
Smooth Muscle Specific Ablation of CXCL12 in Mice Downregulates CXCR7 Associated with Defective Coronary Arteries and Cardiac Hypertrophy

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
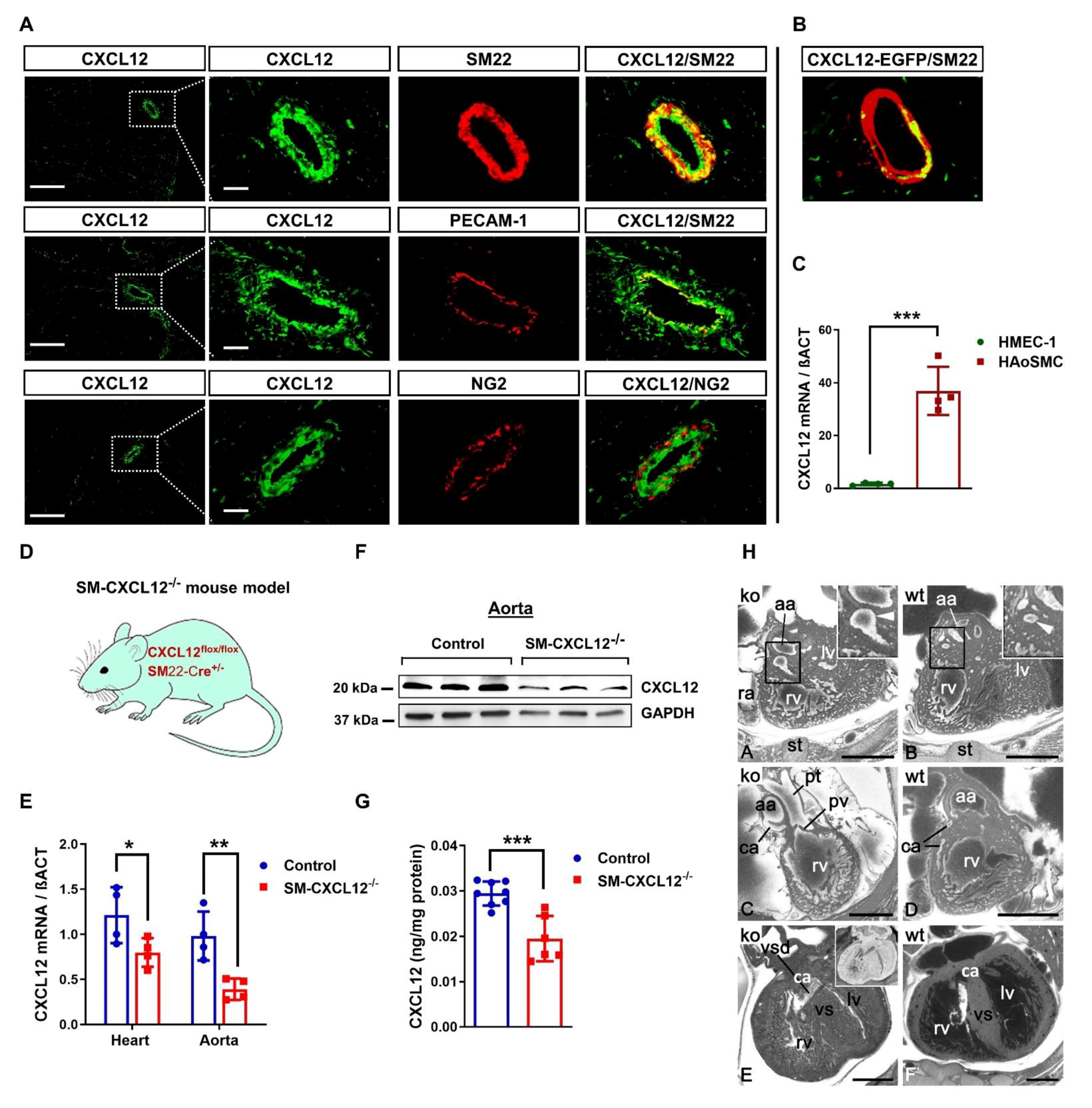
2.1. CXCL12 Is Predominantly Expressed in Vascular Smooth Muscle Cells
2.2. Loss of CXCL12 in SMCs Conferred Substantial Perinatal Mortality and Cardiovascular Abnormalities
2.3. Smooth Muscle-Specific CXCL12 KO Mutants Developed Severe Cardiac Hypertrophy
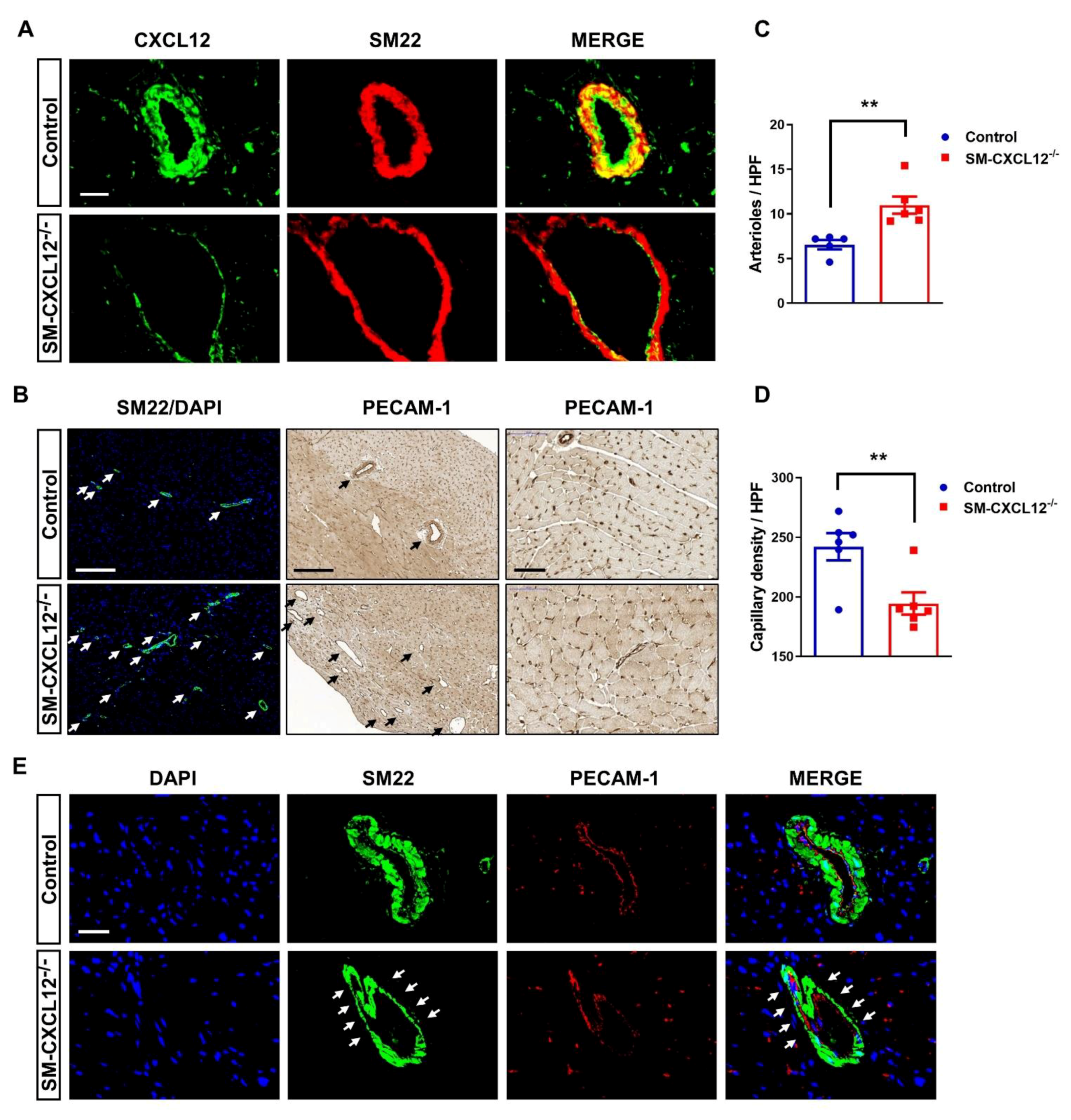
2.4. SM-CXCL12−/− Hearts Revealed Increased Cardiac Fibrosis and Abnormal Coronary Arteries
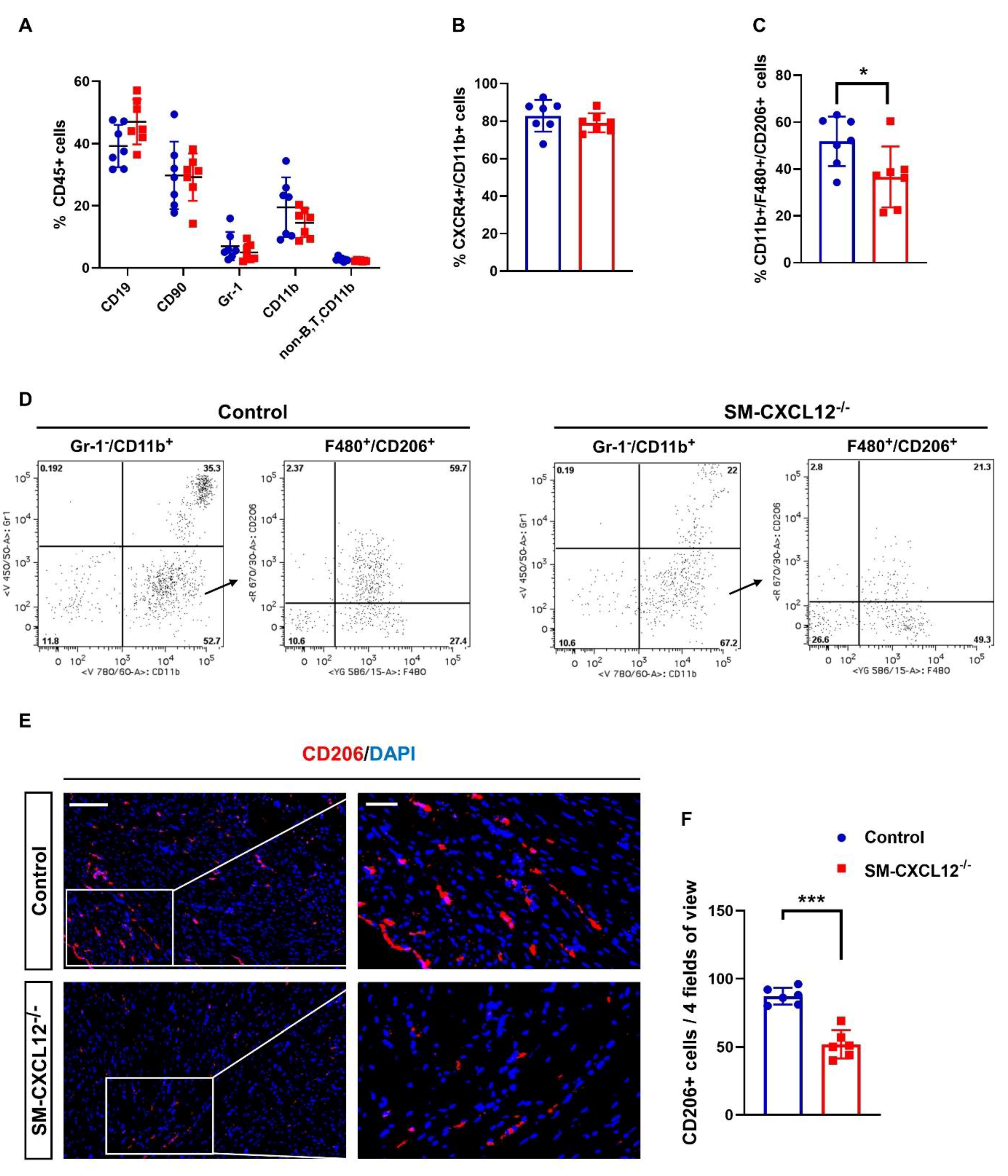
2.5. RNAseq Analysis Revealed Increased Signaling for Hypertrophic Cardiomyopathy and Downregulation of M2 Macrophage Markers RETNLA and CXCL14
2.6. SM-CXCL12−/− Mouse Hearts Showed Decreased M2-Like Macrophages (CD206 + )
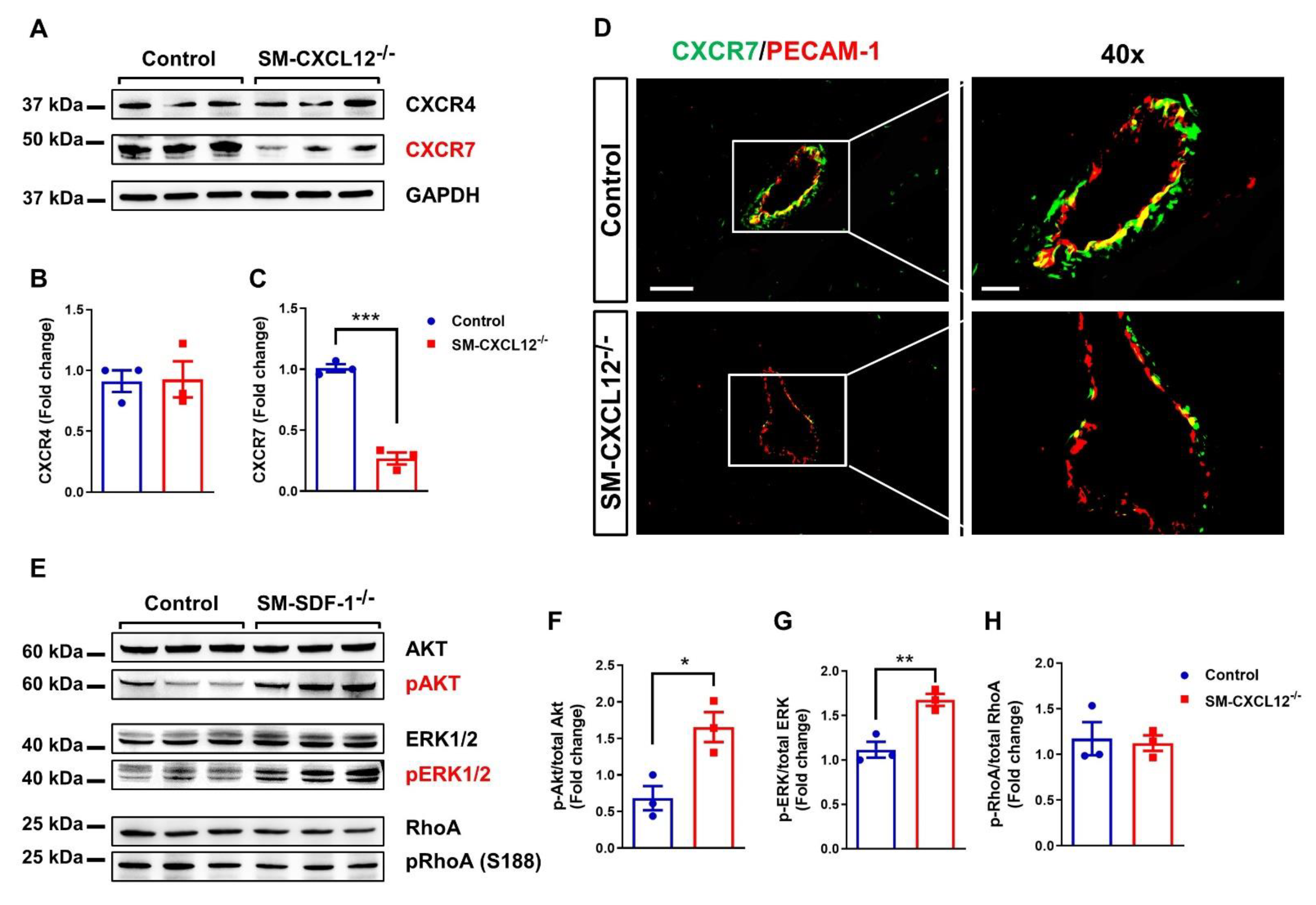
2.7. Downregulation of the CXCL12 Co-Receptor CXCR7 in SM-CXCL12−/− Hearts
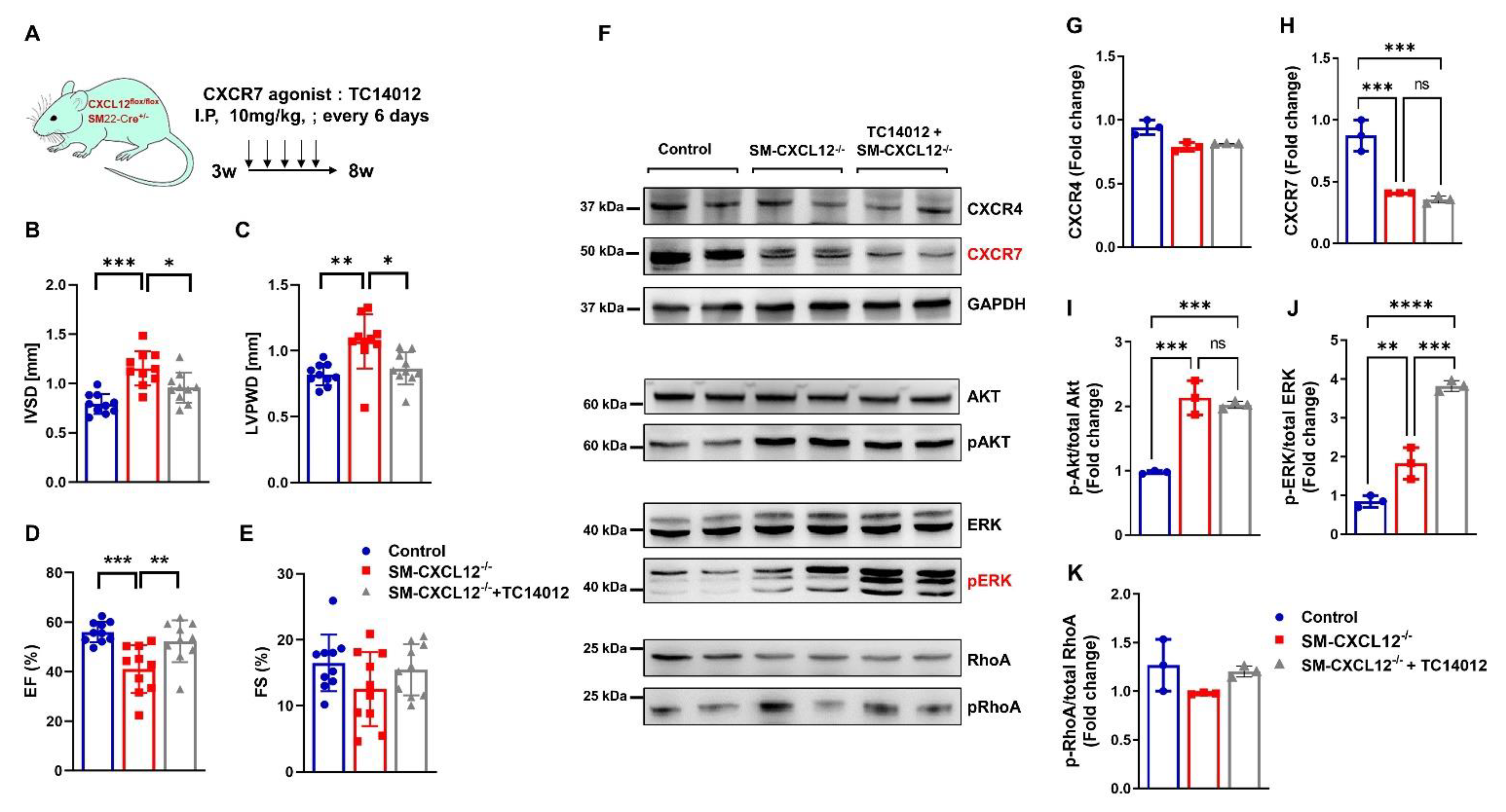
2.8. CXCR7 Agonist Treatment Attenuated Hypertrophic Remodeling in SM-CXCL12−/− Mice Associated with Activation of pERK
3. Discussion
4. Materials and Methods
4.1. Mouse Strains
4.2. High-Resolution Episcopic Microscopy (HREM)
4.3. Echocardiography
4.4. Quantitative RT-PCR in Heart Tissue and Human Cells
4.5. Western Blot
4.6. CXCL12 ELISA
4.7. Histology and Immunostaining
4.8. Flow Cytometric Analyses of Spleen, BM and Heart
4.9. RNA Sequencing
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghadge, S.K.; Muhlstedt, S.; Ozcelik, C.; Bader, M. SDF-1alpha as a therapeutic stem cell homing factor in myocardial infarction. Pharmacol. Ther. 2011, 129, 97–108. [Google Scholar] [CrossRef]
- Doring, Y.; Pawig, L.; Weber, C.; Noels, H. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front. Physiol. 2014, 5, 212. [Google Scholar] [CrossRef]
- Zaruba, M.M.; Franz, W.M. Role of the SDF-1-CXCR4 axis in stem cell-based therapies for ischemic cardiomyopathy. Expert Opin Biol Ther 2010, 10, 321–335. [Google Scholar] [CrossRef]
- Sierro, F.; Biben, C.; Martinez-Munoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M.; et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Crawford, D.; Tsuchihashi, T.; Behrens, T.W.; Srivastava, D. The chemokine receptor CXCR7 functions to regulate cardiac valve remodeling. Dev. Dyn. 2011, 240, 384–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.; Hu, S.; Chen, H.; Bu, D.; Zhu, L.; Xu, C.; Chu, F.; Huo, X.; Tang, Y.; Sun, X.; et al. Loss of Endothelial CXCR7 Impairs Vascular Homeostasis and Cardiac Remodeling After Myocardial Infarction: Implications for Cardiovascular Drug Discovery. Circulation 2017, 135, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Domanska, U.M.; Kruizinga, R.C.; Nagengast, W.B.; Timmer-Bosscha, H.; Huls, G.; de Vries, E.G.; Walenkamp, A.M. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur. J. Cancer 2013, 49, 219–230. [Google Scholar] [CrossRef]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Hirota, S.; Iizasa, H.; Yoshida, H.; Kawabata, K.; Kataoka, Y.; Kitamura, Y.; Matsushima, K.; Yoshida, N.; Nishikawa, S.; et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 1998, 393, 591–594. [Google Scholar] [CrossRef]
- Gerrits, H.; van Ingen Schenau, D.S.; Bakker, N.E.; van Disseldorp, A.J.; Strik, A.; Hermens, L.S.; Koenen, T.B.; Krajnc-Franken, M.A.; Gossen, J.A. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis 2008, 46, 235–245. [Google Scholar] [CrossRef]
- McGrath, K.E.; Koniski, A.D.; Maltby, K.M.; McGann, J.K.; Palis, J. Embryonic expression and function of the chemokine SDF-1 and its receptor, CXCR4. Dev. Biol. 1999, 213, 442–456. [Google Scholar] [CrossRef] [Green Version]
- Farouk, S.S.; Rader, D.J.; Reilly, M.P.; Mehta, N.N. CXCL12: A new player in coronary disease identified through human genetics. Trends Cardiovasc. Med. 2010, 20, 204–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, E.S.; Miller, L.; Patel, A.N.; Anderson, R.D.; Mendelsohn, F.O.; Traverse, J.; Silver, K.H.; Shin, J.; Ewald, G.; Farr, M.J.; et al. Changes in ventricular remodelling and clinical status during the year following a single administration of stromal cell-derived factor-1 non-viral gene therapy in chronic ischaemic heart failure patients: The STOP-HF randomized Phase II trial. Eur. Heart J. 2015, 36, 2228–2238. [Google Scholar] [CrossRef]
- Penn, M.S.; Mendelsohn, F.O.; Schaer, G.L.; Sherman, W.; Farr, M.; Pastore, J.; Rouy, D.; Clemens, R.; Aras, R.; Losordo, D.W. An open-label dose escalation study to evaluate the safety of administration of nonviral stromal cell-derived factor-1 plasmid to treat symptomatic ischemic heart failure. Circ. Res. 2013, 112, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askari, A.T.; Unzek, S.; Popovic, Z.B.; Goldman, C.K.; Forudi, F.; Kiedrowski, M.; Rovner, A.; Ellis, S.G.; Thomas, J.D.; DiCorleto, P.E.; et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 2003, 362, 697–703. [Google Scholar] [CrossRef]
- Zaruba, M.M.; Theiss, H.D.; Vallaster, M.; Mehl, U.; Brunner, S.; David, R.; Fischer, R.; Krieg, L.; Hirsch, E.; Huber, B.; et al. Synergy between CD26/DPP-IV inhibition and G-CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell 2009, 4, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Dai, S.; Wu, W.J.; Tan, W.; Zhu, X.; Mu, J.; Guo, Y.; Bolli, R.; Rokosh, G. Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: Role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation 2007, 116, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.; Fish, J.E.; White, M.D.; Yu, S.; Smyth, J.W.; Shaw, R.M.; DiMaio, J.M.; Srivastava, D. Stromal cell-derived factor-1alpha is cardioprotective after myocardial infarction. Circulation 2008, 117, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.D.; Huang, Y.; Liu, D.; Hickey, R.; Krause, D.S.; Giordano, F.J. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation 2004, 110, 3300–3305. [Google Scholar] [CrossRef] [Green Version]
- Zaruba, M.M.; Zhu, W.; Soonpaa, M.H.; Reuter, S.; Franz, W.M.; Field, L.J. Granulocyte colony-stimulating factor treatment plus dipeptidylpeptidase-IV inhibition augments myocardial regeneration in mice expressing cyclin D2 in adult cardiomyocytes. Eur. Heart J. 2012, 33, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef]
- Seo, J.; Kim, Y.O.; Jo, I. Differential expression of stromal cell-derived factor 1 in human brain microvascular endothelial cells and pericytes involves histone modifications. Biochem. Biophys. Res. Commun. 2009, 382, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Nemenoff, R.A.; Horita, H.; Ostriker, A.C.; Furgeson, S.B.; Simpson, P.A.; VanPutten, V.; Crossno, J.; Offermanns, S.; Weiser-Evans, M.C. SDF-1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1300–1308. [Google Scholar] [CrossRef] [Green Version]
- Muhlstedt, S.; Ghadge, S.K.; Duchene, J.; Qadri, F.; Jarve, A.; Vilianovich, L.; Popova, E.; Pohlmann, A.; Niendorf, T.; Boye, P.; et al. Cardiomyocyte-derived CXCL12 is not involved in cardiogenesis but plays a crucial role in myocardial infarction. J. Mol. Med. 2016, 94, 1005–1014. [Google Scholar] [CrossRef]
- Ivins, S.; Chappell, J.; Vernay, B.; Suntharalingham, J.; Martineau, A.; Mohun, T.J.; Scambler, P.J. The CXCL12/CXCR4 Axis Plays a Critical Role in Coronary Artery Development. Dev. Cell 2015, 33, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Cavallero, S.; Shen, H.; Yi, C.; Lien, C.L.; Kumar, S.R.; Sucov, H.M. CXCL12 Signaling Is Essential for Maturation of the Ventricular Coronary Endothelial Plexus and Establishment of Functional Coronary Circulation. Dev. Cell 2015, 33, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, Y.; Noels, H.; van der Vorst, E.P.C.; Neideck, C.; Egea, V.; Drechsler, M.; Mandl, M.; Pawig, L.; Jansen, Y.; Schröder, K.; et al. Vascular CXCR4 Limits Atherosclerosis by Maintaining Arterial Integrity: Evidence From Mouse and Human Studies. Circulation 2017, 136, 388–403. [Google Scholar] [CrossRef]
- Cheng, W.L.; She, Z.G.; Qin, J.J.; Guo, J.H.; Gong, F.H.; Zhang, P.; Fang, C.; Tian, S.; Zhu, X.Y.; Gong, J.; et al. Interferon Regulatory Factor 4 Inhibits Neointima Formation by Engaging Krüppel-Like Factor 4 Signaling. Circulation 2017, 136, 1412–1433. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, R.; Saddouk, F.Z.; Carrao, A.C.; Krause, D.S.; Greif, D.M.; Martin, K.A. Promoters to Study Vascular Smooth Muscle. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 603–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; Goldstein, B.A.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.E.; et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, N.N.; Li, M.; William, D.; Khera, A.V.; DerOhannessian, S.; Qu, L.; Ferguson, J.F.; McLaughlin, C.; Shaikh, L.H.; Shah, R.; et al. The novel atherosclerosis locus at 10q11 regulates plasma CXCL12 levels. Eur. Heart J. 2011, 32, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, S.; Liu, C.; Aviv, A.; Ho, J.E.; Courchesne, P.; Muntendam, P.; Larson, M.G.; Cheng, S.; Wang, T.J.; Mehta, N.N.; et al. Stromal cell-derived factor 1 as a biomarker of heart failure and mortality risk. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2100–2105. [Google Scholar] [CrossRef] [Green Version]
- Ghasemzadeh, N.; Hritani, A.W.; De Staercke, C.; Eapen, D.J.; Veledar, E.; Al Kassem, H.; Khayata, M.; Zafari, A.M.; Sperling, L.; Hooper, C.; et al. Plasma stromal cell-derived factor 1alpha/CXCL12 level predicts long-term adverse cardiovascular outcomes in patients with coronary artery disease. Atherosclerosis 2015, 238, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Ara, T.; Tokoyoda, K.; Okamoto, R.; Koni, P.A.; Nagasawa, T. The role of CXCL12 in the organ-specific process of artery formation. Blood 2005, 105, 3155–3161. [Google Scholar] [CrossRef] [Green Version]
- Segers, V.F.; Revin, V.; Wu, W.; Qiu, H.; Yan, Z.; Lee, R.T.; Sandrasagra, A. Protease-resistant stromal cell-derived factor-1 for the treatment of experimental peripheral artery disease. Circulation 2011, 123, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Wang, J.; Song, H.; Huang, Y.; Yang, J.; Kong, X.; Guo, L.; Zheng, F.; Zhang, L. Adenovirus-mediated stromal cell-derived factor-1 alpha gene transfer improves cardiac structure and function after experimental myocardial infarction through angiogenic and antifibrotic actions. Mol. Biol. Rep. 2010, 37, 1957–1969. [Google Scholar] [CrossRef] [Green Version]
- LaRocca, T.J.; Altman, P.; Jarrah, A.A.; Gordon, R.; Wang, E.; Hadri, L.; Burke, M.W.; Haddad, G.E.; Hajjar, R.J.; Tarzami, S.T. CXCR4 Cardiac Specific Knockout Mice Develop a Progressive Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2267. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.R.; Jarrah, A.A.; Benard, L.; Chen, J.; Schwarzkopf, M.; Hadri, L.; Tarzami, S.T. Deletion of CXCR4 in cardiomyocytes exacerbates cardiac dysfunction following isoproterenol administration. Gene Ther. 2014, 21, 496–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Ellims, A.H.; Beale, A.L.; Taylor, A.J.; Murphy, A.; Dart, A.M. Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy. Am. J. Transl. Res. 2017, 9, 5063–5073. [Google Scholar]
- Fang, L.; Beale, A.; Ellims, A.H.; Moore, X.L.; Ling, L.H.; Taylor, A.J.; Chin-Dusting, J.; Dart, A.M. Associations between fibrocytes and postcontrast myocardial T1 times in hypertrophic cardiomyopathy. J. Am. Heart Assoc. 2013, 2, e000270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.; Wu, Z.L.; Gan, Z.; Gui, P.; Yao, S.L. CXCL14 Overexpression Attenuates Sepsis-Associated Acute Kidney Injury by Inhibiting Proinflammatory Cytokine Production. Mediat. Inflamm. 2020, 2020, 2431705. [Google Scholar] [CrossRef]
- Ben-Mordechai, T.; Holbova, R.; Landa-Rouben, N.; Harel-Adar, T.; Feinberg, M.S.; Abd Elrahman, I.; Blum, G.; Epstein, F.H.; Silman, Z.; Cohen, S.; et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J. Am. Coll. Cardiol. 2013, 62, 1890–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblond, A.L.; Klinkert, K.; Martin, K.; Turner, E.C.; Kumar, A.H.; Browne, T.; Caplice, N.M. Systemic and Cardiac Depletion of M2 Macrophage through CSF-1R Signaling Inhibition Alters Cardiac Function Post Myocardial Infarction. PLoS ONE 2015, 10, e0137515. [Google Scholar] [CrossRef]
- Lech, M.; Anders, H.J. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta 2013, 1832, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Agudelo, J.S.; Camara, N.O. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dong, H.; Jiang, L.; Li, Z.; Ma, X. Bleomycin inhibits proliferation and induces apoptosis in TPC-1 cells through reversing M2-macrophages polarization. Oncol. Lett. 2018, 16, 3858–3866. [Google Scholar] [CrossRef] [Green Version]
- Ghadge, S.K.; Messner, M.; Van Pham, T.; Doppelhammer, M.; Petry, A.; Gorlach, A.; Husse, B.; Franz, W.M.; Zaruba, M.M. Prolyl-hydroxylase inhibition induces SDF-1 associated with increased CXCR4+/CD11b+ subpopulations and cardiac repair. J. Mol. Med. 2017, 95, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luker, K.E.; Steele, J.M.; Mihalko, L.A.; Ray, P.; Luker, G.D. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene 2010, 29, 4599–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the "decoy" receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Tong, X.; Xia, W.; Chen, L.; Zhang, X.; Yu, B.; Yang, Z.; Tao, J. CXCR7/p-ERK-Signaling Is a Novel Target for Therapeutic Vasculogenesis in Patients with Coronary Artery Disease. PLoS ONE 2016, 11, e0161255. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, M.; Harada, M.; Nomura, S.; Ko, T.; Ikeda, Y.; Guo, J.; Bujo, S.; Yanagisawa-Murakami, H.; Satoh, M.; Yamada, S.; et al. CXCR7 ameliorates myocardial infarction as a β-arrestin-biased receptor. Sci. Rep. 2021, 11, 3426. [Google Scholar] [CrossRef]
- Li, X.; Zhu, M.; Penfold, M.E.; Koenen, R.R.; Thiemann, A.; Heyll, K.; Akhtar, S.; Koyadan, S.; Wu, Z.; Gremse, F.; et al. Activation of CXCR7 limits atherosclerosis and improves hyperlipidemia by increasing cholesterol uptake in adipose tissue. Circulation 2014, 129, 1244–1253. [Google Scholar] [CrossRef] [Green Version]
- Gravel, S.; Malouf, C.; Boulais, P.E.; Berchiche, Y.A.; Oishi, S.; Fujii, N.; Leduc, R.; Sinnett, D.; Heveker, N. The peptidomimetic CXCR4 antagonist TC14012 recruits beta-arrestin to CXCR7: Roles of receptor domains. J. Biol. Chem. 2010, 285, 37939–37943. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Tripathi, V.; Ahmad, M.; Nath, N.; Mir, R.A.; Chauhan, S.S.; Luthra, K. CXCR7 mediated Giα independent activation of ERK and Akt promotes cell survival and chemotaxis in T cells. Cell Immunol. 2012, 272, 230–241. [Google Scholar] [CrossRef]
- Cao, Z.; Lis, R.; Ginsberg, M.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Fong, G.H.; Sakmar, T.P.; Rafii, S.; Ding, B.S. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 2016, 22, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dembowsky, K.; Chevalier, E.; Stüve, P.; Korf-Klingebiel, M.; Lochner, M.; Napp, L.C.; Frank, H.; Brinkmann, E.; Kanwischer, A.; et al. C-X-C Motif Chemokine Receptor 4 Blockade Promotes Tissue Repair After Myocardial Infarction by Enhancing Regulatory T Cell Mobilization and Immune-Regulatory Function. Circulation 2019, 139, 1798–1812. [Google Scholar] [CrossRef]
- Chu, P.Y.; Zatta, A.; Kiriazis, H.; Chin-Dusting, J.; Du, X.J.; Marshall, T.; Kaye, D.M. CXCR4 antagonism attenuates the cardiorenal consequences of mineralocorticoid excess. Circ. Heart Fail. 2011, 4, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Holtwick, R.; Gotthardt, M.; Skryabin, B.; Steinmetz, M.; Potthast, R.; Zetsche, B.; Hammer, R.E.; Herz, J.; Kuhn, M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc. Natl. Acad. Sci. USA 2002, 99, 7142–7147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weninger, W.J.; Geyer, S.H.; Mohun, T.J.; Rasskin-Gutman, D.; Matsui, T.; Ribeiro, I.; Costa Lda, F.; Izpisúa-Belmonte, J.C.; Müller, G.B. High-resolution episcopic microscopy: A rapid technique for high detailed 3D analysis of gene activity in the context of tissue architecture and morphology. Anat. Embryol. 2006, 211, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Geyer, S.H.; Maurer-Gesek, B.; Reissig, L.F.; Weninger, W.J. High-resolution Episcopic Microscopy (HREM) - Simple and Robust Protocols for Processing and Visualizing Organic Materials. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohun, T.J.; Weninger, W.J. Generation of volume data by episcopic three-dimensional imaging of embryos. Cold Spring Harb. Protoc. 2012, 2012, 681–682. [Google Scholar] [CrossRef] [Green Version]
- Mohun, T.J.; Weninger, W.J. Embedding embryos for high-resolution episcopic microscopy (HREM). Cold Spring Harb. Protoc. 2012, 2012, 678–680. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.R.; Chandran, A.; Rosenthal, N.A.; Godwin, J.W. Isolation and analysis of single cells from the mouse heart. J. Immunol. Methods 2013, 393, 74–80. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghadge, S.K.; Messner, M.; Seiringer, H.; Maurer, T.; Staggl, S.; Zeller, T.; Müller, C.; Börnigen, D.; Weninger, W.J.; Geyer, S.H.; et al. Smooth Muscle Specific Ablation of CXCL12 in Mice Downregulates CXCR7 Associated with Defective Coronary Arteries and Cardiac Hypertrophy. Int. J. Mol. Sci. 2021, 22, 5908. https://doi.org/10.3390/ijms22115908
Ghadge SK, Messner M, Seiringer H, Maurer T, Staggl S, Zeller T, Müller C, Börnigen D, Weninger WJ, Geyer SH, et al. Smooth Muscle Specific Ablation of CXCL12 in Mice Downregulates CXCR7 Associated with Defective Coronary Arteries and Cardiac Hypertrophy. International Journal of Molecular Sciences. 2021; 22(11):5908. https://doi.org/10.3390/ijms22115908
Chicago/Turabian StyleGhadge, Santhosh Kumar, Moritz Messner, Herbert Seiringer, Thomas Maurer, Simon Staggl, Tanja Zeller, Christian Müller, Daniela Börnigen, Wolfgang J. Weninger, Stefan H. Geyer, and et al. 2021. "Smooth Muscle Specific Ablation of CXCL12 in Mice Downregulates CXCR7 Associated with Defective Coronary Arteries and Cardiac Hypertrophy" International Journal of Molecular Sciences 22, no. 11: 5908. https://doi.org/10.3390/ijms22115908
APA StyleGhadge, S. K., Messner, M., Seiringer, H., Maurer, T., Staggl, S., Zeller, T., Müller, C., Börnigen, D., Weninger, W. J., Geyer, S. H., Sopper, S., Krogsdam, A., Pölzl, G., Bauer, A., & Zaruba, M.-M. (2021). Smooth Muscle Specific Ablation of CXCL12 in Mice Downregulates CXCR7 Associated with Defective Coronary Arteries and Cardiac Hypertrophy. International Journal of Molecular Sciences, 22(11), 5908. https://doi.org/10.3390/ijms22115908