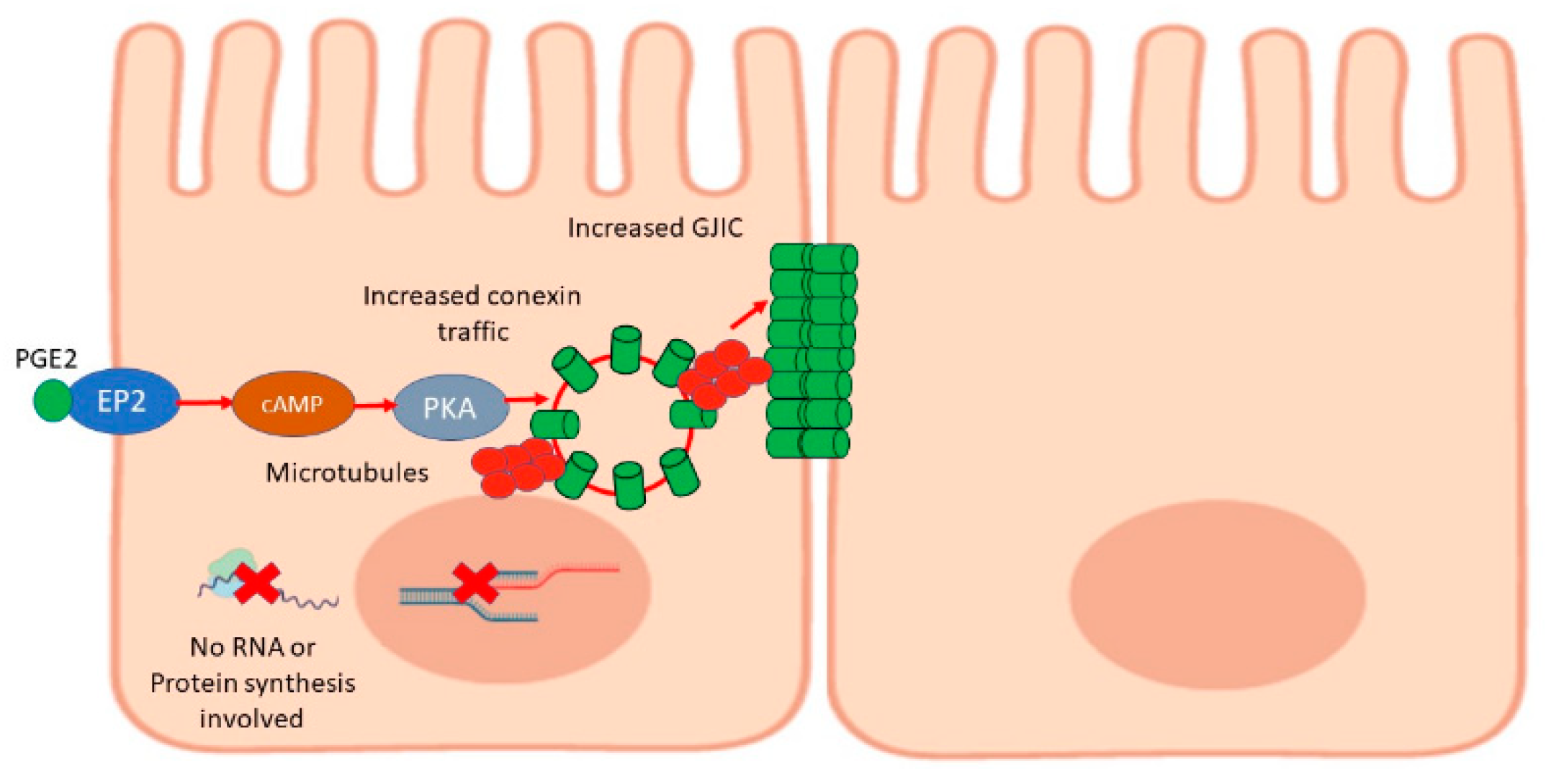
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Measurement of Gap Junctional Intercellular Communication by Dye Transfer Assays
4.3. Chemicals
4.4. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calder, P.C. Eicosanoids. Essays Biochem. 2020, 64, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Oesterling, T.O.; Morozowich, W.; Roseman, T.J. Prostaglandins. J. Pharm. Sci. 1972, 61, 1861–1895. [Google Scholar] [CrossRef]
- Samuelsson, B.; Granstrom, E.; Green, K.; Hamberg, M.; Hammarstrom, S. Prostaglandins. Annu. Rev. Biochem. 1975, 44, 669–695. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, N.; Nakamura, T. Prostaglandins as hormones. Dig. Dis. Sci. 1985, 30, 109S–113S. [Google Scholar] [CrossRef] [PubMed]
- Poyser, N.L. The physiology of prostaglandins. Clin. Endocrinol. Metab. 1973, 2, 393–410. [Google Scholar] [CrossRef]
- Jones, R.L. Functions of prostaglandins. Pathobiol. Annu. 1972, 2, 359–380. [Google Scholar]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Aoki, T.; Narumiya, S. Prostaglandins and chronic inflammation. Trends Pharmacol. Sci. 2012, 33, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Wang, D. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef]
- Fischer, S.M. Prostaglandins and cancer. Front. Biosci. 1997, 2, d482–d500. [Google Scholar] [CrossRef] [Green Version]
- Hirsh, P.D.; Campbell, W.B.; Willerson, J.T.; Hillis, L. Prostaglandins and ischemic heart disease. Am. J. Med. 1981, 71, 1009–1026. [Google Scholar] [CrossRef]
- Pitt, B.; Shea, M.J.; Romson, J.L.; Lucchesi, B.R. Prostaglandins and Prostaglandin Inhibitors in Ischemic Heart Disease. Ann. Intern. Med. 1983, 99, 83–92. [Google Scholar] [CrossRef]
- O’Brien, W.F. The Role of Prostaglandins in Labor and Delivery. Clin. Perinatol. 1995, 22, 973–984. [Google Scholar] [CrossRef]
- Chwalisz, K. The use of progesterone antagonists for cervical ripening and as an adjunct to labour and delivery. Hum. Reprod. 1994, 9, 131–161. [Google Scholar] [CrossRef] [PubMed]
- Yount, S.M.; Lassiter, N. The Pharmacology of Prostaglandins for Induction of Labor. J. Midwifery Women’s Health 2013, 58, 133–144. [Google Scholar] [CrossRef]
- Stoff, J.S. Prostaglandins and hypertension. Am. J. Med. 1986, 80, 56–61. [Google Scholar] [CrossRef]
- Smith, M.C.; Dunn, M.J. The Role of Prostaglandins in Human Hypertension. Am. J. Kidney Dis. 1985, 5, A32–A39. [Google Scholar] [CrossRef]
- Peebles, R.S. Prostaglandins in asthma and allergic diseases. Pharmacol. Ther. 2019, 193, 1–19. [Google Scholar] [CrossRef]
- Antonova, M.; Wienecke, T.; Olesen, J.; Ashina, M. Prostaglandins in migraine. Curr. Opin. Neurol. 2013, 26, 269–275. [Google Scholar] [CrossRef]
- Hayaishi, O.; Matsumura, H. Prostaglandins and sleep. Adv. Neuroimmunol. 1995, 5, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Pilbeam, C.C.; Harrison, J.R.; Raisz, L.G. The role of prostaglandins in the regulation of bone metabolism. Clin. Orthop. Relat. Res. 1995, 36–46. [Google Scholar] [PubMed]
- Miller, S.C.; Marks, S.C., Jr. Effects of prostaglandins on the skeleton. Clin. Plast. Surg. 1994, 21, 393–400. [Google Scholar] [CrossRef]
- Akaogi, J.; Nozaki, T.; Satoh, M.; Yamada, H. Role of PGE2 and EP receptors in the pathogenesis of rheumatoid arthritis and as a novel therapeutic strategy. Endocr. Metab. Immune Disord. Drug Targets 2006, 6, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B.; Folco, G.; Granström, E.; Kindahl, H.; Malmsten, C. Prostaglandins and thromboxanes: Biochemical and physiological considerations. Adv. Prostaglandin Thromboxane Res. 1978, 4, 1–25. [Google Scholar] [PubMed]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B. Success of prostaglandin E2 in structure-function is a challenge for structure-based therapeutics. Proc. Natl. Acad. Sci. USA 2003, 100, 8609–8611. [Google Scholar] [CrossRef] [Green Version]
- Backlund, M.G.; Mann, J.R.; DuBois, R.N. Mechanisms for the Prevention of Gastrointestinal Cancer: The Role of Prostaglandin E2. Oncology 2005, 69, 28–32. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E Receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.-H. Gap Junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar]
- Maeda, S.; Tsukihara, T. Structure of the gap junction channel and its implications for its biological functions. Cell. Mol. Life Sci. 2011, 68, 1115–1129. [Google Scholar] [CrossRef]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef] [Green Version]
- Ek-Vitorín, J.F.; Burt, J.M. Structural basis for the selective permeability of channels made of communicating junction proteins. Biochim. Biophys. Acta BBA Biomembr. 2013, 1828, 51–68. [Google Scholar] [CrossRef] [Green Version]
- Hervé, J.-C.; Derangeon, M. Gap-junction-mediated cell-to-cell communication. Cell Tissue Res. 2012, 352, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Ekurtenbach, S.; Ekurtenbach, S.; Ezoidl, G. Gap junction modulation and its implications for heart function. Front. Physiol. 2014, 5, 82. [Google Scholar] [CrossRef]
- Desplantez, T.; Dupont, E.; Severs, N.J.; Weingart, R. Gap Junction Channels and Cardiac Impulse Propagation. J. Membr. Biol. 2007, 218, 13–28. [Google Scholar] [CrossRef]
- Brink, P.R. Gap junctions in vascular smooth muscle. Acta Physiol. Scand. 1998, 164, 349–356. [Google Scholar] [CrossRef]
- Daniel, E.E.; Wang, Y.-F. Gap junctions in intestinal smooth muscle and interstitial cells of Cajal. Microsc. Res. Tech. 1999, 47, 309–320. [Google Scholar] [CrossRef]
- Belousov, A.B.; Fontes, J.D. Neuronal gap junction coupling as the primary determinant of the extent of glutamate-mediated excitotoxicity. J. Neural Transm. 2014, 121, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Belousov, A.B. The regulation and role of neuronal gap junctions during neuronal injury. Channels 2012, 6, 390–392. [Google Scholar] [CrossRef] [Green Version]
- Meda, P. Gap junction proteins are key drivers of endocrine function. Biochim. Biophys. Acta BBA Biomembr. 2018, 1860, 124–140. [Google Scholar] [CrossRef]
- Serre-Beinier, V.; Mas, C.; Calabrese, A.; Caton, D.; Bauquis, J.; Caille, R.; Charollais, A.; Cirulli, V.; Meda, P. Connexins and secretion. Biol. Cell 2002, 94, 477–492. [Google Scholar] [CrossRef] [Green Version]
- Koval, M. Sharing signals: Connecting lung epithelial cells with gap junction channels. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L875–L893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Vega, L.; O’Shaughnessy, E.; Albuloushi, A.; Martin, P. Connexins and the Epithelial Tissue Barrier: A Focus on Connexin 26. Biology 2021, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Radu, A.; Dahl, G.; Loewenstein, W.R. Hormonal regulation of cell junction permeability: Upregulation by catecholamine and prostaglandin E1. J. Membr. Biol. 1982, 70, 239–251. [Google Scholar] [CrossRef]
- Gregory, M.; Cyr, D.G. Effects of prostaglandin E2 on gap junction protein alpha 1 in the rat epididymis. Biol. Reprod. 2018, 100, 123–132. [Google Scholar] [CrossRef]
- Garfield, R.E.; Kannan, M.S.; Daniel, E.E. Gap junction formation in myometrium: Control by estrogens, progesterone, and prostaglandins. Am. J. Physiol. Physiol. 1980, 238, C81–C89. [Google Scholar] [CrossRef]
- Cheng, B.; Kato, Y.; Zhao, S.; Luo, J.; Sprague, E.; Bonewald, L.F.; Jiang, J.X. PGE2 Is Essential for Gap Junction-Mediated Intercellular Communication between Osteocyte-Like MLO-Y4 Cells in Response to Mechanical Strain. Endocrinology 2001, 142, 3464–3473. [Google Scholar] [CrossRef]
- Bostanci, M.O.; Bağirici, F. The effects of octanol on penicillin induced epileptiform activity in rats: An in vivo study. Epilepsy Res. 2006, 71, 188–194. [Google Scholar] [CrossRef]
- Manjarrez-Marmolejo, J.; Franco-Pérez, J. Gap Junction Blockers: An Overview of their Effects on Induced Seizures in Animal Models. Curr. Neuropharmacol. 2016, 14, 759–771. [Google Scholar] [CrossRef]
- Marandykina, A.; Palacios-Prado, N.; Rimkutė, L.; Skeberdis, V.A.; Bukauskas, F.F. Regulation of connexin36 gap junction channels by n-alkanols and arachidonic acid. J. Physiol. 2013, 591, 2087–2101. [Google Scholar] [CrossRef]
- Pan, F.; Mills, S.L.; Massey, S.C. Screening of gap junction antagonists on dye coupling in the rabbit retina. Vis. Neurosci. 2007, 24, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, M.; Hopperstad, M.G.; Spray, D.C. Quinine blocks specific gap junction channel subtypes. Proc. Natl. Acad. Sci. USA 2001, 98, 10942–10947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramanathan, T.; Vladescu, I.; McCauley, M.J.; Rouzina, I.; Williams, M.C. Force spectroscopy reveals the DNA structural dynamics that govern the slow binding of Actinomycin, D. Nucleic Acids Res. 2012, 40, 4925–4932. [Google Scholar] [CrossRef] [Green Version]
- Sobell, H.M. Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. USA 1985, 82, 5328–5331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choong, M.L.; Yang, H.; Lee, M.A.; Lane, D.P. Specific activation of the p53 pathway by low dose actinomycin D: A new route to p53 based cyclotherapy. Cell Cycle 2009, 8, 2810–2818. [Google Scholar] [CrossRef] [Green Version]
- Ratnadiwakara, M.; Änkö, M.-L. mRNA Stability Assay Using Transcription Inhibition by Actinomycin D in Mouse Pluripotent Stem Cells. Bio-Protocol 2018, 8, 3072. [Google Scholar] [CrossRef]
- Obrig, T.G.; Culp, W.J.; McKeehan, W.L.; Hardesty, B. The mechanism by which cy-cloheximide and related glutarimide antibiotics inhibit peptide synthesis on re-ticulocyte ribosomes. J. Biol. Chem. 1971, 246, 174–181. [Google Scholar] [CrossRef]
- Lee, S.; Liu, B.; Lee, S.; Huang, S.-X.; Shen, B.; Qian, S.-B. Global mapping of translation initiation sites in mam-malian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2012, 109, E2424–E2432. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Coffino, P.; Li, X. Development of a method for screening short-lived proteins using green fluorescent protein. Genome Biol. 2004, 5, R81. [Google Scholar] [CrossRef] [Green Version]
- Hardesty, B.; Obrig, T.; Irvin, J.; Culp, W. The Effect of Sodium Fluoride, Edeine, and Cycloheximide on Peptide Synthesis with Reticulocyte Ribosomes. Gene Expr. Regul. 1973, 1, 377–392. [Google Scholar] [CrossRef]
- Fujiwara, T.; Oda, K.; Yokota, S.; Takatsuki, A.; Ikehara, Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol. Chem. 1988, 263, 18545–18552. [Google Scholar] [CrossRef]
- Ayala, J. Transport and internal organization of membranes: Vesicles, membrane networks and GTP-binding proteins. J. Cell Sci. 1994, 107, 753–763. [Google Scholar] [PubMed]
- Thyberg, J.; Moskalewski, S. Role of Microtubules in the Organization of the Golgi Complex. Exp. Cell Res. 1999, 246, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Mejillano, M.R.; Shivanna, B.D.; Himes, R.H. Studies on the nocodazo-le-induced GTPase activity of tubulin. Arch. Biochem. Biophys. 1996, 336, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Xi, M.; Gerriets, V. Prostaglandin E2 (Dinoprostone). 2020 Jun 22. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Toh, H.; Ichikawa, A.; Narumiya, S. Molecular evolution of receptors for eicosanoids. FEBS Lett. 1995, 361, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Markovič, T.; Jakopin, Ž.; Dolenc, M.S.; Mlinarič-Raščan, I. Structural features of subtype-selective EP receptor modulators. Drug Discov. Today 2017, 22, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Abramovitz, M.; Adam, M.; Boie, Y.; Carrière, M.-C.; Denis, D.; Godbout, C.; Lamontagne, S.; Rochette, C.; Sawyer, N.; Tremblay, N.M.; et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2000, 1483, 285–293. [Google Scholar] [CrossRef]
- Juteau, H.; Gareau, Y.; Labelle, M.; Sturino, C.F.; Sawyer, N.; Tremblay, N.; Lamontagne, S.; Carrière, M.-C.; Denis, D.; Metters, K.M. Structure–activity relationship of cinnamic acylsulfonamide analogues on the human EP3 prostanoid receptor. Bioorg. Med. Chem. 2001, 9, 1977–1984. [Google Scholar] [CrossRef]
- Jiang, J.; Ganesh, T.; Du, Y.; Quan, Y.; Serrano, G.; Qui, M.; Speigel, I.; Rojas, A.; Lelutiu, N.; Dingledine, R. Small molecule antagonist reveals seizu-re-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2. Proc. Natl. Acad. Sci. USA 2012, 109, 3149–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Dingledine, R. Role of Prostaglandin Receptor EP2 in the Regulations of Cancer Cell Proliferation, Invasion, and Inflammation. J. Pharmacol. Exp. Ther. 2012, 344, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Forselles, K.J.A.; Root, J.; Clarke, T.; Davey, D.; Aughton, K.; Dack, K.; Pullen, N. In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP2 receptor antagonist. Br. J. Pharmacol. 2011, 164, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Kiriyama, M.; Ushikubi, F.; Kobayashi, T.; Hirata, M.; Sugimoto, Y.; Narumiya, S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid re-ceptors ex-pressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1997, 122, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.A.; Jones, R.L. Investigation of the prostaglandin E (EP-) receptor subtype mediating relaxation of the rabbit jugular vein. Br. J. Pharmacol. 1992, 105, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, K.; Naganawa, A.; Ishida, A.; Egashira, H.; Sagawa, K.; Harada, H.; Ogawa, M.; Maruyama, T.; Ohuchida, S.; Nakai, H.; et al. Design and Synthesis of a Highly Selective EP2-Receptor Agonist. Bioorganic Med. Chem. Lett. 2001, 11, 2025–2028. [Google Scholar] [CrossRef]
- Woodward, D.F.; Jones, R.L.; Narumiya, S. International Union of Basic and Clinical Pharmacology. LXXXIII: Classification of Prostanoid Receptors, Updating 15 Years of Progress. Pharmacol. Rev. 2011, 63, 471–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haslam, R.J.; Davidson, M.M.L.; Desjardins, J.V. Inhibition of adenylate cyclase by adenosine analogues in preparations of broken and intact human platelets. Evidence for the unidirectional control of platelet function by cyclic AMP. Biochem. J. 1978, 176, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Engh, R.A.; Girod, A.; Kinzel, V.; Huber, R.; Bossemeyer, D. Crystal Structures of Catalytic Subunit of cAMP-dependent Protein Kinase in Complex with Isoquinolinesulfonyl Protein Kinase Inhibitors H7, H8, and H89. J. Biol. Chem. 1996, 271, 26157–26164. [Google Scholar] [CrossRef] [Green Version]
- Kurzrok, R.; Lieb, C.C. Biochemical Studies of Human Semen. II. The Action of Semen on the Human Uterus. Exp. Biol. Med. 1930, 28, 268–272. [Google Scholar] [CrossRef]
- Von Euler, U. History and development of prostaglandins. Gen. Pharmacol. Vasc. Syst. 1983, 14, 3–6. [Google Scholar] [CrossRef]
- Goldblatt, M.W. Properties of human seminal plasma. J. Physiol. 1935, 84, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.E. The Medical Significance of Prostaglandins. Arch. Intern. Med. 1974, 133, 29. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.B. Prostaglandins in Health and Disease: An Overview. Semin. Arthritis Rheum. 2006, 36, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.; Knaus, E.E. Evolution of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs): Cyclooxygenase (COX) Inhibition and Beyond. J. Pharm. Pharm. Sci. 2008, 11, 81s–110s. [Google Scholar] [CrossRef] [Green Version]
- Rainsford, K. Anti-Inflammatory Drugs in the 21st Century. Alzheimer’s Dis. 2007, 42, 3–27. [Google Scholar] [CrossRef]
- Bertolini, A.; Ottani, A.; Sandrini, M. Selective COX-2 Inhibitors and Dual Acting Anti-inflammatory Drugs: Critical Remarks. Curr. Med. Chem. 2002, 9, 1033–1043. [Google Scholar] [CrossRef]
- Cereijido, M.; Ehrenfeld, J.; Meza, I.; Martínez-Palomo, A. Structural and functional membrane polarity in cultured monolayers of MDCK cells. J. Membr. Biol. 1980, 52, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; Stefani, E.; Palomo, A.M. Occluding junctions in a cultured transporting epithelium: Structural and functional heterogeneity. J. Membr. Biol. 1980, 53, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; González-Mariscal, L.; Borboa, L. Occluding junctions and paracellular pathways studied in monolayers of MDCK cells. J. Exp. Biol. 1983, 106, 205–215. [Google Scholar]
- Meza, I.; Ibarra, G.; Sabanero, M.; Martinez-Palomo, A.; Cereijido, M. Occluding junctions and cytoskeletal components in a cultured transporting epithelium. J. Cell Biol. 1980, 87, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Dukes, J.D.; Whitley, P.; Chalmers, A.D. The MDCK variety pack: Choosing the right strain. BMC Cell Biol. 2011, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Abbaci, M.; Barberi-Heyob, M.; Blondel, W.; Guillemin, F.; Didelon, J. Advantages and limitations of commonly used methods to assay the molecular permeability of gap junctional intercellular communication. Biotechniques 2008, 45, 33–62. [Google Scholar] [CrossRef]
- El-Fouly, M.H.; Trosko, J.E.; Chang, C.-C. Scrape-loading and dye transfer. Exp. Cell Res. 1987, 168, 422–430. [Google Scholar] [CrossRef]
- Babica, P.; Sovadinová, I.; Upham, B.L. Scrape Loading/Dye Transfer Assay. Methods Mol. Biol. 2016, 1437, 133–144. [Google Scholar] [PubMed]
- Larre, I.; Ponce, A.; Fiorentino, R.; Shoshani, L.; Contreras, R.G.; Cereijido, M. Contacts and cooperation between cells depend on the hormone ouabain. Proc. Natl. Acad. Sci. USA 2006, 103, 10911–10916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce, A.; Larre, I.; Castillo, A.; Garcia-Villegas, R.; Romero, A.; Flores-Maldonado, C.; Martinez-Rendón, J.; Contreras, R.G.; Cereijido, M. Ouabain Increases Gap Junctional Communication in Epithelial Cells. Cell. Physiol. Biochem. 2014, 34, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Del Toro, A.O.; Jimenez, L.; Hinojosa, L.; Martínez-Rendón, J.; Castillo, A.; Cereijido, M.; Ponce, A. Influence of Endogenous Cardiac Glycosides, Digoxin, and Marinobufagenin in the Physiology of Epithelial Cells. Cardiol. Res. Pr. 2019, 2019, 8646787-15. [Google Scholar] [CrossRef]
- Matlhagela, K.; Taub, M. Involvement of EP1 and EP2 receptors in the regulation of the Na,K-ATPase by prostaglandins in MDCK cells. Prostaglandins Other Lipid Mediat. 2006, 79, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Regan, J.W. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003, 74, 143–153. [Google Scholar] [CrossRef]
- Ponce, A.; Larre, I.; Castillo, A.; Flores-Maldonado, C.; Verdejo-Torres, O.; Contreras, R.G.; Cereijido, M. Ouabain Modulates the Distribution of Connexin 43 in Epithelial Cells. Cell. Physiol. Biochem. 2016, 39, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Shen, V.; Rifas, L.; Kohler, G.; Peck, W.A. Prostaglandins change cell shape and increase intercellular gap junctions in osteoblasts cultured from rat fetal calvaria. J. Bone Miner. Res. 2009, 1, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Schlemmer, S.R.; Kaufman, D.G. Re-establishment of gap junctional intercellular communication (GJIC) between human endometrial carcinomas by prostaglandin E2. Exp. Mol. Pathol. 2012, 93, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Taub, M.; Parker, R.; Mathivanan, P.; Ariff, M.A.M.; Rudra, T. Antagonism of the prostaglandin E2 EP1 receptor in MDCK cells increases growth through activation of Akt and the epidermal growth factor receptor. Am. J. Physiol. Physiol. 2014, 307, F539–F550. [Google Scholar] [CrossRef] [Green Version]
- Karpisheh, V.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Ghalamfarsa, G.; Sabz, G.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat. 2019, 144, 106338. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogazon del Toro, A.; Jimenez, L.; Serrano Rubi, M.; Castillo, A.; Hinojosa, L.; Martinez Rendon, J.; Cereijido, M.; Ponce, A. Prostaglandin E2 Enhances Gap Junctional Intercellular Communication in Clonal Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 5813. https://doi.org/10.3390/ijms22115813
Ogazon del Toro A, Jimenez L, Serrano Rubi M, Castillo A, Hinojosa L, Martinez Rendon J, Cereijido M, Ponce A. Prostaglandin E2 Enhances Gap Junctional Intercellular Communication in Clonal Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(11):5813. https://doi.org/10.3390/ijms22115813
Chicago/Turabian StyleOgazon del Toro, Alejandro, Lidia Jimenez, Mauricio Serrano Rubi, Aida Castillo, Lorena Hinojosa, Jacqueline Martinez Rendon, Marcelino Cereijido, and Arturo Ponce. 2021. "Prostaglandin E2 Enhances Gap Junctional Intercellular Communication in Clonal Epithelial Cells" International Journal of Molecular Sciences 22, no. 11: 5813. https://doi.org/10.3390/ijms22115813