
Catalytic Descriptors to Investigate Catalytic Power in the Reaction of Haloalkane Dehalogenase Enzyme with 1,2-Dichloroethane


Abstract
:1. Introduction
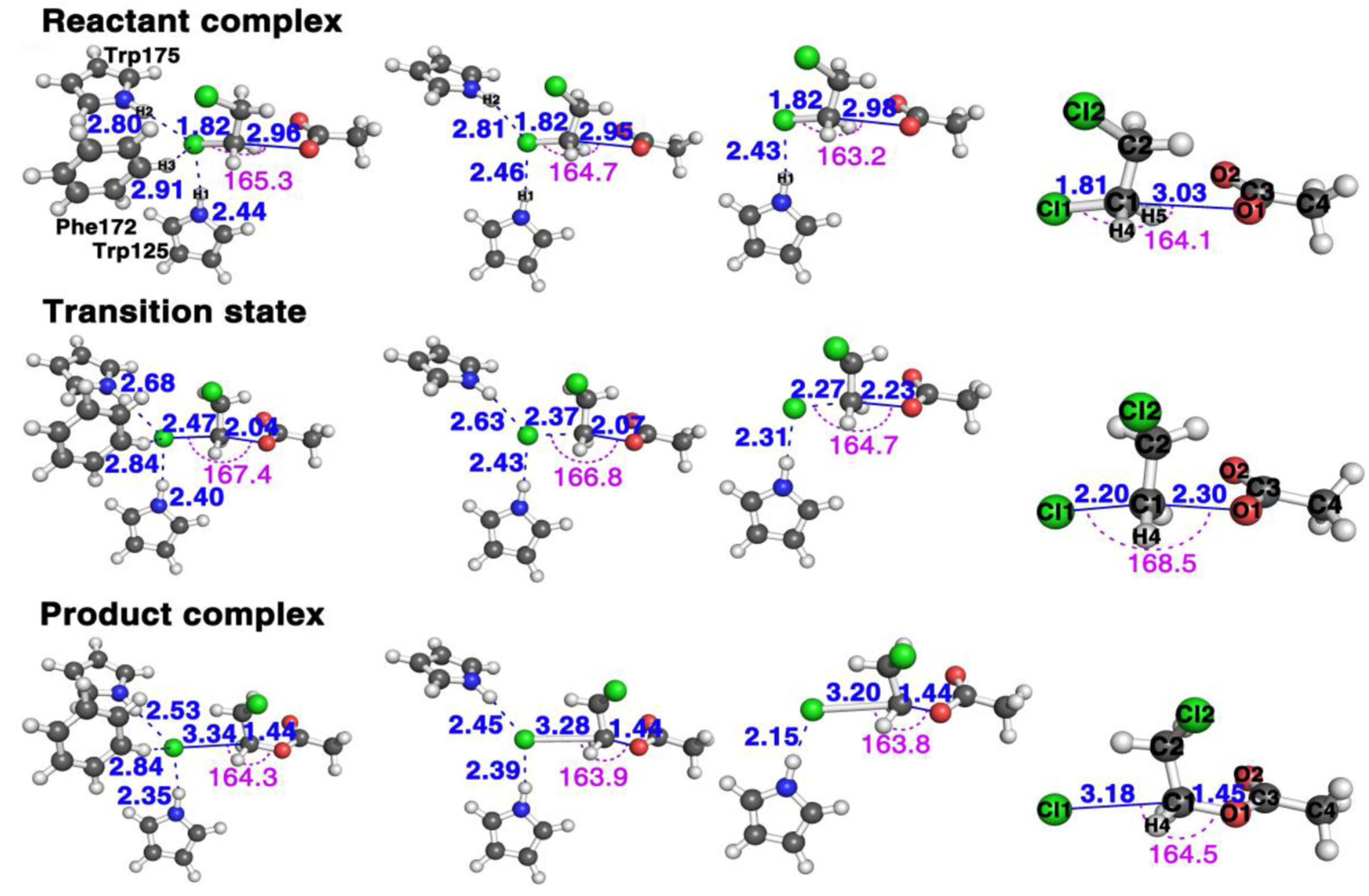
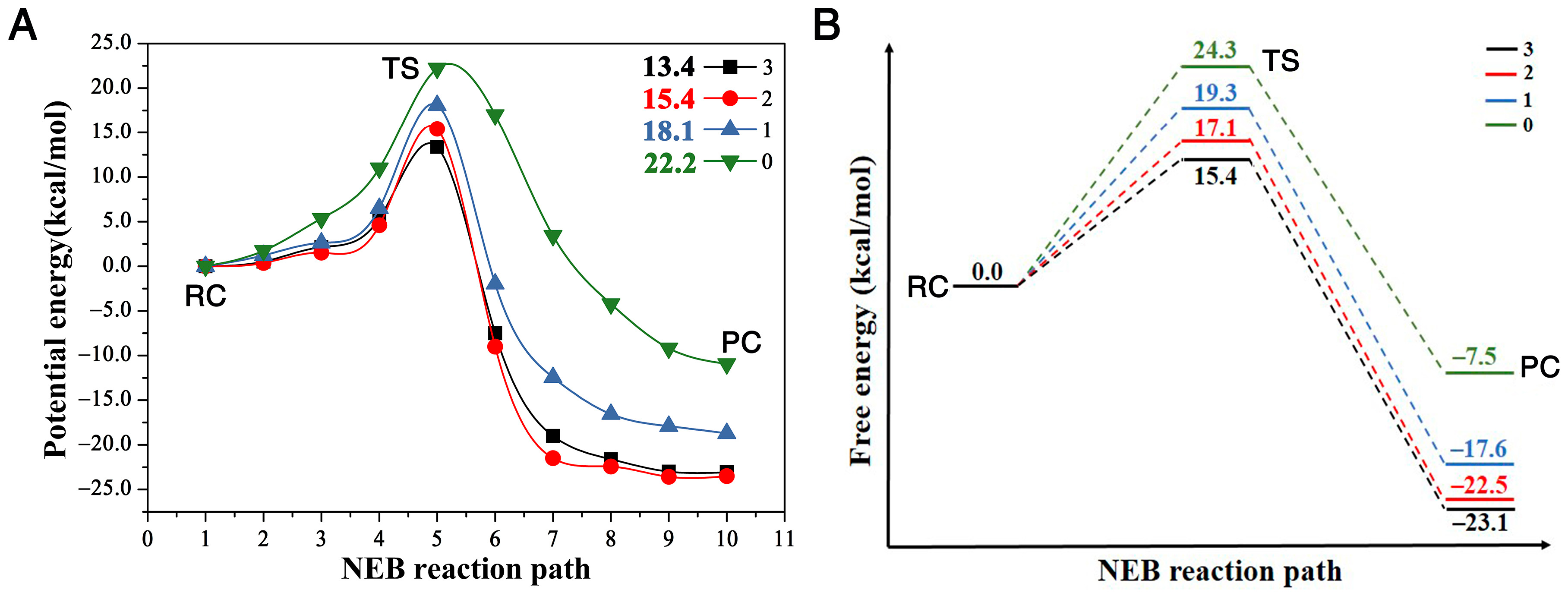
2. Results and Discussion
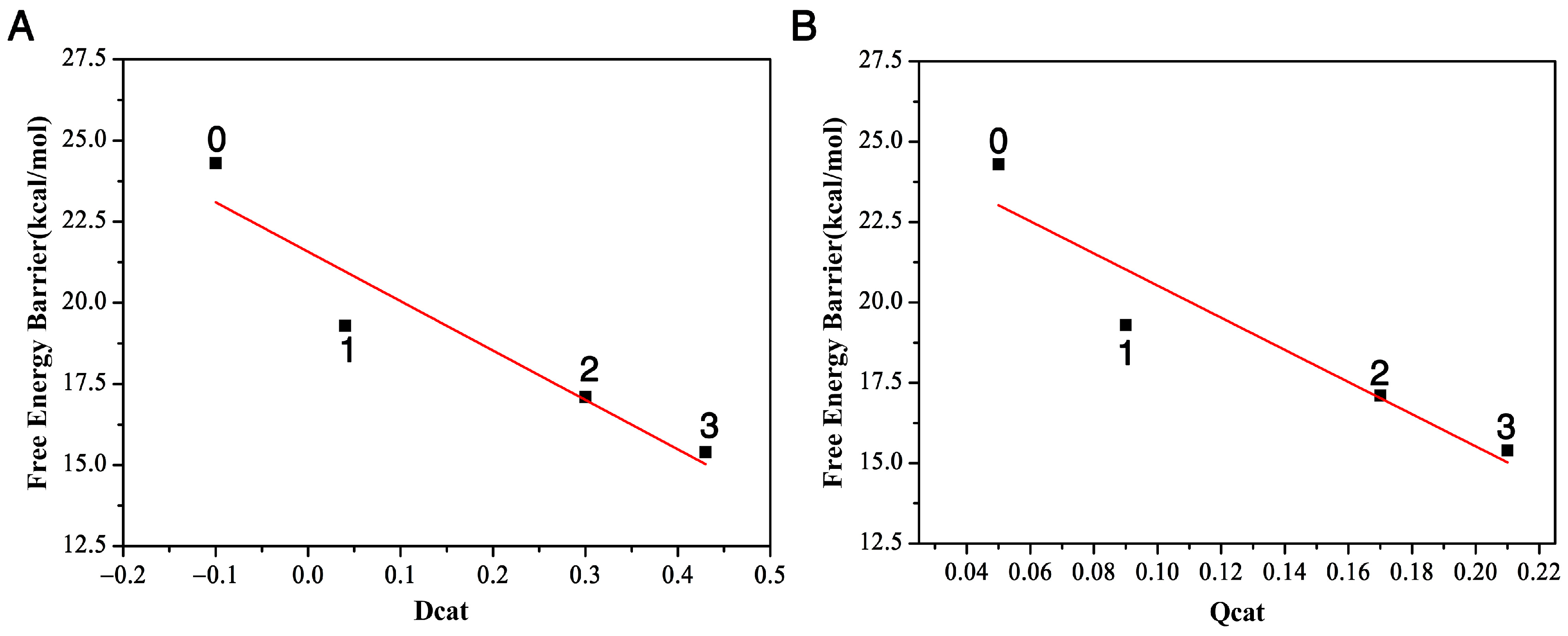
2.1. The Bond Breaking and Formation Descriptor
2.2. The Charge Transfer Descriptor
2.3. Prediction of the Reactivity in Water
3. Methodologies
3.1. Quantum Mechanics Calculation for the AcO− + DCE with Three, Two, One, and Zero Residues
3.2. Quantum Mechanics and Molecular Mechanics Calculation for AcO− + DCE in Water
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warshel, A. Computer Modeling of Chemical Reactions in Enzymes and Solutions; Wiley-Interscience: New York, NY, USA, 1991. [Google Scholar]
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W.H. Freeman and Co.: New York, NY, USA, 1999. [Google Scholar]
- Janssen, D.B.; Scheper, A.; Dijkhuizen, L.; Witholt, B. Degradation of halogenated aliphatic compounds by Xanthobacter autotrophicus GJ10. Appl. Environ. Microb. 1985, 49, 673–677. [Google Scholar] [CrossRef] [Green Version]
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H.M. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. [Google Scholar] [CrossRef] [PubMed]
- Kamerlin, S.C.L.; Warshel, A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins 2010, 78, 1339–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warshel, A.; Bora, R.P. Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys. 2016, 144, 180901. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A. Energetics of enzyme catalysis. Proc. Natl. Acad. Sci. USA 1978, 75, 5250–5254. [Google Scholar] [CrossRef] [Green Version]
- Soriano, A.; Silla, E.; Tunon, I.; Marti, S.; Moliner, V.; Bertran, J. Electrostatic effects in enzyme catalysis: A quantum mechanics/molecular mechanics study of the nucleophilic substitution reaction in haloalkane dehalogenase. Theor. Chem. Acc. 2004, 112, 327–334. [Google Scholar] [CrossRef]
- Soriano, A.; Silla, E.; Tuñón, I.; Ruiz-López, M.F. Dynamic and electrostatic effects in enzymatic processes. An analysis of the nucleophilic substitution reaction in haloalkane dehalogenase. J. Am. Chem. Soc. 2005, 127, 1946–1957. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Parson, W.W.; Warshel, A. Dynamical contributions to enzyme catalysis: Critical tests of a popular hypothesis. Chem. Rev. 2006, 106, 1737–1756. [Google Scholar] [CrossRef]
- Careri, G.; Fasella, P.; Gratton, E. Enzyme dynamics: The statistical physics approach. Ann. Rev. Biophys. Bioeng. 1979, 8, 69–97. [Google Scholar] [CrossRef] [Green Version]
- Neria, E.; Kuplus, M. Molecular dynamics of an enzyme reaction: Proton transfer in TIM. Chem. Phys. Lett. 1997, 267, 23–30. [Google Scholar] [CrossRef]
- Nam, K.; Prat-Resina, X.; Garcia-Viloca, M.; Devi-Kesavan, L.S.; Gao, J. Dynamics of an enzymatic substitution reaction in haloalkane dehalogenase. J. Am. Chem. Soc. 2004, 126, 1369–1376. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Pozharski, E.; Wilson, M.A.; Petsko, G.A.; Karplus, M.; et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838–844. [Google Scholar] [CrossRef]
- Bhabha, G.; Lee, J.; Ekiert, D.C.; Gam, J.; Wilson, L.A.; dyson, H.J.; Benkovic, S.J.; Wright, P.E. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science 2011, 332, 234–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Meseguer, R.; Martí, S.; Ruiz-Pernía, J.J.; Moliner, V.; Tuñón, I. Studying the role of protein dynamics in an SN2 enzyme reaction using free-energy surfaces and solvent coordinates. Nat. Chem. 2013, 5, 566–571. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Francis, K.; Kohen, A. Network of remote and local protein dynamics in dihydrofolate reductase catalysis. ACS Catal. 2015, 5, 3067–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keuning, S.; Janssen, D.B.; Witholt, B. Purification and characterization of hydrolytic haloalkane dehalogenase from Xanthobacter autotrophicus GJ10. J. Bacteriol. 1985, 163, 635–639. [Google Scholar] [CrossRef] [Green Version]
- Janssen, D.B.; Pries, F.; Van der Ploeg, J.; Kazemier, B.; Terpstra, P.; Witholt, B. Cloning of 1,2-dichloroethane degradation genes of Xanthobacter autotrophicus GJ10 and expression and sequencing of the dhlA gene. J. Bacteriol. 1989, 17, 6791–6799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardif, G.; Greer, C.W.; Labbe, D.; Lau, P.C. Involvement of a large plasmid in the degradation of 1,2-dichloroethane by Xanthobacter autotrophicus. Appl. Environ. Microb. 1991, 57, 1853–1857. [Google Scholar] [CrossRef] [Green Version]
- Franken, S.M.; Rozeboom, H.J.; Kalk, K.H.; Dijkstra, B.W. Crystal structure of haloalkane dehalogenase: An enzyme to detoxify halogenated alkanes. EMBO J. 1991, 10, 1297–1302. [Google Scholar] [CrossRef]
- Verschueren, K.H.G.; Franken, S.M.; Rozeboom, H.J.; Kalk, K.H.; Dijkstra, B.W. Refined X-ray Structures of Haloalkane Dehalogenase at pH6.2 and pH8.2 and Implications for the Reaction Mechanism. J. Mol. Biol. 1993, 232, 856–872. [Google Scholar] [CrossRef] [Green Version]
- Schanstra, J.P.; Ridder, I.S.; Heimeriks, G.J.; Rink, R.; Poelarends, G.J.; Kalk, K.H.; Dijkstra, B.W.; Janssen, D.B. Kinetic characterization and X-ray structure of a mutant of haloalkane dehalogenase with higher catalytic activity and modified substrate range. Biochemistry 1996, 35, 13186–13195. [Google Scholar] [CrossRef] [Green Version]
- Damborský, J.; Kutý, M.; Němec, M.; Koca, J. A molecular modeling study of the catalytic mechanism of haloalkane dehalogenase: 1. quantum chemical study of the first reaction step. J. Chem. Inf. Comput. Sci. 1997, 37, 562–568. [Google Scholar] [CrossRef]
- Kutý, M.; Damborský, J.; Prokop, M.; Koca, J. A molecular modeling study of the catalytic mechanism of haloalkane dehalogenase. 2. quantum chemical study of complete reaction mechanism. J. Chem. Inf. Comput. Sci. 1998, 38, 736–741. [Google Scholar] [CrossRef]
- Shurki, A.; Štrajbl, M.; Villa, J.; Warshel, A. How much do enzymes really gain by restraining their reacting fragments? J. Am. Chem. Soc. 2002, 124, 4097–4107. [Google Scholar] [CrossRef] [PubMed]
- Devi-Kesavan, L.S.; Gao, J. Combined QM/MM study of the mechanism and kinetic isotope effect of the nucleophilic substitution reaction in haloalkane dehalogenase. J. Am. Chem. Soc. 2003, 125, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Hur, S.; Kahn, K.; Bruice, T.C. Comparison of formation of reactive conformers for the SN2 displacements by in water and by in a haloalkane dehalogenase. Proc. Natl. Acad. Sci. USA 2003, 100, 2215–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschueren, K.H.G.; Seljée, F.; Rozeboom, H.J.; Kalk, K.H.; Dijkstra, B.W. Crystallographic analysis of the catalytic mechanism of haloalkane dehalogenase. Nature 1993, 363, 693–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosta, E.; Klähn, M.; Warshel, A. Towards accurate ab initio QM/MM calculations of free-energy profiles of enzymatic reactions. J. Phys. Chem. B 2006, 110, 2934–2941. [Google Scholar] [CrossRef]
- Lau, E.Y.; Kahn, K.; Bash, P.A.; Bruice, T.C. The importance of reactant positioning in enzyme catalysis: A hybrid quantum mechanics/molecular mechanics study of a haloalkane dehalogenase. Proc. Natl. Acad. Sci. USA 2000, 97, 9937–9942. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wang, D.; Xu, Y. A new, double-inversion mechanism of the F− + CH3Cl SN2 reaction in aqueous solution. Phys. Chem. Chem. Phys. 2016, 18, 31895–31903. [Google Scholar] [CrossRef]
- Wang, T.; Yin, T.; Wang, T.; Valiev, M. Hybrid Quantum Mechanical and Molecular Mechanics Study of the SN2 Reaction of CCl4 + OH− in Aqueous Solution: The Potential of Mean Force, Reaction Energetics, and Rate Constants. J. Phys. Chem. A 2012, 116, 2371–2376. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Liu, S.; Zha, S.; Cheng, D.; Studt, F.; Henkelman, G.; Gong, J. Theory-guided design of catalytic materials using scaling relationships and reactivity descriptors. Nat. Rev. Mater. 2019, 4, 792–804. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, C.Y.; Zhang, D.; Gong, L.; Zhu, Y.; Zhao, Z.; Xu, Q.; Li, H.; Xia, Z. Guiding Principles for Designing Highly Efficient Metal-Free Carbon Catalysts. Adv. Mater. 2019, 31, 1805252. [Google Scholar] [CrossRef]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. GAUSSIAN 16, Revision A. 03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Levine, R.D. Molecular Reaction Dynamics; Cambridge University Press: New York, NY, USA, 2005; p. 427. [Google Scholar]
- Xu, Y.; Wang, T.; Wang, D. A multilayered-representation quantum mechanical/molecular mechanics study of the SN2 reaction of CH3Br + OH− in aqueous solution. J. Chem. Phys. 2012, 137, 184501. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, J.; Wang, D. Investigation of the CH3Cl + CN− reaction in water: Multilevel quantum mechanics/molecular mechanics study. J. Chem. Phys. 2015, 142, 244505. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Exploring the limit of accuracy of the global hybrid meta density functional for main-group thermochemistry, kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, J.R. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Yang, W. Free energy calculation on enzyme reactions with an efficient iterative procedure to determine minimum energy paths on a combined ab initio QM/MM potential energy surface. J. Chem. Phys. 2000, 112, 3483–3492. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Aprab, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 2 | 1 | 0 | Water | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RC3 | TS3 | RC2 | TS2 | RC1 | TS1 | RC0 | TS0 | RCsol | TSsol | |
| O1 | −0.88 | −0.80 | −0.87 | −0.81 | −0.87 | −0.86 | −0.86 | −0.85 | −0.80 | −0.55 |
| C2H4Cl | 0.33 | 0.53 | 0.34 | 0.52 | 0.33 | 0.52 | 0.31 | 0.47 | 0.27 | 0.42 |
| Cl1 | −0.34 | −0.70 | −0.36 | −0.68 | −0.35 | −0.63 | −0.37 | −0.58 | −0.28 | −0.46 |
| H1 | 0.26 | 0.27 | 0.28 | 0.29 | 0.27 | 0.32 | ||||
| H2 | 0.30 | 0.31 | 0.29 | 0.31 | ||||||
| H3 | 0.11 | 0.09 | ||||||||
| CH3CO2 | −0.94 | −0.73 | −0.94 | −0.77 | −0.94 | −0.85 | −0.94 | −0.89 | −0.66 | −0.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, X.; Li, C.; Gao, D.; Wang, D. Catalytic Descriptors to Investigate Catalytic Power in the Reaction of Haloalkane Dehalogenase Enzyme with 1,2-Dichloroethane. Int. J. Mol. Sci. 2021, 22, 5854. https://doi.org/10.3390/ijms22115854
Xin X, Li C, Gao D, Wang D. Catalytic Descriptors to Investigate Catalytic Power in the Reaction of Haloalkane Dehalogenase Enzyme with 1,2-Dichloroethane. International Journal of Molecular Sciences. 2021; 22(11):5854. https://doi.org/10.3390/ijms22115854
Chicago/Turabian StyleXin, Xin, Chen Li, Delu Gao, and Dunyou Wang. 2021. "Catalytic Descriptors to Investigate Catalytic Power in the Reaction of Haloalkane Dehalogenase Enzyme with 1,2-Dichloroethane" International Journal of Molecular Sciences 22, no. 11: 5854. https://doi.org/10.3390/ijms22115854