
Diacylglycerol Kinase alpha in X Linked Lymphoproliferative Disease Type 1





{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
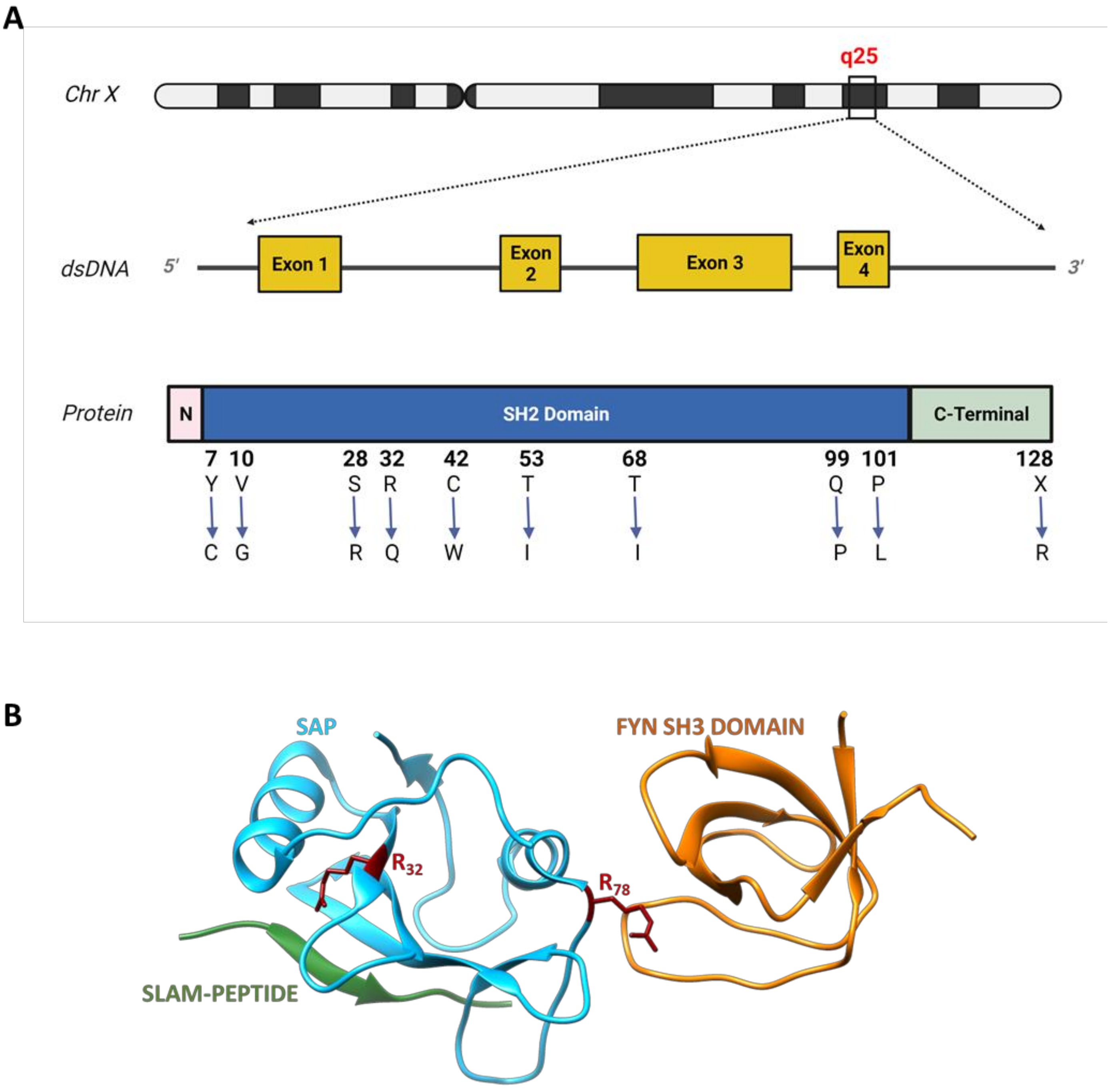
2. X-Linked Lymphoproliferative Disease Type 1
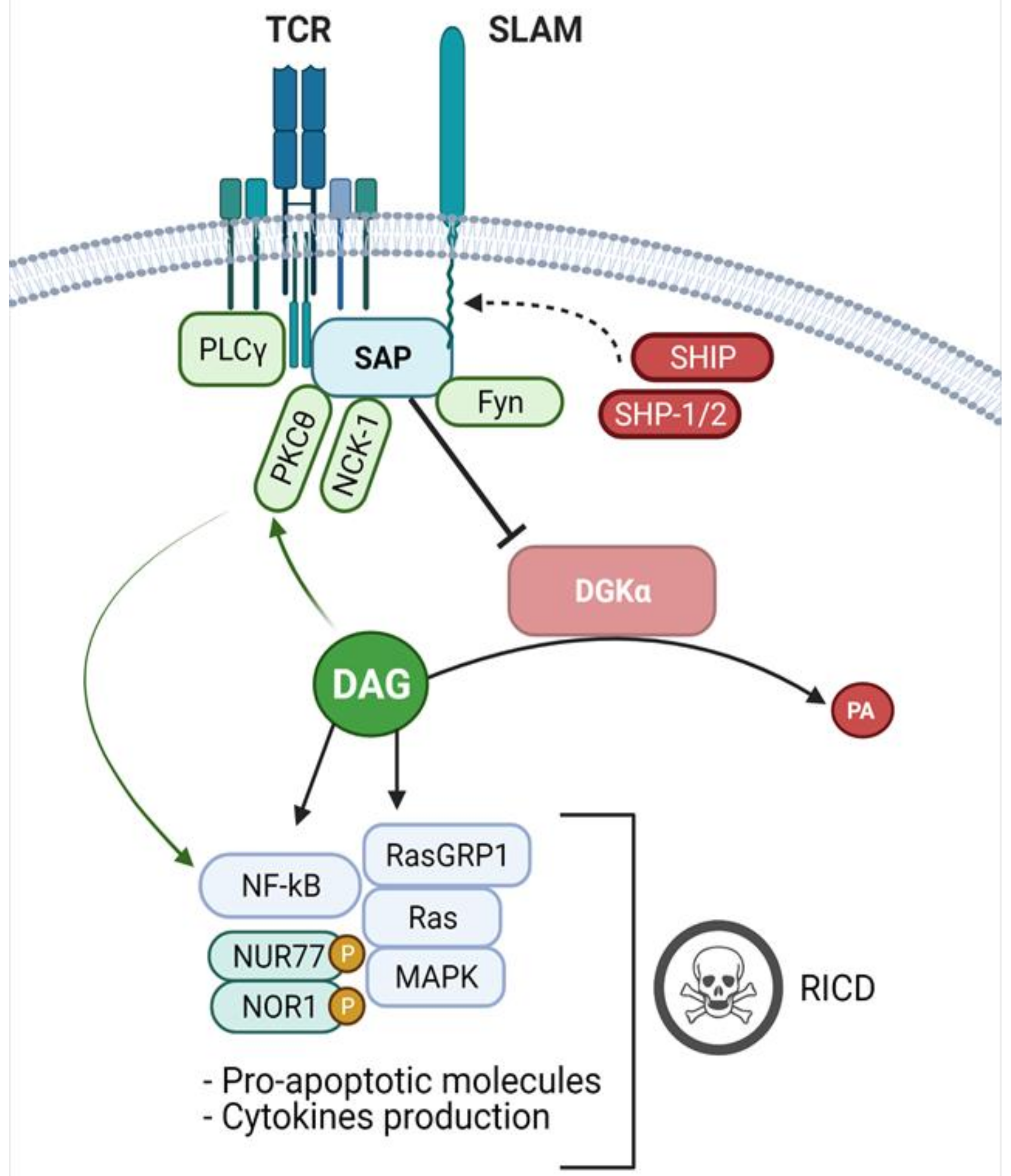
3. Signalling Defects in XLP-1 and Their Biological Effects
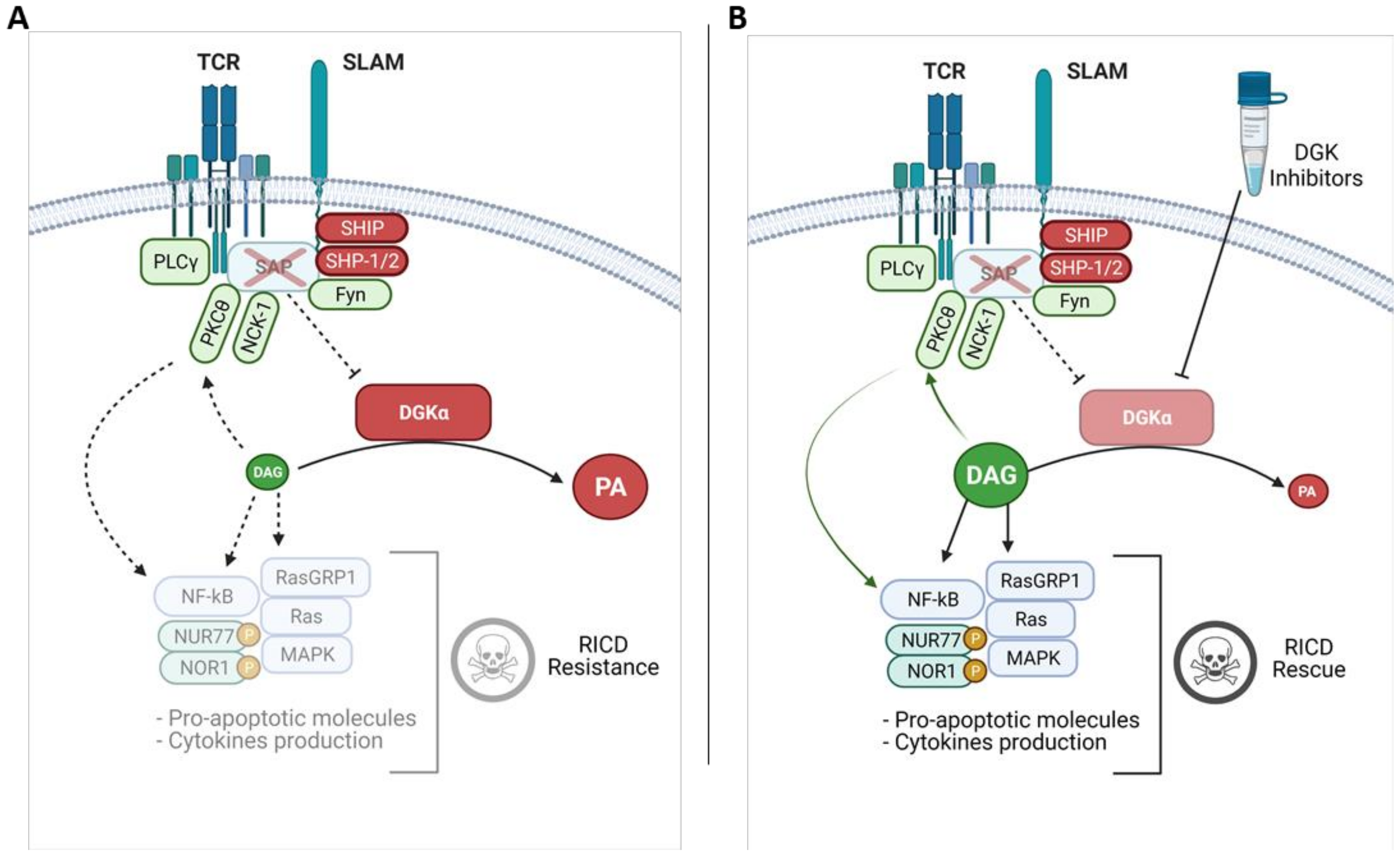
4. SAP Controls DGK Activity and DAG Dependent Signalling
5. DGKα and DGKζ Inhibitors as Potential XLP-1 Therapies
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Snow, A.L.; Pandiyan, P.; Zheng, L.; Krummey, S.M.; Lenardo, M.J. The power and the promise of restimulation-induced cell death in human immune diseases. Immunol. Rev. 2010, 236, 68–82. [Google Scholar] [CrossRef]
- Baldanzi, G.; Pighini, A.; Bettio, V.; Rainero, E.; Traini, S.; Chianale, F.; Porporato, P.E.; Filigheddu, N.; Mesturini, R.; Song, S.; et al. SAP-mediated inhibition of diacylglycerol kinase α regulates TCR-induced diacylglycerol signaling. J. Immunol. 2011, 187, 5941–5951. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, S.; Mérida, I. Diacylglycerol, when simplicity becomes complex. Trends Biochem. Sci. 2007, 32, 27–36. [Google Scholar] [CrossRef]
- Fu, G.; Chen, Y.; Yu, M.; Podd, A.; Schuman, J.; He, Y.; Di, L.; Yassai, M.; Haribhai, D.; North, P.E.; et al. Phospholipase C{gamma}1 is essential for T cell development, activation, and tolerance. J. Exp. Med. 2010, 207, 309–318. [Google Scholar] [CrossRef]
- Huang, Y.H.; Sauer, K. Lipid signaling in T-cell development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a002428. [Google Scholar] [CrossRef] [Green Version]
- Augsten, M.; Pusch, R.; Biskup, C.; Rennert, K.; Wittig, U.; Beyer, K.; Blume, A.; Wetzker, R.; Friedrich, K.; Rubio, I. Live-cell imaging of endogenous Ras-GTP illustrates predominant Ras activation at the plasma membrane. EMBO Rep. 2006, 7, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebinu, J.O.; Stang, S.L.; Teixeira, C.; Bottorff, D.A.; Hooton, J.; Blumberg, P.M.; Barry, M.; Bleakley, R.C.; Ostergaard, H.L.; Stone, J.C. RasGRP links T-cell receptor signaling to Ras. Blood 2000, 95, 3199–3203. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef]
- Krishna, S.; Zhong, X. Role of diacylglycerol kinases in T cell development and function. Crit. Rev. Immunol. 2013, 33, 97–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, Y.; Marks, R.; Ho, A.W.; Peterson, A.C.; Janardhan, S.; Brown, I.; Praveen, K.; Stang, S.; Stone, J.C.; Gajewski, T.F. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat. Immunol. 2006, 7, 1166–1173. [Google Scholar] [CrossRef]
- Arranz-Nicolás, J.; Martin-Salgado, M.; Adán-Barrientos, I.; Liébana, R.; Del Carmen Moreno-Ortíz, M.; Leitner, J.; Steinberger, P.; Ávila-Flores, A.; Merida, I. Diacylglycerol kinase α inhibition cooperates with PD-1-targeted therapies to restore the T cell activation program. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef]
- Olenchock, B.A.; Guo, R.; Carpenter, J.H.; Jordan, M.; Topham, M.K.; Koretzky, G.A.; Zhong, X.P. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol. 2006, 7, 1174–1181. [Google Scholar] [CrossRef]
- Zhong, X.P.; Hainey, E.A.; Olenchock, B.A.; Jordan, M.S.; Maltzman, J.S.; Nichols, K.E.; Shen, H.; Koretzky, G.A. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat. Immunol. 2003, 4, 882–890. [Google Scholar] [CrossRef]
- Baldanzi, G.; Ragnoli, B.; Malerba, M. Potential role of diacylglycerol kinases in immune-mediated diseases. Clin. Sci. 2020, 134, 1637–1658. [Google Scholar] [CrossRef] [PubMed]
- Arranz-Nicolás, J.; Ogando, J.; Soutar, D.; Arcos-Pérez, R.; Meraviglia-Crivelli, D.; Mañes, S.; Mérida, I.; Ávila-Flores, A. Diacylglycerol kinase α inactivation is an integral component of the costimulatory pathway that amplifies TCR signals. Cancer Immunol. Immunother. 2018, 67, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Ruffo, E.; Malacarne, V.; Larsen, S.E.; Das, R.; Patrussi, L.; Wülfing, C.; Biskup, C.; Kapnick, S.M.; Verbist, K.; Tedrick, P.; et al. Inhibition of diacylglycerol kinase α restores restimulation-induced cell death and reduces immunopathology in XLP-1. Sci Transl Med. 2016, 8, 321ra7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangye, S.G. XLP: Clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J. Clin. Immunol. 2014, 34, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Purtilo, D.T.; Cassel, C.K.; Yang, J.P.; Harper, R. X-linked recessive progressive combined variable immunodeficiency (Duncan’s disease). Lancet 1975, 1, 935–940. [Google Scholar] [CrossRef]
- Gaspar, H.B.; Sharifi, R.; Gilmour, K.C.; Thrasher, A.J. X-linked lymphoproliferative disease: Clinical, diagnostic and molecular perspective. Br. J. Haematol. 2002, 119, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Nichols, K.E.; Ma, C.S.; Cannons, J.L.; Schwartzberg, P.L.; Tangye, S.G. Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunol. Rev. 2005, 203, 180–199. [Google Scholar] [CrossRef]
- Coffey, A.J.; Brooksbank, R.A.; Brandau, O.; Oohashi, T.; Howell, G.R.; Bye, J.M.; Cahn, A.P.; Durham, J.; Heath, P.; Wray, P.; et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat. Genet. 1998, 20, 129–135. [Google Scholar] [CrossRef]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef]
- Li, S.C.; Gish, G.; Yang, D.; Coffey, A.J.; Forman-Kay, J.D.; Ernberg, I.; Kay, L.E.; Pawson, T. Novel mode of ligand binding by the SH2 domain of the human XLP disease gene product SAP/SH2D1A. Curr. Biol. 1999, 9, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Poy, F.; Yaffe, M.B.; Sayos, J.; Saxena, K.; Morra, M.; Sumegi, J.; Cantley, L.C.; Terhorst, C.; Eck, M.J. Crystal structures of the XLP protein SAP reveal a class of SH2 domains with extended, phosphotyrosine-independent sequence recognition. Mol. Cell 1999, 4, 555–561. [Google Scholar] [CrossRef]
- Latour, S.; Veillette, A. Molecular and immunological basis of X-linked lymphoproliferative disease. Immunol. Rev. 2003, 192, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Gish, G.; Helgason, C.D.; Humphries, R.K.; Pawson, T.; Veillette, A. Regulation of SLAM-mediated signal transduction by SAP, the X-linked lymphoproliferative gene product. Nat. Immunol. 2001, 2, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.; Lanyi, A.; Song, H.K.; Griesbach, J.; Simarro-Grande, M.; Poy, F.; Howie, D.; Sumegi, J.; Terhorst, C.; Eck, M.J. SAP couples Fyn to SLAM immune receptors. Nat. Cell Biol. 2003, 5, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, M.A.; Mor, A. The SLAM family receptors: Potential therapeutic targets for inflammatory and autoimmune diseases. Autoimmun. Rev. 2018, 17, 674–682. [Google Scholar] [CrossRef]
- Davidson, D.; Shi, X.; Zhang, S.; Wang, H.; Nemer, M.; Ono, N.; Ohno, S.; Yanagi, Y.; Veillette, A. Genetic evidence linking SAP, the X-linked lymphoproliferative gene product, to Src-related kinase FynT in T(H)2 cytokine regulation. Immunity 2004, 21, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Cannons, J.L.; Yu, L.J.; Hill, B.; Mijares, L.A.; Dombroski, D.; Nichols, K.E.; Antonellis, A.; Koretzky, G.A.; Gardner, K.; Schwartzberg, P.L. SAP regulates T(H)2 differentiation and PKC-theta-mediated activation of NF-kappaB1. Immunity 2004, 21, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Davidson, D.; Pérez-Quintero, L.A.; Kurosaki, T.; Swat, W.; Veillette, A. The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity 2012, 36, 974–985. [Google Scholar] [CrossRef] [Green Version]
- Morra, M.; Simarro-Grande, M.; Martin, M.; Chen, A.S.; Lanyi, A.; Silander, O.; Calpe, S.; Davis, J.; Pawson, T.; Eck, M.J.; et al. Characterization of SH2D1A missense mutations identified in X-linked lymphoproliferative disease patients. J. Biol. Chem. 2001, 276, 36809–36816. [Google Scholar] [CrossRef] [Green Version]
- Sylla, B.S.; Murphy, K.; Cahir-McFarland, E.; Lane, W.S.; Mosialos, G.; Kieff, E. The X-linked lymphoproliferative syndrome gene product SH2D1A associates with p62dok (Dok1) and activates NF-kappa B. Proc. Natl. Acad. Sci. USA 2000, 97, 7470–7475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlapatska, L.M.; Mikhalap, S.V.; Berdova, A.G.; Zelensky, O.M.; Yun, T.J.; Nichols, K.E.; Clark, E.A.; Sidorenko, S.P. CD150 association with either the SH2-containing inositol phosphatase or the SH2-containing protein tyrosine phosphatase is regulated by the adaptor protein SH2D1A. J. Immunol. 2001, 166, 5480–5487. [Google Scholar] [CrossRef] [Green Version]
- Cannons, J.L.; Wu, J.Z.; Gomez-Rodriguez, J.; Zhang, J.; Dong, B.; Liu, Y.; Shaw, S.; Siminovitch, K.A.; Schwartzberg, P.L. Biochemical and genetic evidence for a SAP-PKC-theta interaction contributing to IL-4 regulation. J. Immunol. 2010, 185, 2819–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Tangye, S.G.; Sun, X.; Luo, Y.; Lin, Z.; Wu, J. The X-linked lymphoproliferative disease gene product SAP associates with PAK-interacting exchange factor and participates in T cell activation. Proc. Natl. Acad. Sci. USA 2006, 103, 14447–14452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Schibli, D.; Li, S.S. The XLP syndrome protein SAP interacts with SH3 proteins to regulate T cell signaling and proliferation. Cell Signal. 2009, 21, 111–119. [Google Scholar] [CrossRef]
- Wilson, T.J.; Garner, L.I.; Metcalfe, C.; King, E.; Margraf, S.; Brown, M.H. Fine specificity and molecular competition in SLAM family receptor signalling. PLoS ONE 2014, 9, e92184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageyama, R.; Cannons, J.L.; Zhao, F.; Yusuf, I.; Lao, C.; Locci, M.; Schwartzberg, P.L.; Crotty, S. The receptor Ly108 functions as a SAP adaptor-dependent on-off switch for T cell help to B cells and NKT cell development. Immunity 2012, 36, 986–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proust, R.; Bertoglio, J.; Gesbert, F. The adaptor protein SAP directly associates with CD3ζ chain and regulates T cell receptor signaling. PLoS ONE 2012, 7, e43200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proust, R.; Crouin, C.; Gandji, L.Y.; Bertoglio, J.; Gesbert, F. The adaptor protein SAP directly associates with PECAM-1 and regulates PECAM-1-mediated-cell adhesion in T-like cell lines. Mol. Immunol 2014, 58, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.L.; Marsh, R.A.; Krummey, S.M.; Roehrs, P.; Young, L.R.; Zhang, K.; van Hoff, J.; Dhar, D.; Nichols, K.E.; Filipovich, A.H.; et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J. Clin. Invest. 2009, 119, 2976–2989. [Google Scholar] [CrossRef] [Green Version]
- Katz, G.; Krummey, S.M.; Larsen, S.E.; Stinson, J.R.; Snow, A.L. SAP facilitates recruitment and activation of LCK at NTB-A receptors during restimulation-induced cell death. J. Immunol 2014, 192, 4202–4209. [Google Scholar] [CrossRef] [Green Version]
- Peled, M.; Tocheva, A.S.; Sandigursky, S.; Nayak, S.; Philips, E.A.; Nichols, K.E.; Strazza, M.; Azoulay-Alfaguter, I.; Askenazi, M.; Neel, B.G.; et al. Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc. Natl. Acad. Sci. USA 2018, 115, E468–E477. [Google Scholar] [CrossRef] [Green Version]
- Sandigursky, S.; Philips, M.R.; Mor, A. SAP interacts with CD28 to inhibit PD-1 signaling in T lymphocytes. Clin. Immunol. 2020, 217, 108485. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM family receptors and SAP adaptors in immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A. Immune regulation by SLAM family receptors and SAP-related adaptors. Nat. Rev. Immunol. 2006, 6, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Cannons, J.L.; Dutta, M.; Griffiths, G.M.; Schwartzberg, P.L. Positive and negative signaling through SLAM receptors regulate synapse organization and thresholds of cytolysis. Immunity 2012, 36, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Dupré, L.; Andolfi, G.; Tangye, S.G.; Clementi, R.; Locatelli, F.; Aricò, M.; Aiuti, A.; Roncarolo, M.G. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood 2005, 105, 4383–4389. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Palendira, U.; Leese, A.M.; Arkwright, P.D.; Rohrlich, P.S.; Tangye, S.G.; Gaspar, H.B.; Lankester, A.C.; Moretta, A.; Rickinson, A.B. Impaired Epstein-Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood 2010, 116, 3249–3257. [Google Scholar] [CrossRef] [Green Version]
- Palendira, U.; Low, C.; Chan, A.; Hislop, A.D.; Ho, E.; Phan, T.G.; Deenick, E.; Cook, M.C.; Riminton, D.S.; Choo, S.; et al. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol. 2011, 9, e1001187. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Qi, H.; Lu, K.T.; Dutta, M.; Gomez-Rodriguez, J.; Cheng, J.; Wakeland, E.K.; Germain, R.N.; Schwartzberg, P.L. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity 2010, 32, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Cannons, J.L.; Klauschen, F.; Schwartzberg, P.L.; Germain, R.N. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 2008, 455, 764–769. [Google Scholar] [CrossRef]
- Merino, E.; Sanjuán, M.A.; Moraga, I.; Ciprés, A.; Mérida, I. Role of the diacylglycerol kinase alpha-conserved domains in membrane targeting in intact T cells. J. Biol. Chem. 2007, 282, 35396–35404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, M.A.; Pradet-Balade, B.; Jones, D.R.; Martínez, A.C.; Stone, J.C.; Garcia-Sanz, J.A.; Mérida, I. T cell activation in vivo targets diacylglycerol kinase alpha to the membrane: A novel mechanism for Ras attenuation. J. Immunol. 2003, 170, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Gharbi, S.I.; Rincón, E.; Avila-Flores, A.; Torres-Ayuso, P.; Almena, M.; Cobos, M.A.; Albar, J.P.; Mérida, I. Diacylglycerol kinase ζ controls diacylglycerol metabolism at the immunological synapse. Mol. Biol. Cell 2011, 22, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, A.; Le Floc’h, A.; Bantilan, N.S.; Koretzky, G.A.; Huse, M. Diacylglycerol kinase α establishes T cell polarity by shaping diacylglycerol accumulation at the immunological synapse. Sci. Signal. 2014, 7, ra82. [Google Scholar] [CrossRef] [Green Version]
- Merino, E.; Avila-Flores, A.; Shirai, Y.; Moraga, I.; Saito, N.; Mérida, I. Lck-dependent tyrosine phosphorylation of diacylglycerol kinase alpha regulates its membrane association in T cells. J. Immunol. 2008, 180, 5805–5815. [Google Scholar] [CrossRef] [Green Version]
- Ciprés, A.; Carrasco, S.; Merino, E.; Díaz, E.; Krishna, U.M.; Falck, J.R.; Martínez, A.C.; Mérida, I. Regulation of diacylglycerol kinase alpha by phosphoinositide 3-kinase lipid products. J. Biol. Chem. 2003, 278, 35629–35635. [Google Scholar] [CrossRef] [Green Version]
- Ávila-Flores, A.; Arranz-Nicolás, J.; Andrada, E.; Soutar, D.; Mérida, I. Predominant contribution of DGKζ over DGKα in the control of PKC/PDK-1-regulated functions in T cells. Immunol. Cell Biol. 2017, 95, 549–563. [Google Scholar] [CrossRef]
- Joshi, R.P.; Schmidt, A.M.; Das, J.; Pytel, D.; Riese, M.J.; Lester, M.; Diehl, J.A.; Behrens, E.M.; Kambayashi, T.; Koretzky, G.A. The ζ isoform of diacylglycerol kinase plays a predominant role in regulatory T cell development and TCR-mediated ras signaling. Sci. Signal. 2013, 6, ra102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, R.; Mazzeo, C.; Rodriguez, M.C.; Marsh, M.; Fraile-Ramos, A.; Calvo, V.; Avila-Flores, A.; Merida, I.; Izquierdo, M. Diacylglycerol kinase α regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of Fas ligand-containing exosomes in T lymphocytes. Cell Death Differ. 2011, 18, 1161–1173. [Google Scholar] [CrossRef] [Green Version]
- Hurttia, H.; Leino, L. Subcellular localization of diacylglycerol kinase activity in stimulated and unstimulated human peripheral blood lymphocytes and neutrophils. Biochem. Mol. Biol. Int. 1996, 40, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, G.; Cutrupi, S.; Chianale, F.; Gnocchi, V.; Rainero, E.; Porporato, P.; Filigheddu, N.; van Blitterswijk, W.J.; Parolini, O.; Bussolino, F.; et al. Diacylglycerol kinase-alpha phosphorylation by Src on Y335 is required for activation, membrane recruitment and Hgf-induced cell motility. Oncogene 2008, 27, 942–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Rud, J.; Olson, C.M.; Anguita, J.; Osborne, B.A. Phosphorylation of Nur77 by the MEK-ERK-RSK cascade induces mitochondrial translocation and apoptosis in T cells. J. Immunol. 2009, 183, 3268–3277. [Google Scholar] [CrossRef] [Green Version]
- Merida, I.; Arranz-Nicolás, J.; Torres-Ayuso, P.; Ávila-Flores, A. Diacylglycerol Kinase Malfunction in Human Disease and the Search for Specific Inhibitors. Handb. Exp. Pharmacol. 2020, 259, 133–162. [Google Scholar] [CrossRef]
- Jiang, Y.; Sakane, F.; Kanoh, H.; Walsh, J.P. Selectivity of the diacylglycerol kinase inhibitor 3-[2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl]-2, 3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem. Pharmacol. 2000, 59, 763–772. [Google Scholar] [CrossRef]
- Sato, M.; Liu, K.; Sasaki, S.; Kunii, N.; Sakai, H.; Mizuno, H.; Saga, H.; Sakane, F. Evaluations of the selectivities of the diacylglycerol kinase inhibitors r59022 and r59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology 2013, 92, 99–107. [Google Scholar] [CrossRef]
- Boroda, S.; Niccum, M.; Raje, V.; Purow, B.W.; Harris, T.E. Dual activities of ritanserin and R59022 as DGKα inhibitors and serotonin receptor antagonists. Biochem. Pharmacol. 2017, 123, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCloud, R.L.; Franks, C.E.; Campbell, S.T.; Purow, B.W.; Harris, T.E.; Hsu, K.L. Deconstructing Lipid Kinase Inhibitors by Chemical Proteomics. Biochemistry 2018, 57, 231–236. [Google Scholar] [CrossRef]
- Audia, A.; Bhat, K.P. Ritanserin, a novel agent targeting the mesenchymal subtype of glioblastomas. Neuro Oncol. 2018, 20, 151–152. [Google Scholar] [CrossRef]
- Liu, K.; Kunii, N.; Sakuma, M.; Yamaki, A.; Mizuno, S.; Sato, M.; Sakai, H.; Kado, S.; Kumagai, K.; Kojima, H.; et al. A novel diacylglycerol kinase α-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J. Lipid Res. 2016, 57, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaki, A.; Akiyama, R.; Murakami, C.; Takao, S.; Murakami, Y.; Mizuno, S.; Takahashi, D.; Kado, S.; Taketomi, A.; Shirai, Y.; et al. Diacylglycerol kinase α-selective inhibitors induce apoptosis and reduce viability of melanoma and several other cancer cell lines. J. Cell Biochem. 2019, 120, 10043–10056. [Google Scholar] [CrossRef] [PubMed]
- Velnati, S.; Ruffo, E.; Massarotti, A.; Talmon, M.; Varma, K.S.S.; Gesu, A.; Fresu, L.G.; Snow, A.L.; Bertoni, A.; Capello, D.; et al. Identification of a novel DGKα inhibitor for XLP-1 therapy by virtual screening. Eur. J. Med. Chem. 2019, 164, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Velnati, S.; Massarotti, A.; Antona, A.; Talmon, M.; Fresu, L.G.; Galetto, A.S.; Capello, D.; Bertoni, A.; Mercalli, V.; Graziani, A.; et al. Structure activity relationship studies on Amb639752: Toward the identification of a common pharmacophoric structure for DGKα inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Wang, M.; Ge, Y.; Chen, X.P.; Xu, Z.; Sun, Y.; Xiong, X.F. Tyrosine phosphatase SHP2 inhibitors in tumor-targeted therapies. Acta Pharm. Sin. B 2021, 11, 13–29. [Google Scholar] [CrossRef] [PubMed]
- LaMarche, M.J.; Acker, M.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.H.; Chen, Y.N.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 13578–13594. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Cancer Immunotherapy through the Inhibition of Diacylglycerol Kinases Alpha and Zeta. ACS Med. Chem. Lett. 2020, 11, 1083–1085. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Lazetic, S.; Woollatt, E.; Sutherland, G.R.; Lanier, L.L.; Phillips, J.H. Cutting edge: Human 2B4, an activating NK cell receptor, recruits the protein tyrosine phosphatase SHP-2 and the adaptor signaling protein SAP. J. Immunol. 1999, 162, 6981–6985. [Google Scholar]
- Li, C.; Iosef, C.; Jia, C.Y.; Han, V.K.; Li, S.S. Dual functional roles for the X-linked lymphoproliferative syndrome gene product SAP/SH2D1A in signaling through the signaling lymphocyte activation molecule (SLAM) family of immune receptors. J. Biol. Chem. 2003, 278, 3852–3859. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, C.; Huynh, H.; Le, T.B.U.; LaMarche, M.J.; Mohseni, M.; Engelman, J.A.; Hammerman, P.S.; Caponigro, G.; Hao, H.X. Resistance to allosteric SHP2 inhibition in FGFR-driven cancers through rapid feedback activation of FGFR. Oncotarget 2020, 11, 265–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchal, N.; Booth, C.; Cannons, J.L.; Schwartzberg, P.L. X-Linked Lymphoproliferative Disease Type 1: A Clinical and Molecular Perspective. Front. Immunol. 2018, 9, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivat, C.; Booth, C.; Alonso-Ferrero, M.; Blundell, M.; Sebire, N.J.; Thrasher, A.J.; Gaspar, H.B. SAP gene transfer restores cellular and humoral immune function in a murine model of X-linked lymphoproliferative disease. Blood 2013, 121, 1073–1076. [Google Scholar] [CrossRef] [Green Version]
- Shabani, M.; Nichols, K.E.; Rezaei, N. Primary immunodeficiencies associated with EBV-Induced lymphoproliferative disorders. Crit. Rev. Oncol. Hematol. 2016, 108, 109–127. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velnati, S.; Centonze, S.; Girivetto, F.; Baldanzi, G. Diacylglycerol Kinase alpha in X Linked Lymphoproliferative Disease Type 1. Int. J. Mol. Sci. 2021, 22, 5816. https://doi.org/10.3390/ijms22115816
Velnati S, Centonze S, Girivetto F, Baldanzi G. Diacylglycerol Kinase alpha in X Linked Lymphoproliferative Disease Type 1. International Journal of Molecular Sciences. 2021; 22(11):5816. https://doi.org/10.3390/ijms22115816
Chicago/Turabian StyleVelnati, Suresh, Sara Centonze, Federico Girivetto, and Gianluca Baldanzi. 2021. "Diacylglycerol Kinase alpha in X Linked Lymphoproliferative Disease Type 1" International Journal of Molecular Sciences 22, no. 11: 5816. https://doi.org/10.3390/ijms22115816
APA StyleVelnati, S., Centonze, S., Girivetto, F., & Baldanzi, G. (2021). Diacylglycerol Kinase alpha in X Linked Lymphoproliferative Disease Type 1. International Journal of Molecular Sciences, 22(11), 5816. https://doi.org/10.3390/ijms22115816