
Role of Satb1 and Satb2 Transcription Factors in the Glutamate Receptors Expression and Ca2+ Signaling in the Cortical Neurons In Vitro

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
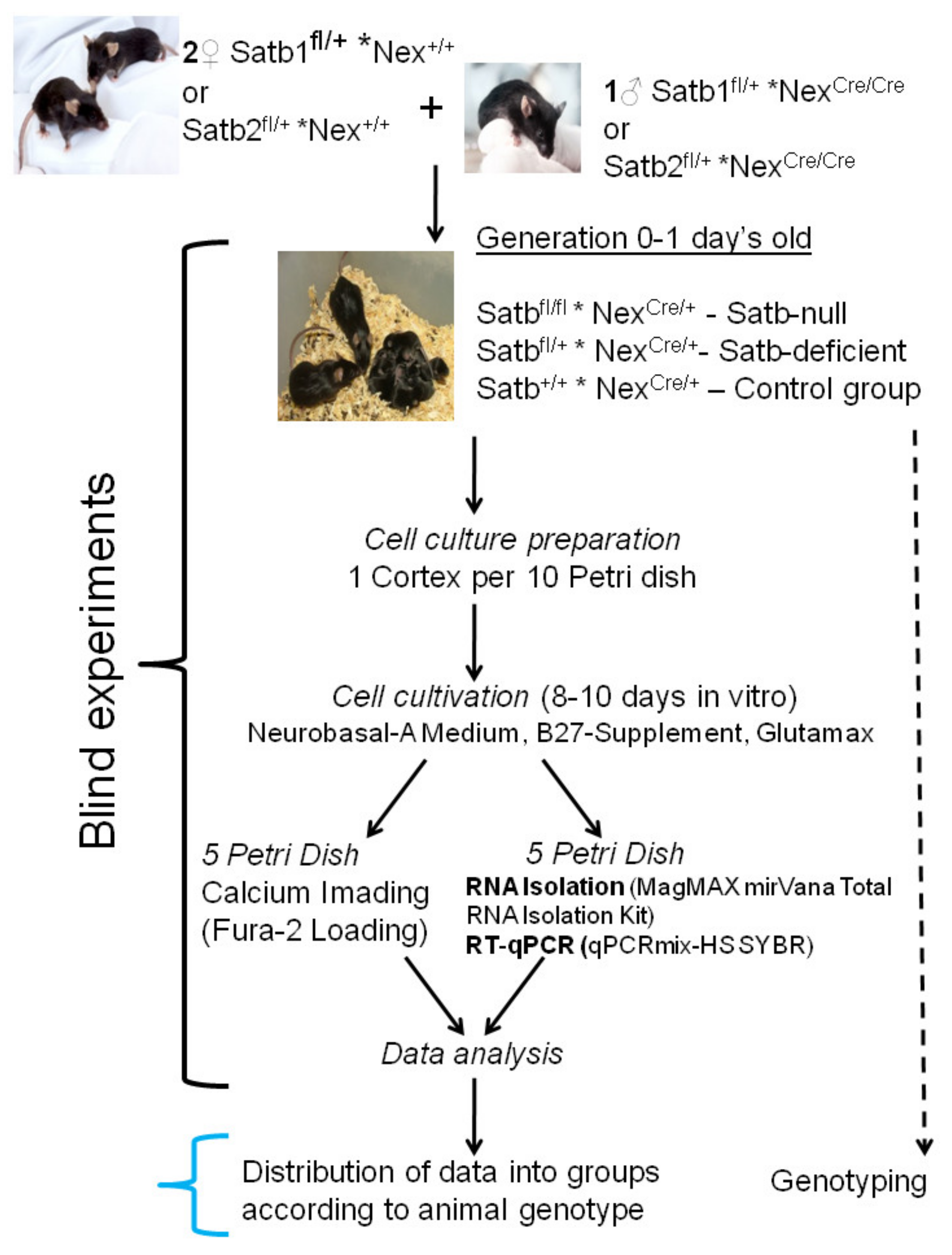
2.1. Animals
2.2. Cell Culture Preparation
2.2.1. Immunocytochemical Method
2.2.2. Assessment of Cell Viability and Apoptosis
2.3. Fluorescent Ca2+ Measurements
2.4. Extraction of RNA
2.5. Real-Time Polymerase Chain Reaction (RT-qPCR)
2.6. Statistical Analysis
3. Results
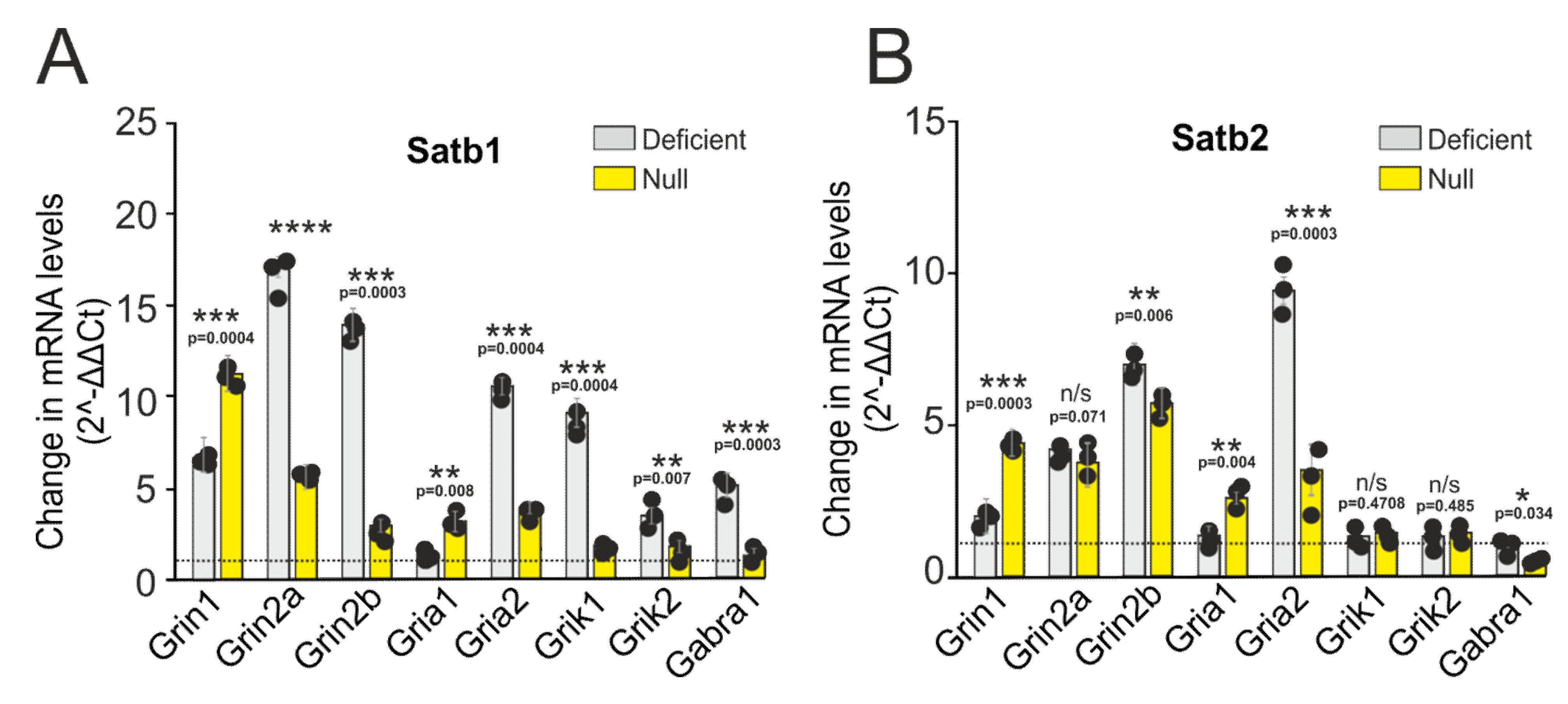
3.1. The Effects of Satb1 and Satb2 Deletions on the Expression of Neurotransmission-Regulating Genes
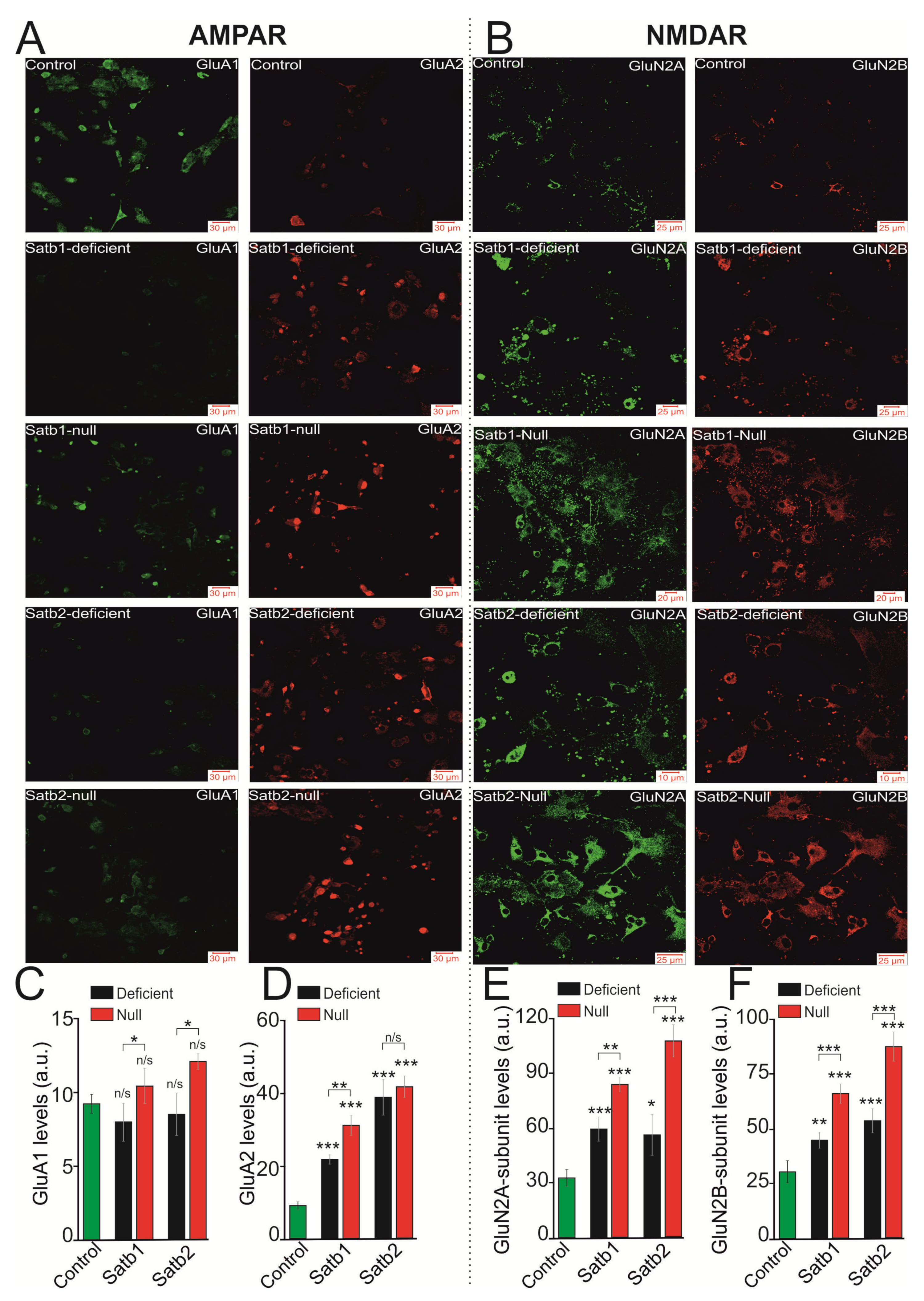
3.2. Deletions in Satb1 and Satb2 Cause an Increase in the Sensitivity of NMDA Receptor to the Agonist and Contribute to Higher Amplitude of Ca2+-Signals Generated by Cortical Neurons
3.3. Deletions in Satb1 and Satb2 Reduce the Sensitivity of the AMPA Receptors of the Cortical Neurons to the Agonist
3.4. Deletions in Satb1 and Satb2 have Different Effects on the Sensitivity of KA Receptors to a Selective Agonist
3.5. Deletions in Satb1 and Satb2 Suppress the Development of the Inhibitory System of Neurotransmission in the Cortical Neurons In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Santiago, C.; Bashaw, G.J. Transcription factors and effectors that regulate neuronal morphology. Development 2014, 141, 4667–4680. [Google Scholar] [CrossRef] [Green Version]
- West, A.E.; Griffith, E.C.; Greenberg, M.E. Regulation of transcription factors by neuronal activity. Nat. Rev. Neurosci. 2002, 3, 921–931. [Google Scholar] [CrossRef]
- Dickinson, L.A.; Joh, T.; Kohwi, Y.; Kohwi-Shigematsu, T. A tissue-specific MAR/SAR DNA-binding protein with unusual binding site recognition. Cell 1992, 70, 631–645. [Google Scholar] [CrossRef]
- Han, H.J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef]
- Cheng, C.; Lu, X.; Wang, G.; Zheng, L.; Shu, X.; Zhu, S.; Liu, K.; Wu, K.; Tong, Q. Expression of SATB1 and heparanase in gastric cancer and its relationship to clinicopathologic features. Apmis 2010, 118, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Takahara, M.; Oba, J.; Xie, L.; Chiba, T.; Takeuchi, S.; Tu, Y.; Nakahara, T.; Uchi, H.; Moroi, Y.; et al. Clinicopathologic and prognostic significance of SATB1 in cutaneous malignant melanoma. J. Dermatol. Sci. 2011, 64, 39–44. [Google Scholar] [CrossRef] [PubMed]
- McInnes, N.; Sadlon, T.J.; Brown, C.Y.; Pederson, S.; Beyer, M.; Schultze, J.L.; McColl, S.; Goodall, G.J.; Barry, S.C. FOXP3 and FOXP3-regulated microRNAs suppress SATB1 in breast cancer cells. Oncogene 2011, 31, 1045–1054. [Google Scholar] [CrossRef] [Green Version]
- Kohwi-Shigematsu, T.; Poterlowicz, K.; Ordinario, E.; Han, H.J.; Botchkarev, V.A.; Kohwi, Y. Genome organizing function of SATB1 in tumor progression. Semin. Cancer Biol. 2013, 23, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, R.; Pradhan, S.J.; Galande, S. Chromatin organizer SATB1 as a novel molecular target for cancer therapy. Curr. Drug Targets 2012, 13, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- De Belle, I.; Cai, S.; Kohwi-Shigematsu, T. The genomic sequences bound to special AT-rich sequence-binding protein 1 (SATB1) in vivo in Jurkat T cells are tightly associated with the nuclear matrix at the bases of the chromatin loops. J. Cell Biol. 1998, 141, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.P.; Bischof, O.; Purbey, P.K.; Notani, D.; Urlaub, H.; Dejean, A.; Galande, S. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat. Cell Biol. 2007, 9, 45–56. [Google Scholar] [PubMed] [Green Version]
- Huang, Y.; Zhang, L.; Song, N.N.; Hu, Z.L.; Chen, J.Y.; Ding, Y.Q. Distribution of Satb1 in the central nervous system of adult mice. Neurosci. Res. 2011, 71, 12–21. [Google Scholar] [CrossRef]
- Fogarty, M.; Grist, M.; Gelman, D.; Marín, O.; Pachnis, V.; Kessaris, N. Spatial genetic patterning of the embryonic neuroepi-thelium generates GABAergic interneuron diversity in the adult cortex. J. Neurosci. 2007, 27, 10935–10946. [Google Scholar] [CrossRef]
- Denaxa, M.; Kalaitzidou, M.; Garefalaki, A.; Achimastou, A.; Lasrado, R.; Maes, T.; Pachnis, V. Maturation-promoting activity of SATB1 in MGE-derived cortical interneurons. Cell Rep. 2012, 2, 1351–1362. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, J.D.; Yasui, D.H.; Niida, H.; Joh, T.; Loh, D.Y.; Kohwi-Shigematsu, T. The MAR-binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T-cell development. Genes Dev. 2000, 14, 521–535. [Google Scholar] [PubMed]
- FitzPatrick, D.R.; Carr, I.M.; McLaren, L.; Leek, J.P.; Wightman, P.; Williamson, K.; Gautier, P.; McGill, N.; Hayward, C.; Firth, H.; et al. Identification of SATB2 as the cleft palate gene on 2q32–q33. Hum. Mol. Genet. 2003, 12, 2491–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britanova, O.; Akopov, S.; Lukyanov, S.; Gruss, P.; Tarabykin, V. Novel transcription factor Satb2 interacts with matrix at-tachment region DNA elements in a tissue-specific manner and demonstrates cell-type-dependent expression in the developing mouse CNS. Eur. J. Neurosci. 2005, 21, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Dobreva, G.; Dambacher, J.; Grosschedl, R. SUMO modification of a novel MAR-binding protein, SATB2, modulates immuno-globulin mu gene expression. Genes. Dev. 2003, 17, 3048–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoyklang, P.; Suphapeetiporn, K.; Siriwan, P.; Desudchit, T.; Chaowanapanja, P.; Gahl, W.A.; Shotelersuk, V. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum. Mutat. 2007, 28, 732–738. [Google Scholar] [CrossRef]
- Aksoy-Aksel, A.; Zampa, F.; Schratt, G. MicroRNAs and synaptic plasticity–a mutual relationship. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, B.; Joilin, G.; Williams, J.M. Plasticity-related microRNA and their potential contribution to the maintenance of long-term potentiation. Front. Mol. Neurosci. 2015, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarate, Y.A.; Fish, J.L. SATB2-associated syndrome: Mechanisms, phenotype, and practical recommendations. Am. J. Med. Genet. A 2017, 173, 327–337. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Perry, H.; Ben-Omran, T.; Sellars, E.A.; Stein, Q.; Almureikhi, M.; Simmons, K.; Klein, O.; Fish, J.; Feingold, M.; et al. Further supporting evidence for the SATB2-associated syndrome found through whole exome sequencing. Am. J. Med. Genet. A 2015, 167A, 1026–1032. [Google Scholar] [CrossRef]
- Docker, D.; Schubach, M.; Menzel, M.; Munz, M.; Spaich, C.; Biskup, S.; Bartholdi, D. Further delineation of the SATB2 phenotype. Eur. J. Hum. Genet. 2014, 22, 1034–1039. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Turovsky, E.A.; Babaev, A.A.; Tarabykin, V.S.; Turovskaya, M.V. Sip1 mutation suppresses the resistance of cerebral cortex neurons to hypoxia through the disturbance of mechanisms of hypoxic preconditioning. Biochem. Suppl. Ser. A Membr. Cell Biol. 2017, 11, 330–337. [Google Scholar] [CrossRef]
- Turovsky, E.; Karagiannis, A.; Abdala, A.P.; Gourine, A.V. Impaired CO2 sensitivity of astrocytes in a mouse model of Rett syndrome. J. Physiol. 2015, 593, 3159–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Close, J.; Xu, H.; De Marco García, N.; Batista-Brito, R.; Rossignol, E.; Rudy, B.; Fishell, G. Satb1 is an activity-modulated tran-scription factor required for the terminal differentiation and connectivity of medial ganglionic eminence-derived cortical in-terneurons. J. Neurosci. 2012, 32, 17690–17705. [Google Scholar] [CrossRef] [Green Version]
- Goebbels, S.; Bormuth, I.; Bode, U.; Hermanson, O.; Schwab, M.H.; Nave, K.-A. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 2006, 44, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Leone, D.P.; Bateson, R.K.; Dobreva, G.; Kohwi, Y.; Kohwi-Shigematsu, T.; Grosschedl, R.; McConnell, S.-K. A network of genetic repression and derepression specifies projection fates in the developing neocortex. Proc. Natl. Acad. Sci. USA 2012, 109, 19071–19078. [Google Scholar] [CrossRef] [Green Version]
- Gaidin, S.G.; Turovskaya, M.V.; Gavrish, M.S.; Babaev, A.A.; Mal’tseva, V.N.; Blinova, E.V.; Turovsky, E.A. The selective BDNF overexpression in neurons protects neuroglial networks against OGD and glutamate-induced excitotoxicity. Int. J. Neurosci. 2020, 130, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Turovskaya, M.V.; Gaidin, S.G.; Vedunova, M.V.; Babaev, A.A.; Turovsky, E.A. BDNF overexpression enhances the precondi-tioning effect of brief episodes of hypoxia, promoting survival of GABAergic neurons. Neurosci Bull. 2020, 36, 733–760. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wang, J. Primary neuron-glia culture from rat cortex as a model to study neuroinflammation in CNS injuries or diseases. Bio Protoc. 2016, 6, e1788. [Google Scholar] [CrossRef]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D human induced pluripotent stem cell-derived cultures for neuro-degenerative disease modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, H.G.; Gray, T.; et al. Design and validation of a human brain endothelial mi-crovessel-on-a-chip open microfluidic model enabling advanced optical imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Schmid, I.; Uittenbogaart, C.; Jamieson, B.D. Live-cell assay for detection of apoptosis by dual-laser flow cytometry using Hoechst 33342 and 7-amino-actino-mycin D. Nat Protoc. 2007, 2, 187–190. [Google Scholar] [CrossRef]
- Gaidin, S.G.; Turovskaya, M.V.; Mal’tseva, V.N.; Zinchenko, V.P.; Blinova, E.V.; Turovsky, E.A. A complex neuroprotective effect of alpha-2-adrenergic receptor agonists in a model of cerebral ischemia–reoxygenation in vitro. Biochem. Suppl. Ser. A Membr. Cell Biol. 2019, 13, 319–333. [Google Scholar] [CrossRef]
- Zinchenko, V.P.; Turovskaya, M.V.; Teplov, I.Y.; Berezhnov, A.V.; Turovsky, E.A. The role of parvalbumin-containing inter-neurons in the regulation of spontaneous synchronous activity of brain neurons in culture. Biophysics 2016, 61, 85–93. [Google Scholar] [CrossRef]
- De Jonge, H.J.M.; Fehrmann, R.S.N.; De Bont, E.S.J.M.; Hofstra, R.M.W.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.E.; van der Zee, A.G.J.; te Meerman, G.J.; ter Elst, A. Evidence based selection of house-keeping genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Turovskaya, M.V.; Babaev, A.A.; Zinchenko, V.P.; Epifanova, E.A.; Borisova, E.V.; Tarabykin, V.S.; Turovsky, E.A. Sip-1 mu-tations cause disturbances in the activity of NMDA- and AMPA-, but not kainate receptors of neurons in the cerebral cortex. Neurosci. Lett. 2017, 650, 180–186. [Google Scholar] [CrossRef]
- Turovskaya, M.V.; Zinchenko, V.P.; Babaev, A.A.; Epifanova, E.A.; Tarabykin, V.S.; Turovsky, E.A. Mutation in the Sip1 tran-scription factor leads to a disturbance of the preconditioning of AMPA receptors by episodes of hypoxia in neurons of the cerebral cortex due to changes in their activity and subunit composition. The protective effects of interleukin-10. Arch. Biochem. Biophys. 2018, 654, 126–135. [Google Scholar]
- Sierra, A.; Martín-Suárez, S.; Valcárcel-Martín, R.; Pascual-Brazo, J.; Aelvoet, S.A.; Abiega, O.; Deudero, J.J.; Brewster, A.L.; Bernales, I.; Anderson, A.E.; et al. Neuronal hyperactivity accelerates depletion of neural stem cells and impairs hippocampal neurogenesis. Cell Stem Cell 2015, 16, 488–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamotis, M.A.; Tamberg, N.; Woo, Y.J.; Li, J.; Davy, B.; Kohwi-Shigematsu, T.; Kohwi, Y. Satb1 ablation alters temporal ex-pression of immediate early genes and reduces dendritic spine density during postnatal brain development. Mol. Cell. Biol. 2012, 32, 333–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Brichta, L.; Shin, W.; Jackson-Lewis, V.; Blesa, J.; Yap, E.L.; Walker, Z.; Zhang, J.; Roussarie, J.-P.; Alvarez, M.J.; Califano, A.; et al. Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat. Neurosci. 2015, 18, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, D.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons. Cell Stem Cell. 2019, 25, 514–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilde, K.L.; Levine, A.J.; Hinckley, C.A.; Hayashi, M.; Montgomery, J.M.; Gullo, M.; Driscoll, S.P.; Grosschedl, R.; Kohwi, Y.; Kohwi-Shigematsu, T.; et al. Satb2 is required for the development of a spinal exteroceptive microcircuit that modulates limb position. Neuron 2016, 91, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, N.; Shin, S.; Lee, K.-A. First Korean case of SATB2-associated 2q32-q33 microdeletion syndrome. Ann. Lab. Med. 2015, 35, 275–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, D.; Shimada, S.; Shimojima, K.; Sugawara, M.; Kawasaki, H.; Shigematu, H.; Takahashi, Y.; Inoue, Y.; Imai, K.; Yamamoto, T. Interstitial duplication of 2q32.1–q33.3 in a patient with epilepsy, developmental delay, and autistic behavior. Am. J. Med. Genet. A 2013, 161A, 1078–1084. [Google Scholar] [CrossRef]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Lobsiger, C.; Cleveland, D. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 2007, 10, 1355–1360. [Google Scholar] [CrossRef]
- Heneka, M.T.; Rodríguez, J.J.; Verkhratsky, A. Neuroglia in neurodegeneration. Brain Res. Rev. 2010, 63, 189–211. [Google Scholar] [CrossRef]
- Alcamo, E.A.; Chirivella, L.; Dautzenberg, M.; Dobreva, G.; Fariñas, I.; Grosschedl, R.; McConnell, S.K. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 2008, 57, 364–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britanova, O.; de Juan Romero, C.; Cheung, A.; Kwan, K.Y.; Schwark, M.; Gyorgy, A.; Vogel, T.; Akopov, S.; Mitkovski, M.; Agoston, D.; et al. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 2008, 57, 378–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaitner, C.; Reddy, C.; Abentung, A.; Whittle, N.; Rieder, D.; Delekate, A.; Korte, M.; Jain, G.; Fischer, A.; Sananbenesi, F.; et al. Satb2 determines miRNA expression and long-term memory in the adult central nervous system. eLife 2016, 5, e17361. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; You, Q.-L.; Zhang, S.-R.; Huang, W.-Y.; Zou, W.-J.; Jie, W.; Li, S.-J.; Liu, J.-H.; Lv, C.-Y.; Cong, J.; et al. Satb2 ablation impairs hippocampus-based long-term spatial memory and short-term working memory and immediate early genes (IEGs)-mediated hippocampal synaptic plasticity. Mol. Neurobiol. 2017, 1–16. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Peter, C.J.; Jiang, Y.; Roussos, P.; Vogel-Ciernia, A.; Shen, E.Y.; Mitchell, A.C.; Mao, W.; Whittle, C.; Dincer, A.; et al. Conserved high-er-order chromatin regulates NMDA receptor gene expression and cognition. Neuron 2014, 84, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Nestler, E.J.; Peñ a, C.J.; Kundakovic, M.; Mitchell, A.; Akbarian, S. Epigenetic basis of mental Illness. Neuroscientist 2016, 22, 447–463. [Google Scholar] [CrossRef]
- Rajarajan, P.; Gil, S.E.; Brennand, K.J.; Akbarian, S. Spatial genome organization and cognition. Nat. Rev. Neurosci. 2016, 17, 681–691. [Google Scholar] [CrossRef] [Green Version]
- García-Gallo, M.; Renart, J.; Díaz-Guerra, M. The NR1 subunit of the N-methyl-D-aspartate receptor can be efficiently expressed alone in the cell surface of mammalian cells and is required for the transport of the NR2A subunit. Biochem. J. 2001, 356, 539–547. [Google Scholar] [CrossRef]
- Jonas, P. AMPA-type glutamate receptors-nonselective cation channels mediating fast excitatory transmission in the CNS. EXS 1993, 66, 61–76. [Google Scholar]
- Sommer, B.; Keinanen, K.; Verdoorn, T.A.; Wisden, W.; Burnashev, N.; Herb, A.; Kohler, M.; Takagi, T.; Sakmann, B.; Seeburg, P.H. Flip and flop: A cell-specific functional switch in glutamate-operated channels of the CNS. Science 1990, 249, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.; Kelly, L.; Farrant, M. Regulation of Ca2+-permeable AMPA receptors: Synaptic plasticity and beyond. Curr. Opin. Neurobiol. 2006, 16, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Jessberger, S.; Kempermann, G. Adult-born hippocampal neurons mature into activity-dependent responsiveness. Eur. J. Neuorosci. 2003, 18, 2707–2712. [Google Scholar] [CrossRef]
- Li, Y.; Huang, W.Y.; Lv, C.Y.; Cong, J.; Jie, W.; Li, S.J.; Li, J.M. Satb2 ablation decreases PTZ-induced seizure susceptibility and pyramidal neuronal excitability. Brain Res. 2018, 15, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Rellos, P.; Pike, A.C.; Niesen, F.H.; Salah, E.; Lee, W.H.; von Delft, F.; Knapp, S. Structure of the CaMKIIδ/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol. 2010, 8, e1000426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giese, K.P.; Fedorov, N.B.; Filipkowski, R.K.; Silva, A.J. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 1998, 279, 870–873. [Google Scholar] [CrossRef]
- Zhou, Y.; Takahashi, E.; Li, W.; Halt, A.; Wiltgen, B.; Ehninger, D.; Li, G.-D.; Hell, J.W.; Kennedy, M.B.; Silva, A.J. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J. Neurosci. 2007, 27, 13843–13853. [Google Scholar] [CrossRef] [Green Version]
- Pinto, J.G.A.; Jones, D.G.; Murphy, K.M. Comparing development of synaptic proteins in rat visual, somatosensory, and frontal cortex. Front Neural Circuits 2013, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Kornau, H.C.; Schenker, L.T.; Kennedy, M.B.; Seeburg, P.H. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 1995, 269, 1737–1740. [Google Scholar] [CrossRef]
- Cook, D.J.; Teves, L.; Tymianski, M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 2012, 483, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell 2020, 181, 784–799. [Google Scholar] [CrossRef]
- Ozdener, M.H.; Subramaniam, S.; Sundaresan, S.; Sery, O.; Hashimoto, T.; Asakawa, Y.; Besnard, P.; Abumrad, N.A.; Khan, N.A. CD36- and GPR120-mediated Ca(2)(+) signaling in human taste bud cells mediates differential responses to fatty acids and is altered in obese mice. Gastroenterology 2014, 146, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurokawa, M.; Yoon, S.Y.; Alfandari, D.; Fukami, K.; Sato, K.; Fissore, R.A. Proteolytic processing of phospholipase Czeta and [Ca2+]i oscillations during mammalian fertilization. Dev. Biol. 2007, 312, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Soontornniyomkij, V.; Risbrough, V.B.; Young, J.W.; Soontornniyomkij, B.; Jeste, D.V.; Achim, C.L. Hippocampal calbindin-1 immunoreactivity correlate of recognition memory performance in aged mice. Neurosci. Lett. 2012, 516, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Bouschet, T.; Martin, S.; Henley, J.M. Receptor-activity-modifying proteins are required for forward trafficking of the calci-um-sensing receptor to the plasma membrane. J. Cell Sci. 2005, 118, 4709–4720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ari, Y.; Tremblay, E.; Ottersen, O.P.; Meldrum, B.S. The role of epileptic activity in hippocampal and "remote" cerebral lesions induced by kainic acid. Brain Res. 1980, 191, 79–97. [Google Scholar] [CrossRef]
- Carta, M.; Opazo, P.; Veran, J.; Athané, A.; Choquet, D.; Coussen, F.; Mulle, C. CaMKII-dependent phosphorylation of GluK5 mediates plasticity of kainate receptors. EMBO J. 2013, 32, 496–510. [Google Scholar] [CrossRef] [Green Version]
- Selak, S.; Paternain, A.V.; Alle3r, M.I.; Picó, E.; Rivera, R.; Lerma, J. A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron 2009, 63, 357–371. [Google Scholar] [CrossRef]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ari, Y.; Spitzer, N.C. Phenotypic checkpoints regulate neuronal development. Trends Neurosci. 2010, 33, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Katsarou, A.M.; Moshé, S.L.; Galanopoulou, A.S. Interneuronopathies and their role in early life epilepsies and neurodevelop-mental disorders. Epilepsia Open 2017, 2, 284–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couillard-Despres, S.; Quehl, E.; Altendorfer, K.; Karl, C.; Ploetz, S.; Bogdahn, U.; Winkler, J.; Aigner, L. Human in vitro reporter model of neuronal devel-opment and early differentiation processes. BMC Neurosci. 2008, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstraelen, P.; Van Dyck, M.; Verschuuren, M.; Kashikar, N.D.; Nuydens, R.; Timmermans, J.-P.; De Vos, W.H. Image-based profiling of synaptic connectivity in primary neuronal cell culture. Front. Neurosci. 2018, 12, 389. [Google Scholar] [CrossRef]
- Savarese, F.; Dávila, A.; Nechanitzky, R.; De La Rosa-Velazquez, I.; Pereira, C.F.; Engelke, R.; Takahashi, K.; Jenuwein, T.; Kohwi-Shigematsu, T.; Fisher, A.G.; et al. Satb1 and Satb2 regulate embryonic stem cell differentiation and Nanog expression. Genes Dev. 2009, 23, 2625–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobreva, G.; Chahrour, M.; Dautzenberg, M.; Chirivella, L.; Kanzler, B.; Fariñas, I.; Karsenty, G.; Grosschedl, R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 2006, 125, 971–986. [Google Scholar] [CrossRef] [Green Version]
- Baranek, C.; Dittrich, M.; Parthasarathy, S.; Bonnon, C.G.; Britanova, O.; Lanshakov, D.; Boukhtouche, F.; Sommer, J.E.; Colmenares, C.; Tarabykin, V.; et al. Protooncogene Ski cooperates with the chroma-tin-remodeling factor Satb2 in specifying callosal neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 3546–3551. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Song, N.-N.; Lan, W.; Hu, L.; Su, C.-J.; Ding, Y.-Q.; Zhang, L. Expression of transcription factor Satb2 in adult mouse brain. Anat Rec. 2013, 296, 452–461. [Google Scholar] [CrossRef]
- Lee, J.S.; Yoo, Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J.H. SATB2-associated syndrome presenting with Rett-like phenotypes. Clin. Gen. 2016, 89, 728–732. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Ballif, B.C.; Lucas, A.; Spence, E.J.; Powell, C.; Aylsworth, A.S.; Torchia, B.A.; Shaffer, L.G. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS ONE 2009, 4, e6568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, W.J.; Bosma, M.M. Ion channel development, spontaneous activity, and activity-dependent development in nerve and muscle cells. Physiol. Rev. 2005, 85, 883–941. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Swann, J.W. A role for L-type calcium channels in the maturation of parvalbumin-containing hippocampal inter-neurons. Neuroscience 2005, 135, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Batista-Brito, R.; Rossignol, E.; Hjerling-Leffler, J.; Denaxa, M.; Wegner, M.; Lefebvre, V.; Pachnis, V.; Fishell, G. The cell-intrinsic requirement of Sox6 for cortical interneuron development. Neuron 2009, 63, 466–481. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in applying computer-aided drug design for neurodegenerative diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef] [PubMed]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-throughput screening platforms in the discovery of novel drugs for neurodegenerative diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gapdh | Forward 5′-ccacggcaagttcaacggcac-3′ Reverse 5′-gatgatgacccttttggccccacc-3′ |
| Grik1 | Forward 5′-ggaggatgaggcggggacc-3′ Reverse 5′-gcatgctcttcgggaggcttcaaaac-3′ |
| Grik2 | Forward 5′-ggatgggaaatatggagcccaggatgat-3′ Reverse 5′-tcaggggagagaggattcaggaaggag-3′ |
| Grin1 | Forward 5′-tgacggtgagatggaagagctg-3′ Reverse 5′-ctgccatgttgtcgatgtccag-3′ |
| Grin2a | Forward 5′- cagtaaaccaggccaataagcga-3′ Reverse 5′- atctgtatggcgttgggcttgt-3′ |
| Grin2b | Forward 5′-gtcctccaaagacacgagcac-3′ Reverse 5′-gccctcctccctctcaatagc-3′ |
| Gria1 | Forward 5′-aggggaatgtggaagcaaggac-3′ Reverse 5′-ccaatcccagccctccaatcag-3′ |
| Gria2 | Forward 5′-agccaaggactcgggaagtaagg-3′ Reverse 5′-caccagcattgccaaaccaagg-3′ |
| Gabra1 | Forward 5′- tatctttgggcctggaccctcattctg-3′ Reverse 5′-ccataaggttgtttagccggagcactg-3′ |
| Dcx | Forward 5′- gcaatggggaccgttacttcaa-3′ Reverse 5′- agccagcaacgcatcaaaactac-3′ |
| Syp | Forward 5′- aggtgctgcagtgggtctttg-3′ Reverse 5′- actctccgtcttgttggcacact-3′ |
| Dlg3 | Forward 5′- aggagatcacattggaaaggggtaa-3′ Reverse 5′- tggtgataaagatggatgggtcgt-3′ |
| Pik3ca | Forward 5′-ctgagatgggagctgggactgc-3′ Reverse 5′-gtgtccacgtgttagacagaacactg-3′ |
| Pik3cb | Forward 5′-gaggttatgagtgtgcttccgccctat-3′ Reverse 5′-agtcttcgtgtttcgtcttccagttcctc-3′ |
| Pik3cg | Forward 5′-gctgcggagttctaccaccgattg-3′ Reverse 5′-caggtagtctgggagaggtttggacg-3′ |
| Camk2a | Forward 5′-gagcagcaggcatggtttgggt-3′ Reverse 5′-ggtgcttgagagcctcagcgg-3′ |
| Prkca | Forward 5′-ccaacgactccacggcgtctc-3′ Reverse 5′-tgcttgtgaacattcatgtcgcaggtgt-3′ |
| Prkce | Forward 5′-tgatcatcgatctctcgggatcatcggg-3′ Reverse 5′-gcccacctcgtcaggggtttc-3′ |
| Prkcg | Forward 5′-tggttcaccgccgatgccac-3′ Reverse 5′-ccgcaaagggagggcacg-3′ |
| Prkcg | Forward 5′-gccccccaacatggactgtctct-3′ Reverse 5′-gggttgtcagtgccacctgcgat-3′ |
| EC50/ΔF(Fmax–Fmin) | |||
|---|---|---|---|
| NMDAR | AMPAR | KAR | |
| Control | 6.39 µM/0.139 | 0.004 µM/0.285 | 0.32 µM/0.181 |
| Satb1fl/+ * NexCre/+ | 9.94 µM/0.153 * | 0.018 µM/0.222 *** | 0.39 µM/0.137 n/s |
| Satb1fl/fl * NexCre/+ | 1.26 µM/0.254 ** | 0.002 µM/0.171 n/s | 0.27 µM/0.239 n/s |
| Satb2fl/+ * NexCre/+ | 6.19 µM/0.173 * | 0.025 µM/0.221 *** | 0.176 µM/0.179 *** |
| Satb2fl/fl * NexCre/+ | 2.83 µM/0.184 *** | 0.092 µM/0.222 *** | 0.49 µM/0.126 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turovsky, E.A.; Turovskaya, M.V.; Fedotova, E.I.; Babaev, A.A.; Tarabykin, V.S.; Varlamova, E.G. Role of Satb1 and Satb2 Transcription Factors in the Glutamate Receptors Expression and Ca2+ Signaling in the Cortical Neurons In Vitro. Int. J. Mol. Sci. 2021, 22, 5968. https://doi.org/10.3390/ijms22115968
Turovsky EA, Turovskaya MV, Fedotova EI, Babaev AA, Tarabykin VS, Varlamova EG. Role of Satb1 and Satb2 Transcription Factors in the Glutamate Receptors Expression and Ca2+ Signaling in the Cortical Neurons In Vitro. International Journal of Molecular Sciences. 2021; 22(11):5968. https://doi.org/10.3390/ijms22115968
Chicago/Turabian StyleTurovsky, Egor A., Maria V. Turovskaya, Evgeniya I. Fedotova, Alexey A. Babaev, Victor S. Tarabykin, and Elena G. Varlamova. 2021. "Role of Satb1 and Satb2 Transcription Factors in the Glutamate Receptors Expression and Ca2+ Signaling in the Cortical Neurons In Vitro" International Journal of Molecular Sciences 22, no. 11: 5968. https://doi.org/10.3390/ijms22115968