
Inherited Defects of the ASC-1 Complex in Congenital Neuromuscular Diseases

 and
and
Abstract
:1. Introduction
2. TRIP4 Mutations Are Associated with a Large Spectrum of Clinical and Histological Phenotypes, Potentially Affecting Cardiac Muscles
3. ASCC1 Mutations: Lethal Involvement of Central Nervous System, Skeletal Muscles and Bones
4. Genetic Bases of the ASC-1 and ASCC1 Related Disorders
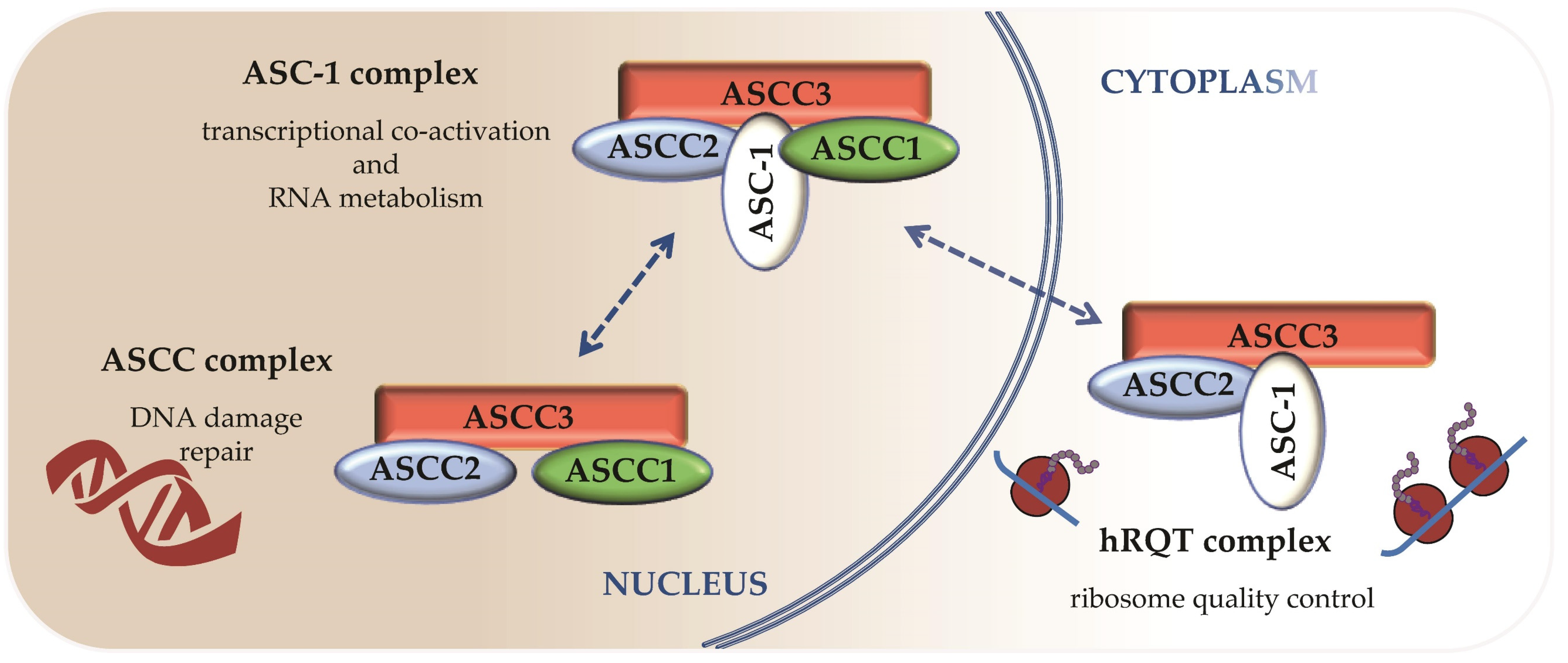
5. Different ASC-1 Complex Conformations Reveal Specific Roles of ASC-1 and ASCC1
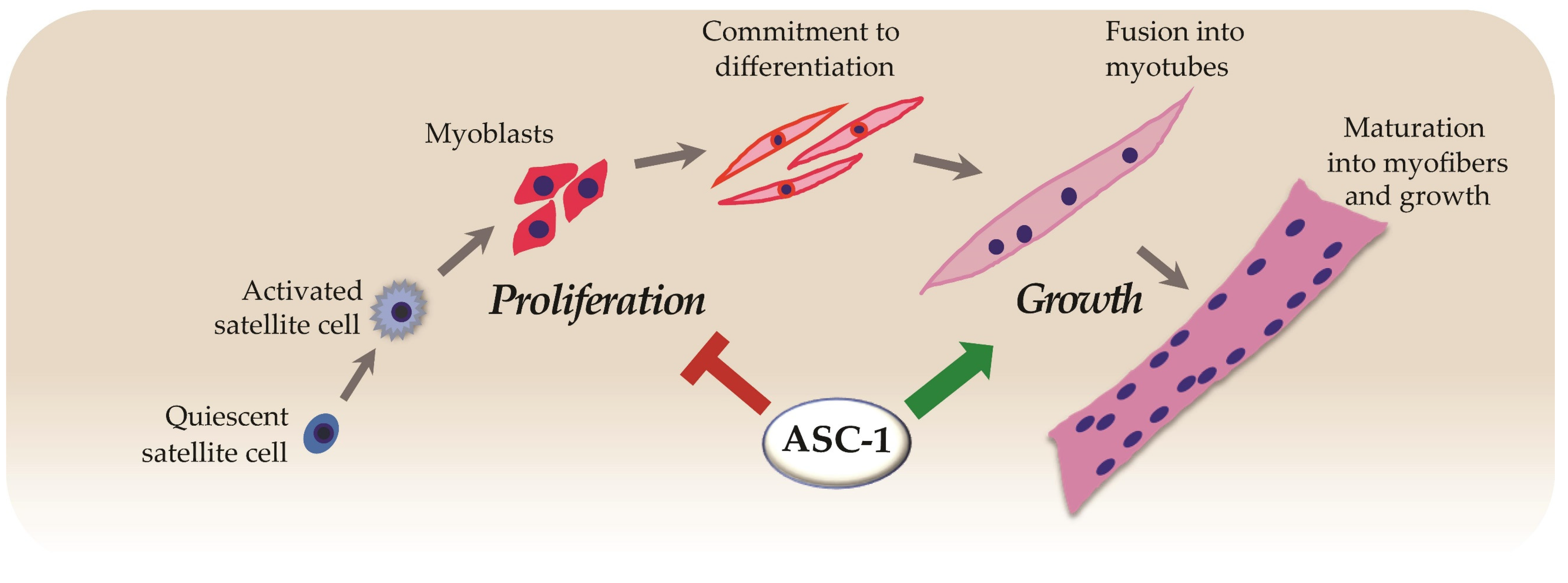
6. ASC-1 Is a Novel Regulator of Cell Proliferation, Growth and Cell Cycle
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALKBH3 | Alpha-ketoglutarate-dependent Dioxygenase alkB Homolog 3 |
| ALS | Amyotrophic Lateral Sclerosis |
| AP-1 | Activator Protein 1 |
| ASC1-RM | ASC-1 Related Myopathy |
| ASC-1 | Activating Signal Cointegrator-1 |
| ASCC1 | Activating Signal Cointegrator-1 complex subunit 1 |
| ASCC2 | Activating Signal Cointegrator-1 complex subunit 2 |
| ASCC3 | Activating Signal Cointegrator-1 complex subunit 3 |
| ASCC | Activation Signal Cointegrator Complex |
| ASCH domain | ASC-1 homology domain |
| CDK4/6 | Cyclin-dependent kinase 4 and 6 |
| CM | Congenital Myopathy |
| CNS | Central Nervous System |
| COX-2 | Cyclooxygenase 2 |
| CUE | Coupling of ubiquitin to ER degradation |
| DDR | DNA Damage Response |
| EM | Electron Microscopy |
| EMG | Electromyogram |
| ER | Endoplasmic Reticulum |
| G0 | quiescent phase of the cell cycle |
| G1 | gap1 phase of the cell cycle |
| KH domain | K-homology domain |
| MAPK | Mitogen Activated Protein Kinase |
| MHC | Myosin Heavy Chain |
| MRI | Magnetic Resonance Imaging |
| mRNA | messenger RNA |
| NADH-TR | NADH-tetrazolium reductase |
| NF-kB | nuclear factor-kappa B |
| NOS | Nitric Oxide Synthase |
| PI3K | Phosphoinositide 3 Kinase |
| pRb | Retinoblastoma protein |
| RNAP II | RNA polymerase II |
| ROS | Reactive Oxygen Species |
| RQC | Ribosome-associated Quality Control |
| hRQT | human RQC-trigger complex |
| SMA | Spinal Muscular Atrophy |
| snRNP | small nuclear ribonucleoprotein |
| SRF | Serum Response Factor |
| TRIP4 | Thyroid Hormone Receptor Interactor 4 |
References
- Böhm, J.; Vasli, N.; Malfatti, E.; Le Gras, S.; Feger, C.; Jost, B.; Monnier, N.; Brocard, J.; Karasoy, H.; Gérard, M.; et al. An Integrated Diagnosis Strategy for Congenital Myopathies. PLoS ONE 2013, 8, e0067527. [Google Scholar] [CrossRef]
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Diagnostic Approach to the Congenital Muscular Dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Schorling, D.C.; Kirschner, J.; Bönnemann, C.G. Congenital Muscular Dystrophies and Myopathies: An Overview and Update. Neuropediatrics 2017, 48, 247–261. [Google Scholar] [CrossRef] [PubMed]
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.H.; Sewry, C.; et al. Approach to the Diagnosis of Congenital Myopathies. Neuromuscul. Disord. 2014, 24, 97–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungbluth, H.; Ochala, J.; Treves, S.; Gautel, M. Current and Future Therapeutic Approaches to the Congenital Myopathies. Semin. Cell Dev. Biol. 2017, 64, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treves, S.; Jungbluth, H.; Voermans, N.; Muntoni, F.; Zorzato, F. Ca2+ Handling Abnormalities in Early-Onset Muscle Diseases: Novel Concepts and Perspectives. Semin. Cell Dev. Biol. 2017, 64, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Horstick, E.J.; Linsley, J.W.; Dowling, J.J.; Hauser, M.A.; McDonald, K.K.; Ashley-Koch, A.; Saint-Amant, L.; Satish, A.; Cui, W.W.; Zhou, W.; et al. Stac3 Is a Component of the Excitation-Contraction Coupling Machinery and Mutated in Native American Myopathy. Nat. Commun. 2013, 4, 1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungbluth, H.; Gautel, M. Pathogenic Mechanisms in Centronuclear Myopathies. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Fujise, K.; Okubo, M.; Abe, T.; Yamada, H.; Nishino, I.; Noguchi, S.; Takei, K.; Takeda, T. Mutant BIN1-Dynamin 2 Complexes Dysregulate Membrane Remodeling in the Pathogenesis of Centronuclear Myopathy. J. Biol. Chem. 2020, 296. [Google Scholar] [CrossRef]
- Lawlor, M.W.; Ottenheijm, C.A.; Lehtokari, V.-L.; Cho, K.; Pelin, K.; Wallgren-Pettersson, C.; Granzier, H.; Beggs, A.H. Novel Mutations in NEB Cause Abnormal Nebulin Expression and Markedly Impaired Muscle Force Generation in Severe Nemaline Myopathy. Skelet. Muscle 2011, 1, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, K.J.; Ravenscroft, G.; Laing, N.G. Skeletal Muscle α-Actin Diseases (Actinopathies): Pathology and Mechanisms. Acta Neuropathol. 2013, 125, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Gonchar, A.D.; Kopylova, G.V.; Kochurova, A.M.; Berg, V.Y.; Shchepkin, D.V.; Koubasova, N.A.; Tsaturyan, A.K.; Kleymenov, S.Y.; Matyushenko, A.M.; Levitsky, D.I. Effects of Myopathy-Causing Mutations R91P and R245G in the TPM3 Gene on Structural and Functional Properties of Slow Skeletal Muscle Tropomyosin. Biochem. Biophys. Res. Commun. 2021, 534, 8–13. [Google Scholar] [CrossRef]
- Jungbluth, H.; Treves, S.; Zorzato, F.; Sarkozy, A.; Ochala, J.; Sewry, C.; Phadke, R.; Gautel, M.; Muntoni, F. Congenital Myopathies: Disorders of Excitation-Contraction Coupling and Muscle Contraction. Nat. Rev. Neurol. 2018, 14, 151–167. [Google Scholar] [CrossRef]
- Kaplan, J.-C.; Hamroun, D. The 2016 Version of the Gene Table of Monogenic Neuromuscular Disorders (Nuclear Genome). Neuromuscul. Disord. 2015, 25, 991–1020. [Google Scholar] [CrossRef] [Green Version]
- Gonorazky, H.D.; Bönnemann, C.G.; Dowling, J.J. The Genetics of Congenital Myopathies. Handb. Clin. Neurol. 2018, 148, 549–564. [Google Scholar] [CrossRef]
- Majczenko, K.; Davidson, A.E.; Camelo-Piragua, S.; Agrawal, P.B.; Manfready, R.A.; Li, X.; Joshi, S.; Xu, J.; Peng, W.; Beggs, A.H.; et al. Dominant Mutation of CCDC78 in a Unique Congenital Myopathy with Prominent Internal Nuclei and Atypical Cores. Am. J. Hum. Genet. 2012, 91, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravenscroft, G.; Bryson-Richardson, R.J.; Nowak, K.J.; Laing, N.G. Recent Advances in Understanding Congenital Myopathies. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Davignon, L.; Chauveau, C.; Julien, C.; Dill, C.; Duband-Goulet, I.; Cabet, E.; Buendia, B.; Lilienbaum, A.; Rendu, J.; Minot, M.C.; et al. The Transcription Coactivator ASC-1 Is a Regulator of Skeletal Myogenesis, and Its Deficiency Causes a Novel Form of Congenital Muscle Disease. Hum. Mol. Genet. 2016, 25, 1559–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.-J.; Sung, H.-S.; Goo, Y.-W.; Lee, H.M.; Park, O.K.; Jung, S.-Y.; Lim, J.; Kim, H.-J.; Lee, S.-K.; Kim, T.S.; et al. Novel Transcription Coactivator Complex Containing Activating Signal Cointegrator 1. Mol. Cell. Biol. 2002, 22, 5203–5211. [Google Scholar] [CrossRef] [Green Version]
- Auboeuf, D.; Hönig, A.; Berget, S.M.; O’Malley, B.W. Coordinate Regulation of Transcription and Splicing by Steroid Receptor Coregulators. Science 2002, 298, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Burroughs, A.M.; Aravind, L. The ASCH Superfamily: Novel Domains with a Fold Related to the PUA Domain and a Potential Role in RNA Metabolism. Bioinformatics 2006, 22, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Villar-Quiles, R.N.; Catervi, F.; Cabet, E.; Juntas-Morales, R.; Genetti, C.A.; Gidaro, T.; Koparir, A.; Yüksel, A.; Coppens, S.; Deconinck, N.; et al. ASC-1 Is a Cell Cycle Regulator Associated with Severe and Mild Forms of Myopathy. Ann. Neurol. 2020, 87, 217–232. [Google Scholar] [CrossRef]
- Knierim, E.; Hirata, H.; Wolf, N.I.; Morales-Gonzalez, S.; Schottmann, G.; Tanaka, Y.; Rudnik-Schöneborn, S.; Orgeur, M.; Zerres, K.; Vogt, S.; et al. Mutations in Subunits of the Activating Signal Cointegrator 1 Complex Are Associated with Prenatal Spinal Muscular Atrophy and Congenital Bone Fractures. Am. J. Hum. Genet. 2016, 98, 473–489. [Google Scholar] [CrossRef] [Green Version]
- Böhm, J.; Malfatti, E.; Oates, E.; Jones, K.; Brochier, G.; Boland, A.; Deleuze, J.-F.; Romero, N.B.; Laporte, J. Novel ASCC1 Mutations Causing Prenatal-Onset Muscle Weakness with Arthrogryposis and Congenital Bone Fractures. J. Med. Genet. 2019, 56, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.; O’Connell, J.D.; Iocolano, A.D.; Coady, J.A.; Yu, Y.; Gangopadhyay, J.; Gygi, S.P.; Reed, R. The Neurodegenerative Diseases ALS and SMA Are Linked at the Molecular Level via the ASC-1 Complex. Nucleic Acids Res. 2018, 46, 11939–11951. [Google Scholar] [CrossRef]
- Bönnemann, C.G. The Collagen VI-Related Myopathies: Muscle Meets Its Matrix. Nat. Rev. Neurol. 2011, 7, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadaszadeh, B.; Petit, N.; Jaillard, C.; Brockington, M.; Quijano Roy, S.; Merlini, L.; Romero, N.; Estournet, B.; Desguerre, I.; Chaigne, D.; et al. Mutations in SEPN1 Cause Congenital Muscular Dystrophy with Spinal Rigidity and Restrictive Respiratory Syndrome. Nat. Genet. 2001, 29, 17–18. [Google Scholar] [CrossRef]
- Ferreiro, A.; Quijano-Roy, S.; Pichereau, C.; Moghadaszadeh, B.; Goemans, N.; Bönnemann, C.; Jungbluth, H.; Straub, V.; Villanova, M.; Leroy, J.-P.; et al. Mutations of the Selenoprotein N Gene, Which Is Implicated in Rigid Spine Muscular Dystrophy, Cause the Classical Phenotype of Multiminicore Disease: Reassessing the Nosology of Early-Onset Myopathies. Am. J. Hum. Genet. 2002, 71, 739–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, N.F.; Kidson, W.; Quijano-Roy, S.; Estournet, B.; Ferreiro, A.; Guicheney, P.; Manson, J.I.; Kornberg, A.J.; Shield, L.K.; North, K.N. SEPN1: Associated with Congenital Fiber-Type Disproportion and Insulin Resistance. Ann. Neurol. 2006, 59, 546–552. [Google Scholar] [CrossRef]
- Scoto, M.; Cirak, S.; Mein, R.; Feng, L.; Manzur, A.Y.; Robb, S.; Childs, A.-M.; Quinlivan, R.M.; Roper, H.; Jones, D.H.; et al. SEPN1-Related Myopathies: Clinical Course in a Large Cohort of Patients. Neurology 2011, 76, 2073–2078. [Google Scholar] [CrossRef]
- Feng, J.-J.; Marston, S. Genotype-Phenotype Correlations in ACTA1 Mutations That Cause Congenital Myopathies. Neuromuscul. Disord. 2009, 19, 6–16. [Google Scholar] [CrossRef]
- Sparrow, J.C.; Nowak, K.J.; Durling, H.J.; Beggs, A.H.; Wallgren-Pettersson, C.; Romero, N.; Nonaka, I.; Laing, N.G. Muscle Disease Caused by Mutations in the Skeletal Muscle Alpha-Actin Gene (ACTA1). Neuromuscul. Disord. 2003, 13, 519–531. [Google Scholar] [CrossRef]
- Oliveira, J.; Martins, M.; Pinto Leite, R.; Sousa, M.; Santos, R. The New Neuromuscular Disease Related with Defects in the ASC-1 Complex: Report of a Second Case Confirms ASCC1 Involvement. Clin. Genet. 2017, 92, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.G.; Mastromoro, G.; Guida, V.; Truglio, M.; Fabbretti, M.; Torres, B.; Mazza, T.; De Luca, A.; Roggini, M.; Bernardini, L.; et al. A New Case of SMABF2 Diagnosed in Stillbirth Expands the Prenatal Presentation and Mutational Spectrum of ASCC1. Am. J. Med. Genet. A 2020, 182, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Liang, M.; Su, J.; Wang, J.; Li, L.; Zhang, S.; Qin, Z.; Huang, L.; Lu, Y.; Yi, S.; et al. Novel Compound Heterozygous Pathogenic Variants in ASCC1 in a Chinese Patient with Spinal Muscular Atrophy with Congenital Bone Fractures 2: Evidence Supporting a “Definitive” Gene-Disease Relationship. Mol. Genet. Genom. Med. 2020, 8, e1212. [Google Scholar] [CrossRef] [Green Version]
- Soll, J.M.; Brickner, J.R.; Mudge, M.C.; Mosammaparast, N. RNA Ligase-like Domain in Activating Signal Cointegrator 1 Complex Subunit 1 (ASCC1) Regulates ASCC Complex Function during Alkylation Damage. J. Biol. Chem. 2018, 293, 13524–13533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brickner, J.R.; Soll, J.M.; Lombardi, P.M.; Vågbø, C.B.; Mudge, M.C.; Oyeniran, C.; Rabe, R.; Jackson, J.; Sullender, M.E.; Blazosky, E.; et al. A Ubiquitin-Dependent Signalling Axis Specific for ALKBH-Mediated DNA Dealkylation Repair. Nature 2017, 551, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Dango, S.; Mosammaparast, N.; Sowa, M.E.; Xiong, L.-J.; Wu, F.; Park, K.; Rubin, M.; Gygi, S.; Harper, J.W.; Shi, Y. DNA Unwinding by ASCC3 Helicase Is Coupled to ALKBH3-Dependent DNA Alkylation Repair and Cancer Cell Proliferation. Mol. Cell 2011, 44, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Sugiyama, T.; Yamazaki, R.; Nobuta, R.; Inada, T. Identification of a Novel Trigger Complex That Facilitates Ribosome-Associated Quality Control in Mammalian Cells. Sci. Rep. 2020, 10, 3422. [Google Scholar] [CrossRef] [Green Version]
- Juszkiewicz, S.; Speldewinde, S.H.; Wan, L.; Svejstrup, J.Q.; Hegde, R.S. The ASC-1 Complex Disassembles Collided Ribosomes. Mol. Cell 2020, 79, 603–614.e8. [Google Scholar] [CrossRef]
- Schmoller, K.M.; Skotheim, J.M. The Biosynthetic Basis of Cell Size Control. Trends Cell Biol. 2015, 25, 793–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruijtenberg, S.; van den Heuvel, S. Coordinating Cell Proliferation and Differentiation: Antagonism between Cell Cycle Regulators and Cell Type-Specific Gene Expression. Cell Cycle 2016, 15, 196–212. [Google Scholar] [CrossRef] [Green Version]
- Ginzberg, M.B.; Kafri, R.; Kirschner, M. Cell Biology. On Being the Right (Cell) Size. Science 2015, 348, 1245075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegelman, B.M.; Heinrich, R. Biological Control through Regulated Transcriptional Coactivators. Cell 2004, 119, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Baudet, C.; Pozas, E.; Adameyko, I.; Andersson, E.; Ericson, J.; Ernfors, P. Retrograde Signaling onto Ret during Motor Nerve Terminal Maturation. J. Neurosci. 2008, 28, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia Induce Motor Neuron Death via the Classical NF-ΚB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 Activates Microglia through NF-ΚB and NLRP3 Inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant IPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA Damage and Its Links to Neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.J.; Mordes, D.A.; Cameron, L.A.; Neuberg, D.S.; Landini, S.; Eggan, K.; Livingston, D.M. Two Familial ALS Proteins Function in Prevention/Repair of Transcription-Associated DNA Damage. Proc. Natl. Acad. Sci. USA 2016, 113, E7701–E7709. [Google Scholar] [CrossRef] [Green Version]
- McMurray, C.T. To Die or Not to Die: DNA Repair in Neurons. Mutat. Res. 2005, 577, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA Repair, Genome Stability, and Aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedgwick, B. Repairing DNA-Methylation Damage. Nat. Rev. Mol. Cell Biol. 2004, 5, 148–157. [Google Scholar] [CrossRef]
- Ryan, M.M.; Schnell, C.; Strickland, C.D.; Shield, L.K.; Morgan, G.; Iannaccone, S.T.; Laing, N.G.; Beggs, A.H.; North, K.N. Nemaline Myopathy: A Clinical Study of 143 Cases. Ann. Neurol. 2001, 50, 312–320. [Google Scholar] [CrossRef]
- Garcia-Angarita, N.; Kirschner, J.; Heiliger, M.; Thirion, C.; Walter, M.C.; Schnittfeld-Acarlioglu, S.; Albrecht, M.; Müller, K.; Wieczorek, D.; Lochmüller, H.; et al. Severe Nemaline Myopathy Associated with Consecutive Mutations E74D and H75Y on a Single ACTA1 Allele. Neuromuscul. Disord. 2009, 19, 481–484. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Miyatake, S.; Lehtokari, V.-L.; Todd, E.J.; Vornanen, P.; Yau, K.S.; Hayashi, Y.K.; Miyake, N.; Tsurusaki, Y.; Doi, H.; et al. Mutations in KLHL40 Are a Frequent Cause of Severe Autosomal-Recessive Nemaline Myopathy. Am. J. Hum. Genet. 2013, 93, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.F. Functions of AP1 (Fos/Jun) in Bone Development. Ann. Rheum. Dis. 2002, 61, ii40–ii42. [Google Scholar] [CrossRef]
- Kawamata, A.; Izu, Y.; Yokoyama, H.; Amagasa, T.; Wagner, E.F.; Nakashima, K.; Ezura, Y.; Hayata, T.; Noda, M. JunD Suppresses Bone Formation and Contributes to Low Bone Mass Induced by Estrogen Depletion. J. Cell. Biochem. 2008, 103, 1037–1045. [Google Scholar] [CrossRef]
- Bozec, A.; Bakiri, L.; Jimenez, M.; Schinke, T.; Amling, M.; Wagner, E.F. Fra-2/AP-1 Controls Bone Formation by Regulating Osteoblast Differentiation and Collagen Production. J. Cell Biol. 2010, 190, 1093–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerbs, T.; Cui, L.; Muscat, C.; Saleem, A.; van Neste, C.; Domizi, P.; Chan, C.; Wernig, G. Expansion of Bone Precursors through Jun as a Novel Treatment for Osteoporosis-Associated Fractures. Stem Cell Rep. 2020, 14, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Drissi, H.; Hushka, D.; Aslam, F.; Nguyen, Q.; Buffone, E.; Koff, A.; van Wijnen, A.; Lian, J.B.; Stein, J.L.; Stein, G.S. The Cell Cycle Regulator P27kip1 Contributes to Growth and Differentiation of Osteoblasts. Cancer Res. 1999, 59, 3705–3711. [Google Scholar] [PubMed]
- Ogasawara, T.; Mori, Y.; Abe, M.; Suenaga, H.; Kawase-Koga, Y.; Saijo, H.; Takato, T. Role of Cyclin-Dependent Kinase (Cdk)6 in Osteoblast, Osteoclast, and Chondrocyte Differentiation and Its Potential as a Target of Bone Regenerative Medicine. Oral Sci. Int. 2011, 8, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Orloff, M.; Peterson, C.; He, X.; Ganapathi, S.; Heald, B.; Yang, Y.; Bebek, G.; Romigh, T.; Song, J.H.; Wu, W.; et al. Germline Mutations in MSR1, ASCC1, and CTHRC1 in Patients with Barrett Esophagus and Esophageal Adenocarcinoma. JAMA 2011, 306, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Van Nistelrooij, A.M.J.; Dinjens, W.N.M.; Wagner, A.; Spaander, M.C.W.; van Lanschot, J.J.B.; Wijnhoven, B.P.L. Hereditary Factors in Esophageal Adenocarcinoma. Gastrointest. Tumors 2014, 1, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Xu, H.; Luo, M.; Yu, W.; Chen, M.; Liao, Y.; Zhang, C.; Zhao, X.; Jiang, W.; Hou, S.; et al. The Tumor-Promoting Role of TRIP4 in Melanoma Progression and Its Involvement in Response to BRAF-Targeted Therapy. J. Investig. Dermatol. 2018, 138, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, Y.; Li, Y.; Zheng, F.; Zou, K.; Li, Z.; Chen, M.; Hu, S.; Tian, C.; Yu, W.; Guo, W.; et al. TRIP4 Promotes Tumor Growth and Metastasis and Regulates Radiosensitivity of Cervical Cancer by Activating MAPK, PI3K/AKT, and HTERT Signaling. Cancer Lett. 2019, 452, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ren, J.; Bai, Y.; Jiang, J.; Xiao, S. MicroRNA-518-3p Suppresses Cell Proliferation, Invasiveness, and Migration in Colorectal Cancer via Targeting TRIP4. Biochem. Cell Biol. 2020, 98, 575–582. [Google Scholar] [CrossRef]
- Yoo, H.M.; Kang, S.H.; Kim, J.Y.; Lee, J.E.; Seong, M.W.; Lee, S.W.; Ka, S.H.; Sou, Y.-S.; Komatsu, M.; Tanaka, K.; et al. Modification of ASC1 by UFM1 Is Crucial for ERα Transactivation and Breast Cancer Development. Mol. Cell 2014, 56, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Publications | TRIP4 Exon | cDNA Change | Protein Change | Variation Type | Predicted Effect | Primary Involvement Reported |
|---|---|---|---|---|---|---|
| Davignon et al., 2016 | 7 | c.950G>A (homozygous) | p.Trp297Ter | substitution | nonsense | muscle |
| Knierim et al., 2016 | 6 | 7 | c.760C>T (hom or comp het) | p.Arg254Ter | deletion | nonsense | motor neuron + bone |
| c.832C>T (hom or comp het) | p.Arg278Ter | deletion | nonsense | motor neuron + bone | ||
| Villar-Quiles et al., 2020 | 2 | c.141_142delAT (homozygous) | p.Tyr48CysfsTer3 | deletion | frameshift | muscle |
| 4 | 11 | c.534C>G (comp het) | p.His178Gln | substitution | missense | muscle | |
| c.1544_1547delACTG (comp het) | p.Asp515AlafsTer34 | deletion | frameshift | muscle | ||
| 8 | c.1065delC (homozygous) | p.Ile356LeufsTer6 | deletion | frameshift | muscle | |
| 1 | 9 | c.55_56insCT (comp het) | p.Gln19ProfsTer47 | insertion | frameshift | muscle | |
| c.1197delA (comp het) | p.Ser399SerfsTer12 | deletion | frameshift | muscle | ||
| 8 + 9 | homozygous deletion exons 8 and 9 | deletion | in-frame deletion | muscle | ||
| Publications | ASCC1 Exon | cDNA Change | Protein Change | Variation Type | Predict Effect | Primary Involvement Reported |
|---|---|---|---|---|---|---|
| Knierim et al., 2016 | 3a | c.157dupG (homozygous) | p.Glu53GlyfsTer19 | single base pair duplication | frameshift | motor neuron/CNS + bone |
| Oliveira et al., 2017 | 3a | c.157dupG (homozygous) | p.Glu53GlyfsTer19 | single base pair duplication | frameshift | n.c. muscle? + bone |
| Böhm et al., 2019 | 3a | 5 | c.157dupG (hom or comp het) | p.Glu53GlyfsTer19 | single base pair duplication | frameshift | muscle + bone |
| c.466C>T (comp het) | p.Arg156Ter | substitution | nonsense | muscle + bone | ||
| 6 | c.667C>T (homozygous) | p.Glu223Ter | substitution | nonsense | muscle +bone | |
| Giuffrida et al., 2019 | 9a | 6–9a | c.1027C>T (comp het) | p.Arg343Ter | substitution | nonsense | bone + ? |
| hemizygous deletion exons 6 to 9a | microdeletion | in-frame deletion | bone + ? | |||
| Lu et al., 2020 | 5 | 9 | hemizygous deletion in exon 5 | microdeletion | frameshift | CNS + bone | |
| c.932C>G (comp het) | p.Ser311Ter | substitution | nonsense | CNS + bone | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meunier, J.; Villar-Quiles, R.-N.; Duband-Goulet, I.; Ferreiro, A. Inherited Defects of the ASC-1 Complex in Congenital Neuromuscular Diseases. Int. J. Mol. Sci. 2021, 22, 6039. https://doi.org/10.3390/ijms22116039
Meunier J, Villar-Quiles R-N, Duband-Goulet I, Ferreiro A. Inherited Defects of the ASC-1 Complex in Congenital Neuromuscular Diseases. International Journal of Molecular Sciences. 2021; 22(11):6039. https://doi.org/10.3390/ijms22116039
Chicago/Turabian StyleMeunier, Justine, Rocio-Nur Villar-Quiles, Isabelle Duband-Goulet, and Ana Ferreiro. 2021. "Inherited Defects of the ASC-1 Complex in Congenital Neuromuscular Diseases" International Journal of Molecular Sciences 22, no. 11: 6039. https://doi.org/10.3390/ijms22116039