
Valproic Acid Reduces Vasospasm through Modulation of Akt Phosphorylation and Attenuates Neuronal Apoptosis in Subarachnoid Hemorrhage Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Mortality
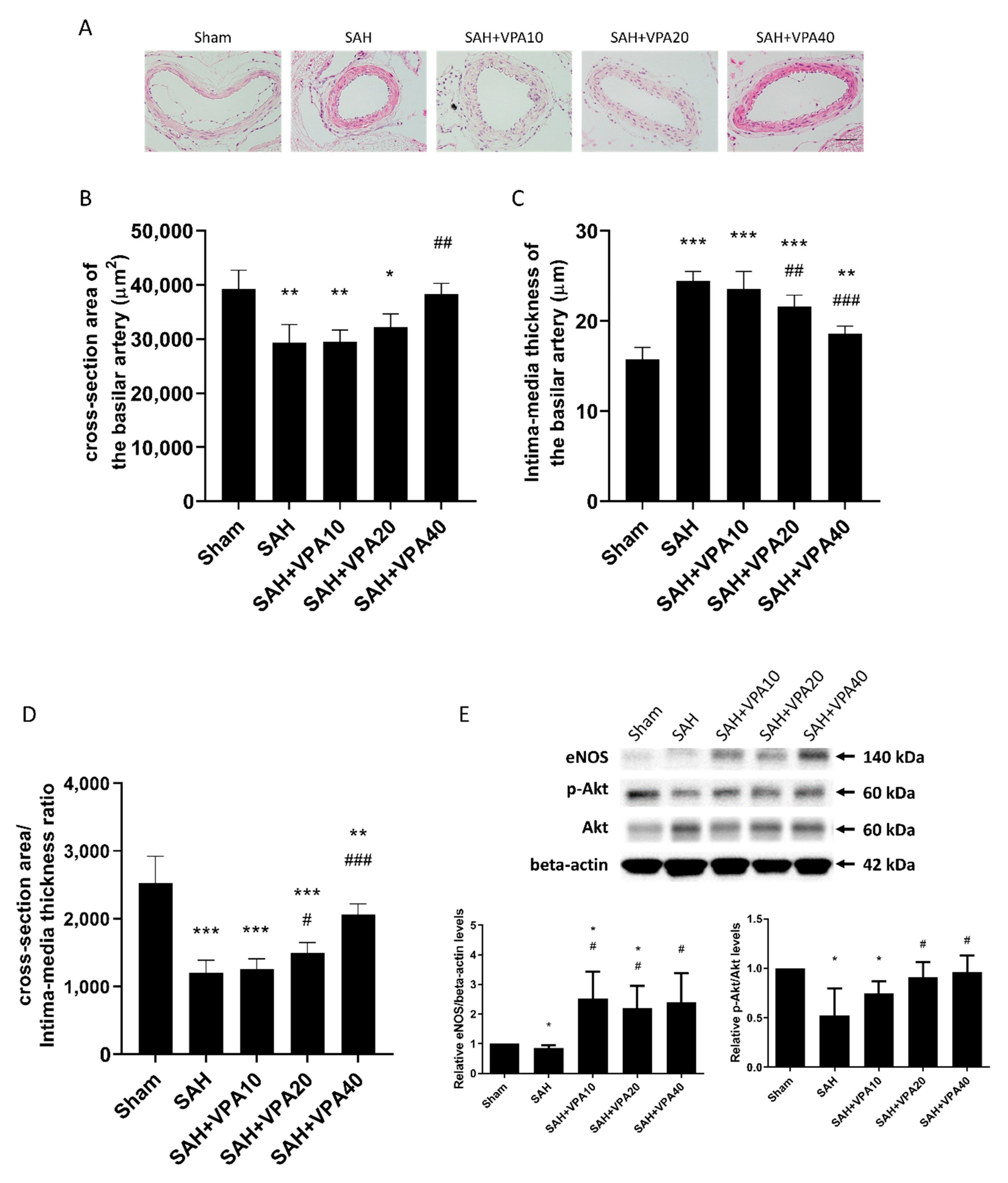
2.2. VPA Attenuated Vasospasm after SAH
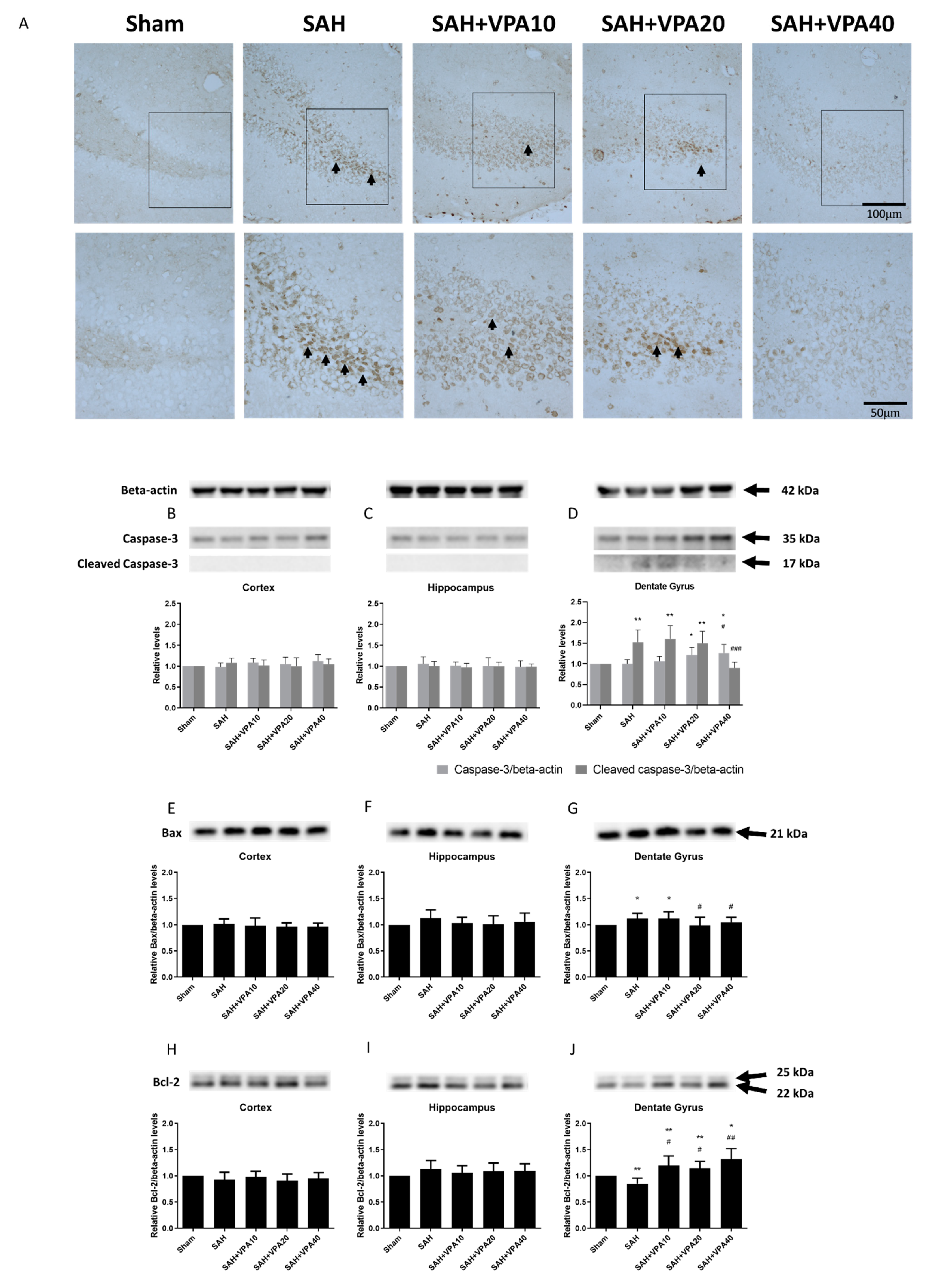
2.3. VPA Reversed SAH-Induced Mitochondrial Apoptosis Signaling in Dentate Gyrus
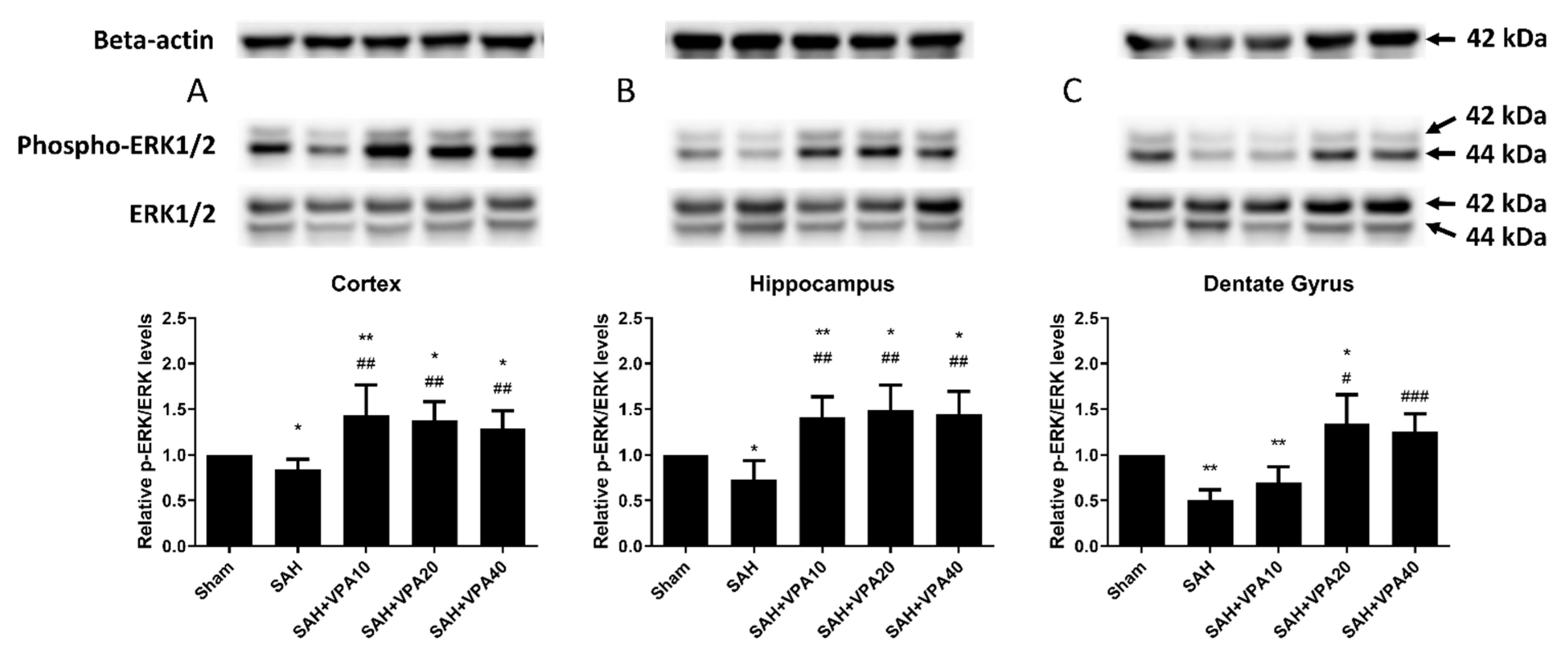
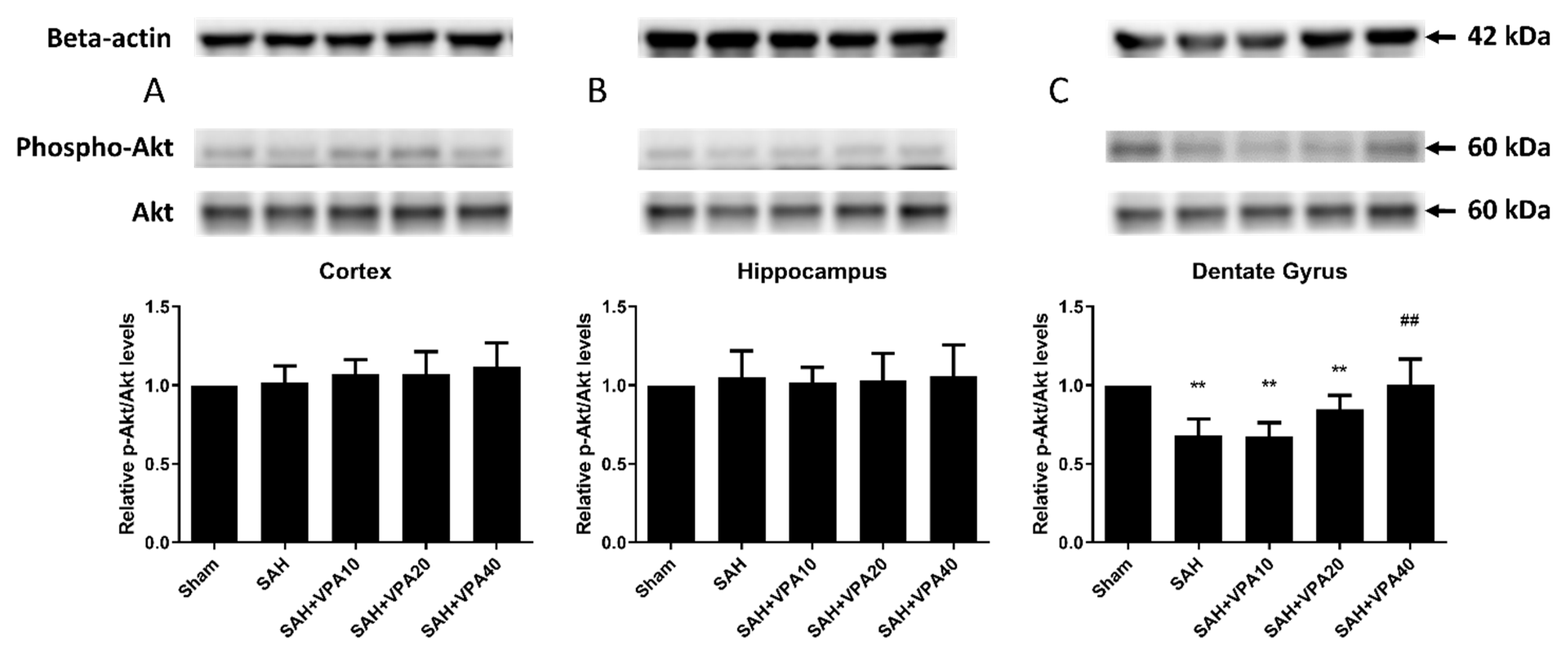
2.4. VPA Enhanced ERK1/2 and Akt Activation in the Dentate Gyrus
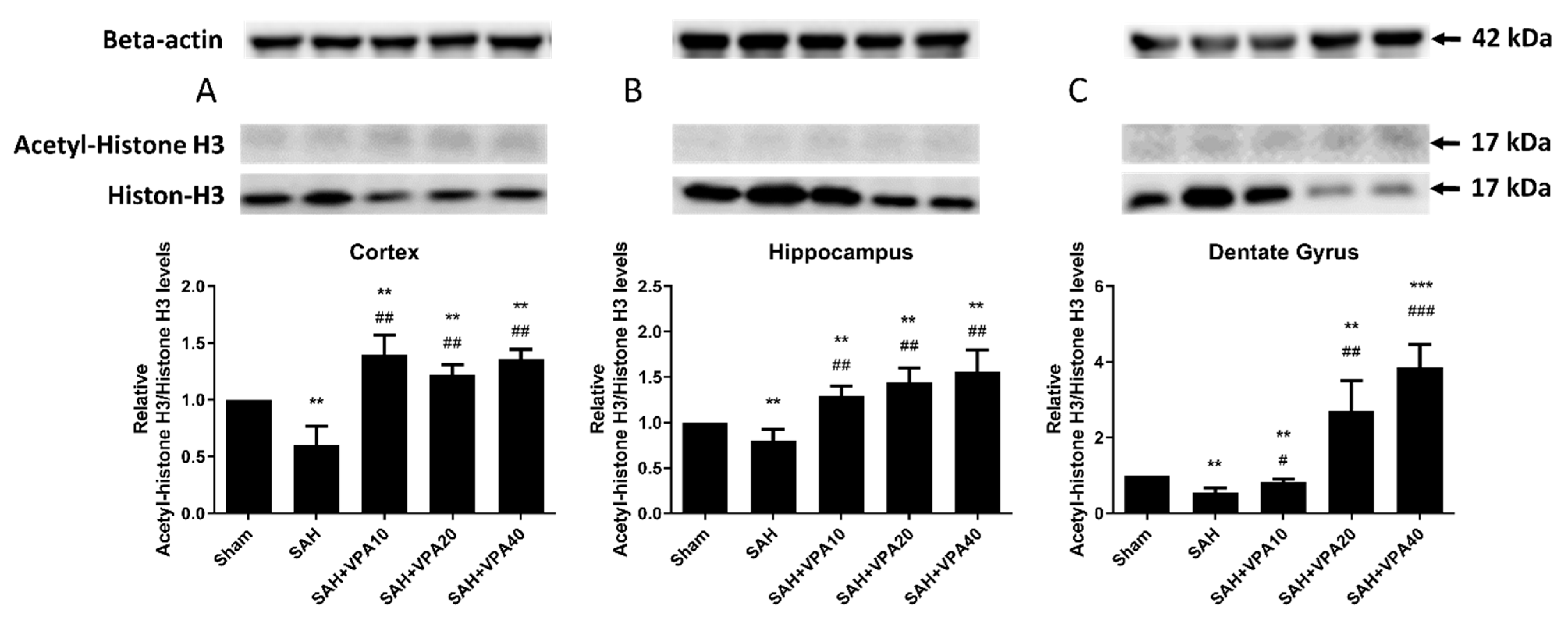
2.5. VPA Up-Regulated Histone H3 Acetylation in Brain Tissues after SAH
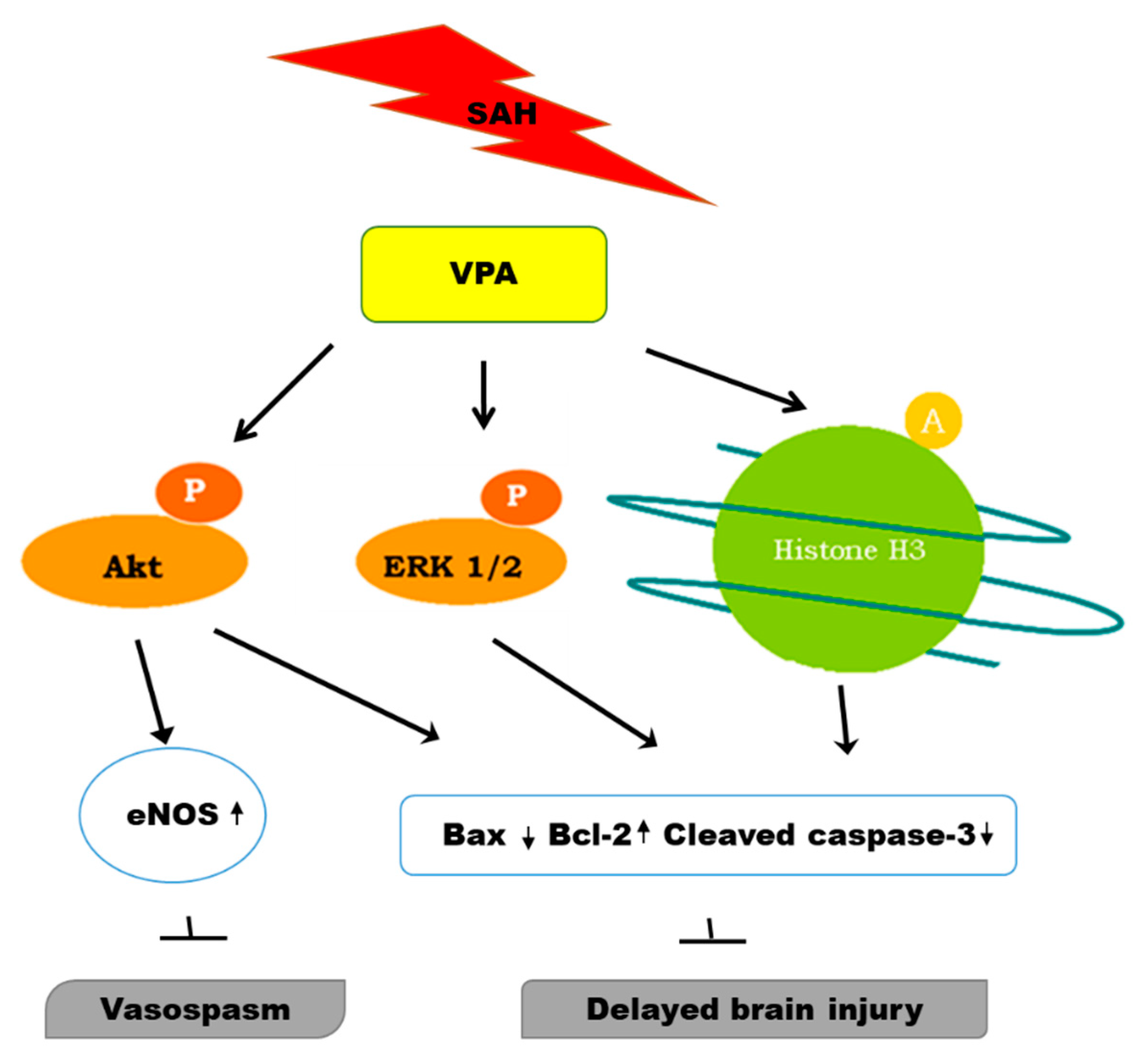
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animal Preparation
4.3. Experimental SAH Model
4.4. Sample Preparation
4.5. Basilar Artery Morphometric Studies
4.6. TUNEL Staining
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feigin, V.L.; Lawes, C.M.; Bennett, D.A.; Anderson, C.S. Stroke epidemiology: A review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003, 2, 43–53. [Google Scholar] [CrossRef]
- Van Gijn, J.; Rinkel, G.J. Subarachnoid haemorrhage: Diagnosis, causes and management. Brain 2001, 124, 249–278. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart disease and stroke statistics-2017 update: A report from the american heart association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Van Gijn, J.; Kerr, R.S.; Rinkel, G.J. Subarachnoid haemorrhage. Lancet 2007, 369, 306–318. [Google Scholar] [CrossRef]
- Broderick, J.P.; Brott, T.G.; Duldner, J.E.; Tomsick, T.; Leach, A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994, 25, 1342–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoerle, T.; Ilodigwe, D.C.; Wan, H.; Lakovic, K.; Sabri, M.; Ai, J.; Macdonald, R.L. Pharmacologic reduction of angiographic vasospasm in experimental subarachnoid hemorrhage: Systematic review and meta-analysis. J. Cereb. Blood Flow Metab. 2012, 32, 1645–1658. [Google Scholar] [CrossRef] [Green Version]
- Kassell, N.F.; Sasaki, T.; Colohan, A.R.; Nazar, G. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke 1985, 16, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.A. Mechanisms of cerebral vasospasm in subarachnoid haemorrhage. Pharmacol. Ther. 1995, 66, 259–284. [Google Scholar] [CrossRef]
- Al-Khindi, T.; Macdonald, R.L.; Schweizer, T.A. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010, 41, e519–e536. [Google Scholar] [CrossRef] [Green Version]
- Kreiter, K.T.; Copeland, D.; Bernardini, G.L.; Bates, J.E.; Peery, S.; Claassen, J.; Du, Y.E.; Stern, Y.; Connolly, E.S.; Mayer, S.A. Predictors of cognitive dysfunction after subarachnoid hemorrhage. Stroke 2002, 33, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Ecker, A.; Riemenschneider, P.A. Arteriographic demonstration of spasm of the intracranial arteries, with special reference to saccular arterial aneurysms. J. Neurosurg. 1951, 8, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Vergouwen, M.D.; Ilodigwe, D.; Macdonald, R.L. Cerebral infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke 2011, 42, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Dankbaar, J.W.; Rijsdijk, M.; van der Schaaf, I.C.; Velthuis, B.K.; Wermer, M.J.; Rinkel, G.J. Relationship between vasospasm, cerebral perfusion, and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neuroradiology 2009, 51, 813–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.C.; Lin, C.L.; Lee, T.Y.; Lee, W.S.; Hsu, C. 17beta-estradiol inhibits subarachnoid hemorrhage-induced inducible nitric oxide synthase gene expression by interfering with the nuclear factor kappa b transactivation. Stroke 2006, 37, 3025–3031. [Google Scholar] [CrossRef] [Green Version]
- Kondo, S.; Hashimoto, N.; Kikuchi, H.; Hazama, F.; Nagata, I.; Kataoka, H. Apoptosis of medial smooth muscle cells in the development of saccular cerebral aneurysms in rats. Stroke 1998, 29, 181–188, discussion 189. [Google Scholar] [CrossRef] [Green Version]
- Hara, A.; Yoshimi, N.; Mori, H. Evidence for apoptosis in human intracranial aneurysms. Neurol. Res. 1998, 20, 127–130. [Google Scholar] [CrossRef]
- Nau, R.; Haase, S.; Bunkowski, S.; Bruck, W. Neuronal apoptosis in the dentate gyrus in humans with subarachnoid hemorrhage and cerebral hypoxia. Brain Pathol. 2002, 12, 329–336. [Google Scholar]
- Cahill, J.; Calvert, J.W.; Zhang, J.H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2006, 26, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Yamaguchi, M.; Kusaka, G.; Schonholz, C.; Nanda, A.; Zhang, J.H. Caspase inhibitors prevent endothelial apoptosis and cerebral vasospasm in dog model of experimental subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2004, 24, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Schievink, W.I. Intracranial aneurysms. N. Engl. J. Med. 1997, 336, 28–40. [Google Scholar] [CrossRef]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of hdac inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chateauvieux, S.; Morceau, F.; Dicato, M.; Diederich, M. Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol. 2010, 2010, 479364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, C.L.; Karren, N.U. Anticonvulsants in bipolar disorder. Aust. N. Z. J. Psychiatry 2006, 40, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G. The mechanisms of action of valproate in neuropsychiatric disorders: Can we see the forest for the trees? Cell Mol. Life Sci. 2007, 64, 2090–2103. [Google Scholar] [CrossRef]
- Monti, B.; Polazzi, E.; Contestabile, A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr. Mol. Pharmacol. 2009, 2, 95–109. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, J.; Zhang, J.; Li, H.; Zhao, Z.; Liao, Y.; Wang, X.; Su, J.; Sang, S.; Yuan, X.; et al. Neuroprotective and anti-apoptotic effects of valproic acid on adult rat cerebral cortex through erk and akt signaling pathway at acute phase of traumatic brain injury. Brain Res. 2014, 1555, 1–9. [Google Scholar] [CrossRef]
- Ren, M.; Leng, Y.; Jeong, M.; Leeds, P.R.; Chuang, D.M. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: Potential roles of histone deacetylase inhibition and heat shock protein induction. J. Neurochem. 2004, 89, 1358–1367. [Google Scholar] [CrossRef]
- Kim, H.J.; Rowe, M.; Ren, M.; Hong, J.S.; Chen, P.S.; Chuang, D.M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J. Pharmacol. Exp. Ther. 2007, 321, 892–901. [Google Scholar] [CrossRef]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of hdac inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Kautu, B.B.; Carrasquilla, A.; Hicks, M.L.; Caldwell, K.A.; Caldwell, G.A. Valproic acid ameliorates C. elegans dopaminergic neurodegeneration with implications for erk-mapk signaling. Neurosci. Lett. 2013, 541, 116–119. [Google Scholar] [CrossRef] [Green Version]
- Bonita, R.; Thomson, S. Subarachnoid hemorrhage: Epidemiology, diagnosis, management, and outcome. Stroke 1985, 16, 591–594. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.D.; Roy, E.J. Dentate gyrus destruction and spatial learning impairment after corticosteroid removal in young and middle-aged rats. Hippocampus 1995, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kanamaru, K.; Tsunoda, H.; Inada, H.; Kuroki, M.; Sun, H.; Waga, S.; Tanaka, T. Heme oxygenase-1 gene induction as an intrinsic regulation against delayed cerebral vasospasm in rats. J. Clin. Investig. 1999, 104, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Calisaneller, T.; Ukita, N.; Dumont, A.S.; Kassell, N.F.; Lee, K.S. A murine model of subarachnoid hemorrhage-induced cerebral vasospasm. J. Neurosci. Methods 2003, 123, 89–97. [Google Scholar] [CrossRef]
- Dudhani, R.V.; Kyle, M.; Dedeo, C.; Riordan, M.; Deshaies, E.M. A low mortality rat model to assess delayed cerebral vasospasm after experimental subarachnoid hemorrhage. J. Vis. Exp. 2013, 71, e4157. [Google Scholar] [CrossRef] [Green Version]
- Gules, I.; Satoh, M.; Clower, B.R.; Nanda, A.; Zhang, J.H. Comparison of three rat models of cerebral vasospasm. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2551–H2559. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.Z.; Wu, S.C.; Lin, C.L.; Kwan, A.L. Valproic acid attenuates intercellular adhesion molecule-1 and e-selectin through a chemokine ligand 5 dependent mechanism and subarachnoid hemorrhage induced vasospasm in a rat model. J. Inflamm. 2015, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Matz, P.; Weinstein, P.; States, B.; Honkaniemi, J.; Sharp, F.R. Subarachnoid injections of lysed blood induce the hsp70 stress gene and produce DNA fragmentation in focal areas of the rat brain. Stroke 1996, 27, 504–512, discussion 513. [Google Scholar] [CrossRef]
- Zhou, C.; Yamaguchi, M.; Colohan, A.R.; Zhang, J.H. Role of p53 and apoptosis in cerebral vasospasm after experimental subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2005, 25, 572–582. [Google Scholar] [CrossRef] [Green Version]
- Prunell, G.F.; Svendgaard, N.A.; Alkass, K.; Mathiesen, T. Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. J. Neurosurg. 2005, 102, 1046–1054. [Google Scholar] [CrossRef]
- Cahill, J.; Calvert, J.W.; Solaroglu, I.; Zhang, J.H. Vasospasm and p53-induced apoptosis in an experimental model of subarachnoid hemorrhage. Stroke 2006, 37, 1868–1874. [Google Scholar] [CrossRef]
- Yuksel, S.; Tosun, Y.B.; Cahill, J.; Solaroglu, I. Early brain injury following aneurysmal subarachnoid hemorrhage: Emphasis on cellular apoptosis. Turk. Neurosurg. 2012, 22, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, K.; Zubkov, A.Y.; Ross, I.B.; Zhang, J.H. Therapeutic effect of caspase inhibitors in the prevention of apoptosis and reversal of chronic cerebral vasospasm. J. Clin. Neurosci. 2002, 9, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Wei, L.; Zhi-Dan, S.; Shi-Guang, Z.; Xiang-Zhen, L. Atorvastatin ameliorates cerebral vasospasm and early brain injury after subarachnoid hemorrhage and inhibits caspase-dependent apoptosis pathway. BMC Neurosci. 2009, 10, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- Yakovlev, A.G.; Knoblach, S.M.; Fan, L.; Fox, G.B.; Goodnight, R.; Faden, A.I. Activation of cpp32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J. Neurosci. 1997, 17, 7415–7424. [Google Scholar] [CrossRef] [Green Version]
- Merry, D.E.; Korsmeyer, S.J. Bcl-2 gene family in the nervous system. Annu. Rev. Neurosci. 1997, 20, 245–267. [Google Scholar] [CrossRef]
- Endo, H.; Nito, C.; Kamada, H.; Yu, F.; Chan, P.H. Akt/gsk3beta survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke 2006, 37, 2140–2146. [Google Scholar] [CrossRef] [Green Version]
- Tibbs, R.; Zubkov, A.; Aoki, K.; Meguro, T.; Badr, A.; Parent, A.; Zhang, J. Effects of mitogen-activated protein kinase inhibitors on cerebral vasospasm in a double-hemorrhage model in dogs. J. Neurosurg. 2000, 93, 1041–1047. [Google Scholar] [CrossRef]
- Chang, C.M.; Su, Y.F.; Chang, C.Z.; Chung, C.L.; Tsai, Y.J.; Loh, J.K.; Lin, C.L. Progesterone attenuates experimental subarachnoid hemorrhage-induced vasospasm by upregulation of endothelial nitric oxide synthase via akt signaling pathway. Biomed. Res. Int. 2014, 2014, 207616. [Google Scholar] [CrossRef]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef]
- Noshita, N.; Lewen, A.; Sugawara, T.; Chan, P.H. Evidence of phosphorylation of akt and neuronal survival after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2001, 21, 1442–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, H.; Nito, C.; Kamada, H.; Nishi, T.; Chan, P.H. Activation of the akt/gsk3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2006, 26, 1479–1489. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Armstead, W.M.; Riley, J.; Vavilala, M.S. Norepinephrine protects cerebral autoregulation and reduces hippocampal necrosis after traumatic brain injury via blockade of erk mapk and il-6 in juvenile pigs. J. Neurotrauma 2016, 33, 1761–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstead, W.M.; Bohman, L.E.; Riley, J.; Yarovoi, S.; Higazi, A.A.; Cines, D.B. Tpa-S481A prevents impairment of cerebrovascular autoregulation by endogenous tpa after traumatic brain injury by upregulating p38 mapk and inhibiting et-1. J. Neurotrauma 2013, 30, 1898–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303, 701–704. [Google Scholar] [CrossRef]
- Clausen, F.; Lundqvist, H.; Ekmark, S.; Lewen, A.; Ebendal, T.; Hillered, L. Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J. Neurotrauma 2004, 21, 1168–1182. [Google Scholar] [CrossRef]
- De Sarno, P.; Li, X.; Jope, R.S. Regulation of akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 2002, 43, 1158–1164. [Google Scholar] [CrossRef]
- Hamming, A.M.; van der Toorn, A.; Rudrapatna, U.S.; Ma, L.; van Os, H.J.; Ferrari, M.D.; van den Maagdenberg, A.M.; van Zwet, E.; Poinsatte, K.; Stowe, A.M.; et al. Valproate reduces delayed brain injury in a rat model of subarachnoid hemorrhage. Stroke 2017, 48, 452–458. [Google Scholar] [CrossRef]
- Bertelsen, F.; Landau, A.M.; Vase, K.H.; Jacobsen, J.; Scheel-Krüger, J.; Møller, A. Acute in vivo effect of valproic acid on the GABAergic system in rat brain: A [11 C]Ro15-4513 microPET study. Brain Res. 2018, 1680, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Tso, M.K.; Lass, E.; Ai, J.; Macdonald, R.L. Valproic Acid Treatment after Experimental Subarachnoid Hemorrhage. Funct. Rehabil. Neurosurg. Neurotraumatol. 2014, 120, 81–85. [Google Scholar]
- Dubois, F.; Caby, S.; Oger, F.; Cosseau, C.; Capron, M.; Grunau, C.; Dissous, C.; Pierce, R.J. Histone deacetylase inhibitors induce apoptosis, histone hyperacetylation and up-regulation of gene transcription in Schistosoma mansoni. Mol. Biochem. Parasitol. 2009, 168, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Han, X.; Ren, H.; Han, X.; Sun, W.; Gu, Y.; Qiao, J.; Dong, Q. Levels of histone H3 acetylation in peripheral blood mononuclear cells of acute cerebral infarction patients. Zhonghua Yi Xue Za Zhi 2014, 94, 2123–2128. [Google Scholar]
- Kuboyama, T.; Wahane, S.; Huang, Y.; Zhou, X.; Wong, J.; Koemeter-Cox, A.; Martini, M.; Friedel, R.H.; Zou, H. HDAC3 inhibition ameliorates spinal cord injury by immunomodulation. Sci. Rep. 2017, 7, 8641. [Google Scholar] [CrossRef]
- Chi, J.-H.; Seo, G.S.; Cheon, J.H.; Lee, S.H. Isoliquiritigenin inhibits TNF-α-induced release of high-mobility group box 1 through activation of HDAC in human intestinal epithelial HT-29 cells. Eur. J. Pharmacol. 2017, 796, 101–109. [Google Scholar] [CrossRef]
- Michaelis, M.; Suhan, T.; Michaelis, U.R.; Beek, K.; Rothweiler, F.; Tausch, L.; Werz, O.; Eikel, D.; Zornig, M.; Nau, H.; et al. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. 2006, 13, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Kostrouch, Z.; Kostrouchová, M. Valproic acid, a molecular lead to multiple regulatory pathways. Folia Biol. 2007, 53, 37–49. [Google Scholar]
- Mora, A.; González-Polo, R.A.; Fuentes, J.M.; Soler, G.; Centeno, F. Different mechanisms of protection against apoptosis by valproate and Li+. JBIC J. Biol. Inorg. Chem. 1999, 266, 886–891. [Google Scholar] [CrossRef]
- Lin, X.P.; Feng, L.; Xie, C.G.; Chen, D.B.; Pei, Z.; Liang, X.L.; Xie, Q.Y.; Li, X.H.; Pan, S.Y. Valproic acid attenuates the sup-pression of acetyl histone h3 and creb activity in an inducible cell model of machado-joseph disease. Int. J. Dev. Neurosci. 2014, 38, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Piao, Y.; Liu, Y.; Xie, X. Change Trends of Organ Weight Background Data in Sprague Dawley Rats at Different Ages. J. Toxicol. Pathol. 2013, 26, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar] [PubMed]
- Lin, C.-L.; Shih, H.-C.; Lieu, A.-S.; Lee, K.-S.; Dumont, A.S.; Kassell, N.F.; Howng, S.-L.; Kwan, A.-L. Attenuation of experimental subarachnoid hemorrhage–induced cerebral vasospasm by the adenosine A2A receptor agonist CGS 21680. J. Neurosurg. 2007, 106, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meguro, T.; Clower, B.R.; Carpenter, R.; Parent, A.D.; Zhang, J.H. Improved rat model for cerebral vasospasm studies. Neurol. Res. 2001, 23, 761–766. [Google Scholar] [CrossRef]
- Lin, C.L.; Dumont, A.S.; Su, Y.F.; Tsai, Y.J.; Huang, J.H.; Chang, K.P.; Howng, S.L.; Kwan, A.L.; Kassell, N.F.; Kao, C.H. Attenuation of cerebral vasospasm and secondary injury by 17beta-estradiol following experimental subarachnoid hemor-rhage. J. Neurosurg. 2009, 110, 457–461. [Google Scholar] [CrossRef]
- Lin, C.L.; Su, Y.F.; Dumont, A.S.; Shih, H.C.; Lieu, A.S.; Howng, S.L.; Lee, K.S.; Kwan, A.L. The effect of an adenosine A1 receptor agonist in the treatment of experimental subarachnoid hemorrhage-induced cerebrovasospasm. Acta Neurochir. 2006, 148, 873–879. [Google Scholar] [CrossRef]
- Emmez, H.; Borcek, A.O.; Gonul, I.I.; Belen, H.B.; Solaroglu, I.; Baykaner, M.K. The effect of hydrogen sulphide on experimental cerebral vasospasm. Turk. Neurosurg. 2017, 27, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Kamat, P.K.; Ahmad, A.S.; Doré, S. Carbon monoxide attenuates vasospasm and improves neurobehavioral function after subarachnoid hemorrhage. Arch. Biochem. Biophys. 2019, 676, 108117. [Google Scholar] [CrossRef] [PubMed]
- Echigo, R.; Shimohata, N.; Karatsu, K.; Yano, F.; Kayasuga-Kariya, Y.; Fujisawa, A.; Ohto, T.; Kita, Y.; Nakamura, M.; Suzuki, S.; et al. Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. J. Transl. Med. 2012, 10, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cai, H.; Wang, Z.; Li, J.; Wang, K.; Yu, Z.; Chen, G. Induction of autophagy by cystatin C: A potential mechanism for prevention of cerebral vasospasm after experimental subarachnoid hemorrhage. Eur. J. Med Res. 2013, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-H.; Tsai, Y.-C.; Tsai, T.-H.; Kuo, K.-L.; Su, Y.-F.; Chang, C.-H.; Lin, C.-L. Valproic Acid Reduces Vasospasm through Modulation of Akt Phosphorylation and Attenuates Neuronal Apoptosis in Subarachnoid Hemorrhage Rats. Int. J. Mol. Sci. 2021, 22, 5975. https://doi.org/10.3390/ijms22115975
Wu C-H, Tsai Y-C, Tsai T-H, Kuo K-L, Su Y-F, Chang C-H, Lin C-L. Valproic Acid Reduces Vasospasm through Modulation of Akt Phosphorylation and Attenuates Neuronal Apoptosis in Subarachnoid Hemorrhage Rats. International Journal of Molecular Sciences. 2021; 22(11):5975. https://doi.org/10.3390/ijms22115975
Chicago/Turabian StyleWu, Chieh-Hsin, Yi-Cheng Tsai, Tai-Hsin Tsai, Keng-Liang Kuo, Yu-Feng Su, Chih-Hui Chang, and Chih-Lung Lin. 2021. "Valproic Acid Reduces Vasospasm through Modulation of Akt Phosphorylation and Attenuates Neuronal Apoptosis in Subarachnoid Hemorrhage Rats" International Journal of Molecular Sciences 22, no. 11: 5975. https://doi.org/10.3390/ijms22115975
APA StyleWu, C. -H., Tsai, Y. -C., Tsai, T. -H., Kuo, K. -L., Su, Y. -F., Chang, C. -H., & Lin, C. -L. (2021). Valproic Acid Reduces Vasospasm through Modulation of Akt Phosphorylation and Attenuates Neuronal Apoptosis in Subarachnoid Hemorrhage Rats. International Journal of Molecular Sciences, 22(11), 5975. https://doi.org/10.3390/ijms22115975