
Is PTSD-Phenotype Associated with HPA-Axis Sensitivity? Feedback Inhibition and Other Modulating Factors of Glucocorticoid Signaling Dynamics

Abstract
:1. Introduction
2. Results
2.1. Behavioral Responses at Day 7 Post-PSS Exposure
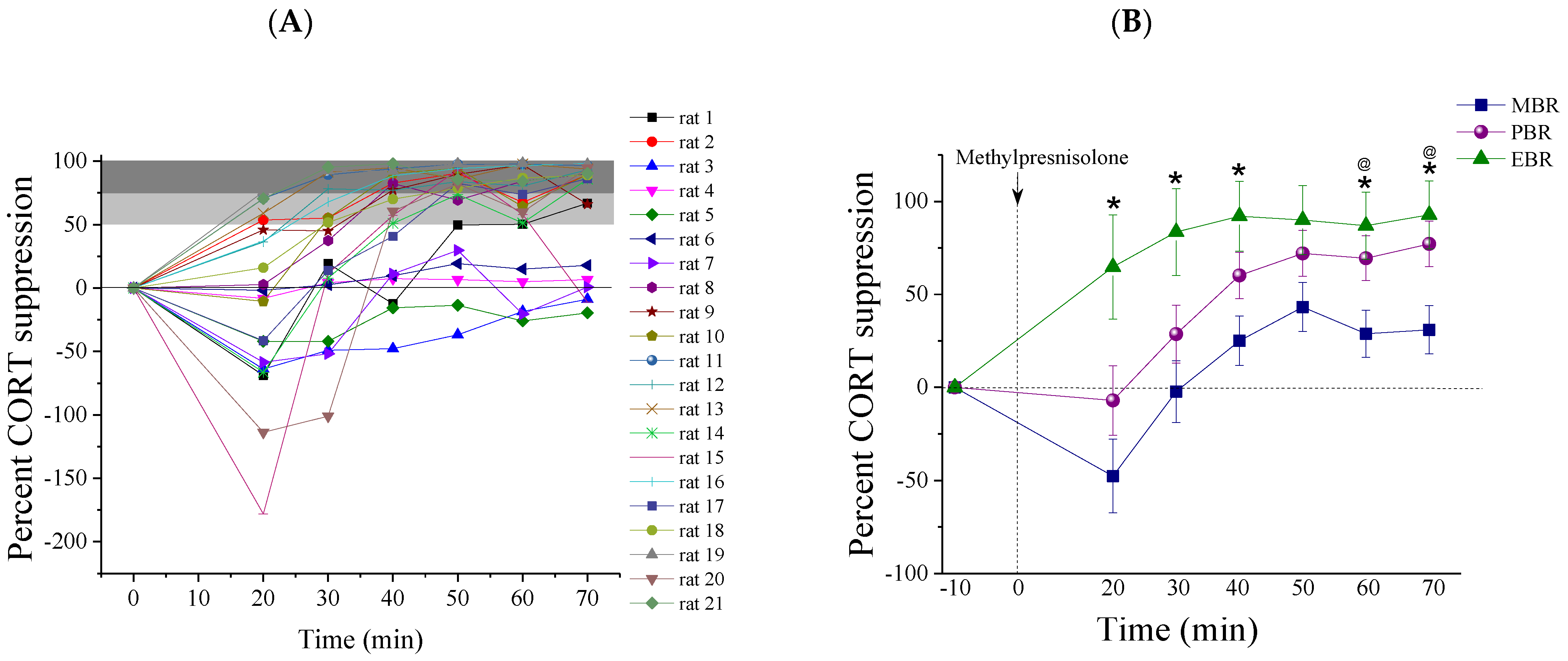
2.2. Methylprednisolone Suppression Test (MPST)
2.3. Pulsatile Activity during MPST
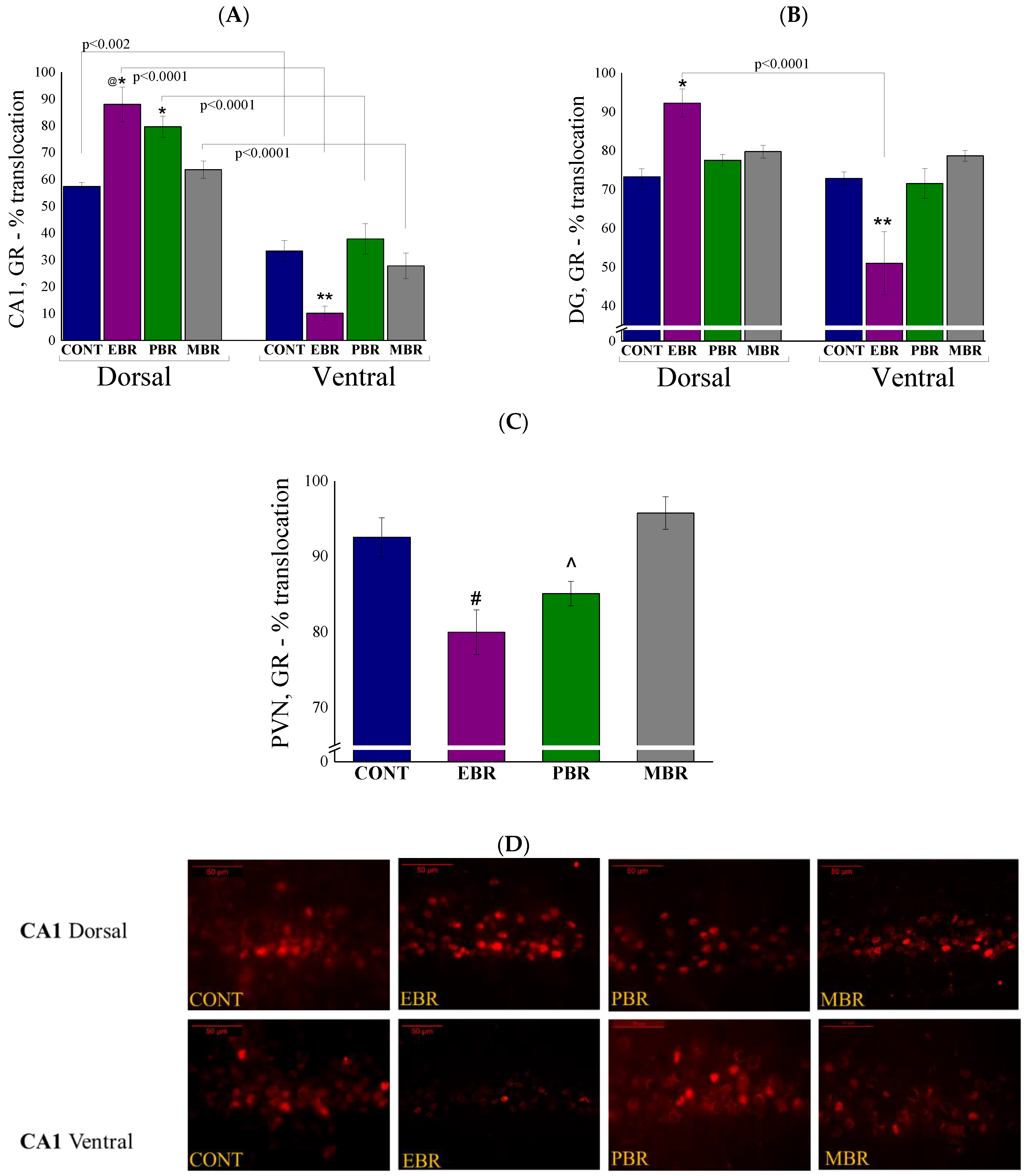
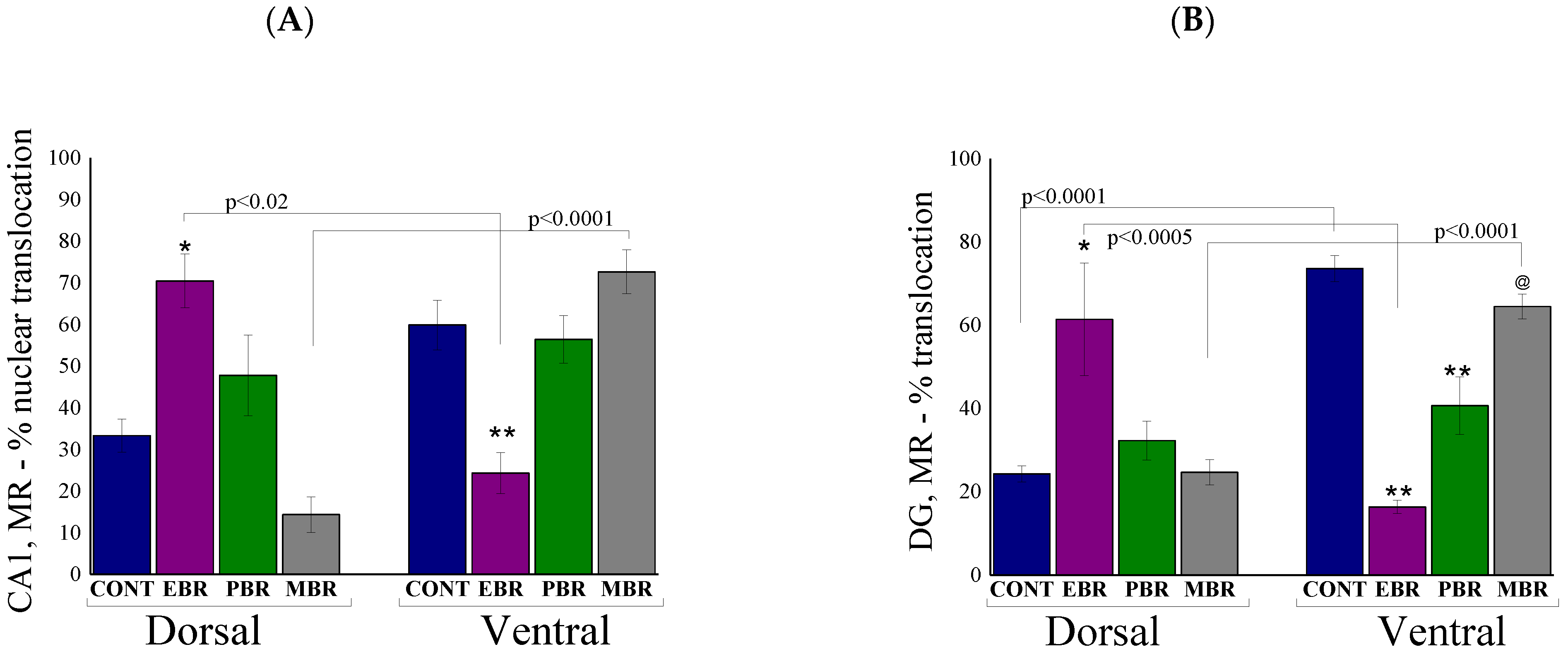
2.4. Brain Immunohistochemistry
2.5. Brain GR-ir Cells 8 Days Post-PSS Exposure
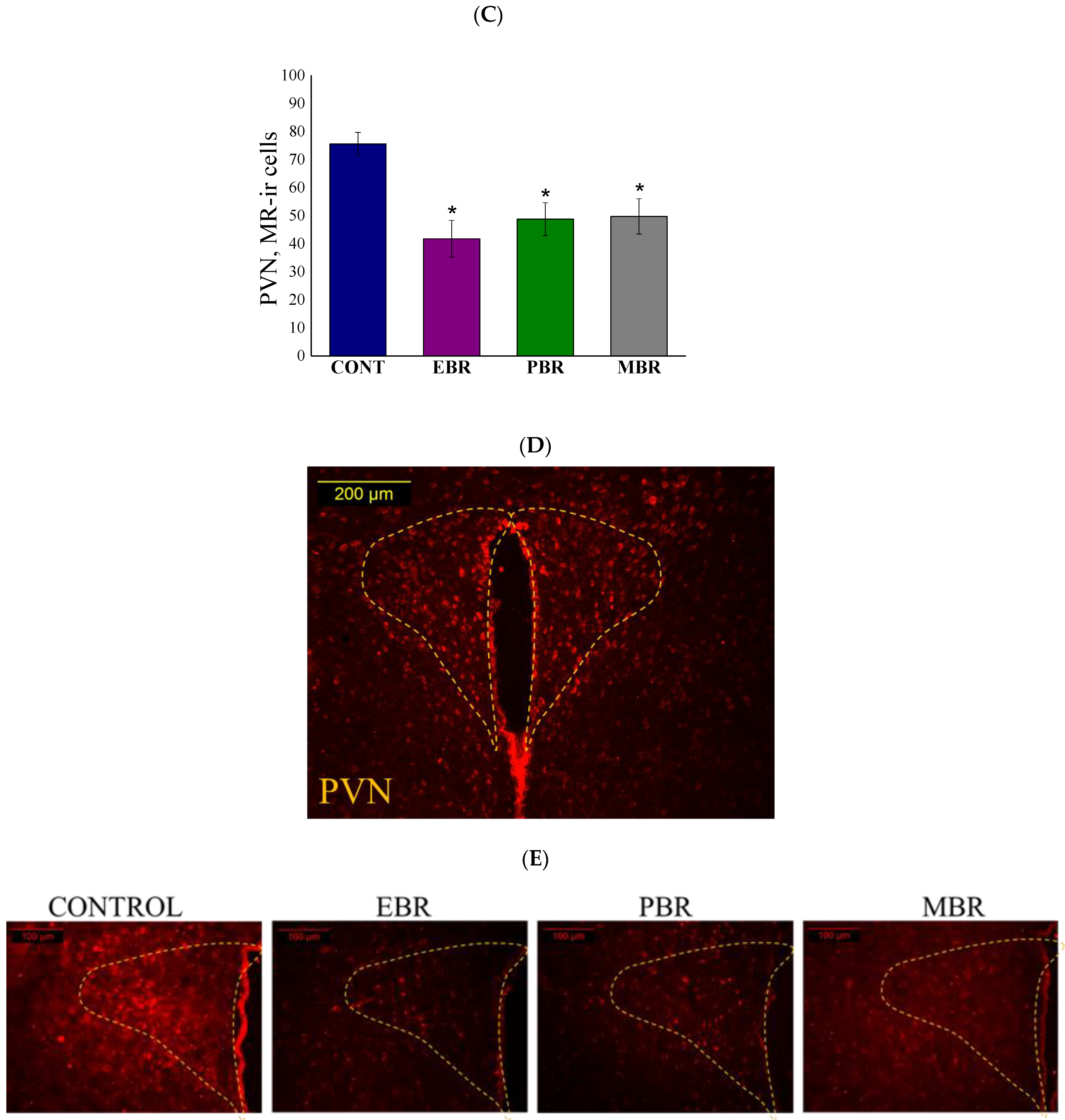
2.6. Brain MR-ir Cells 8 Days Post-PSS Exposure
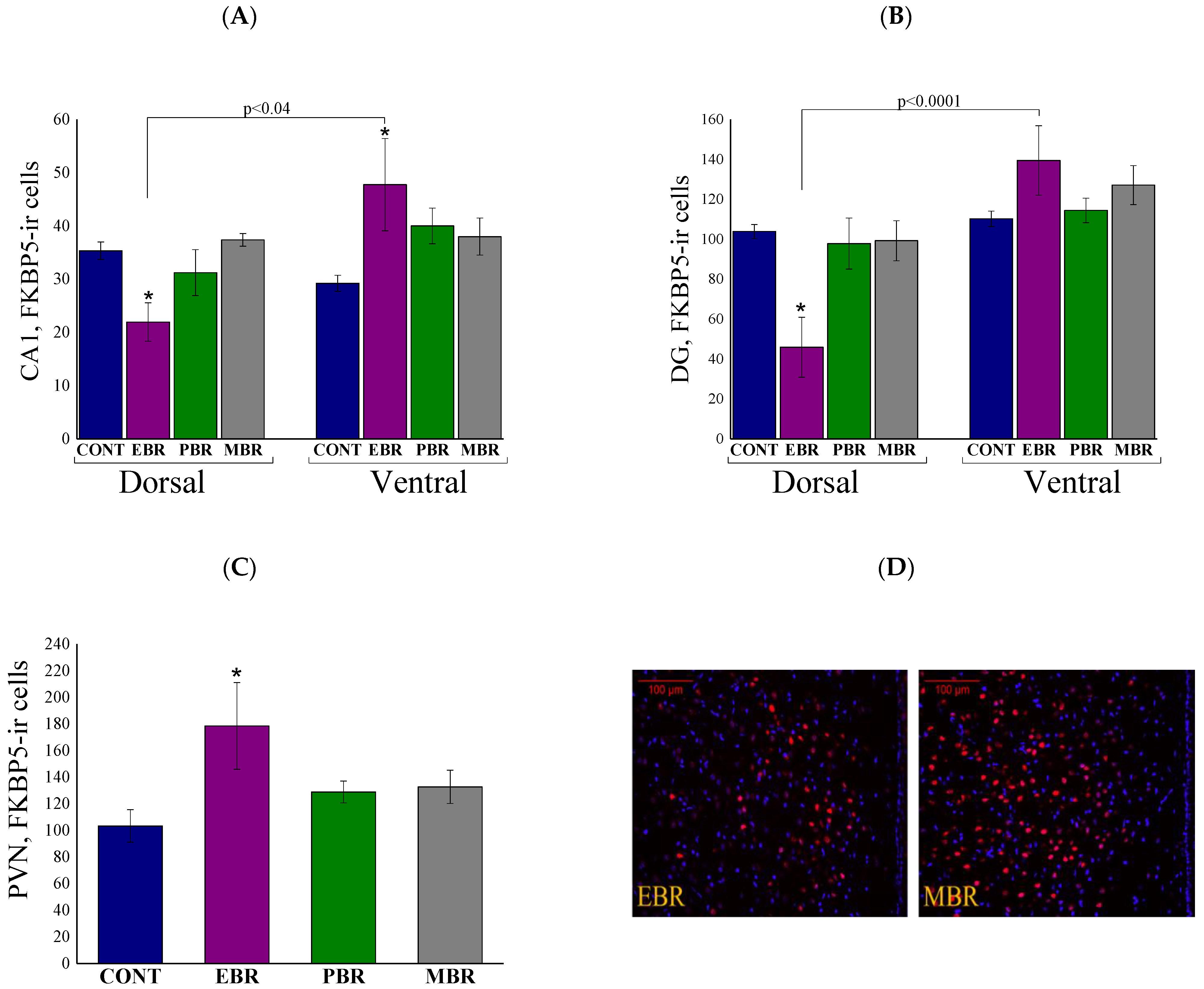
2.7. Brain FKBP5 Levels 8 Days Post-PSS Exposure
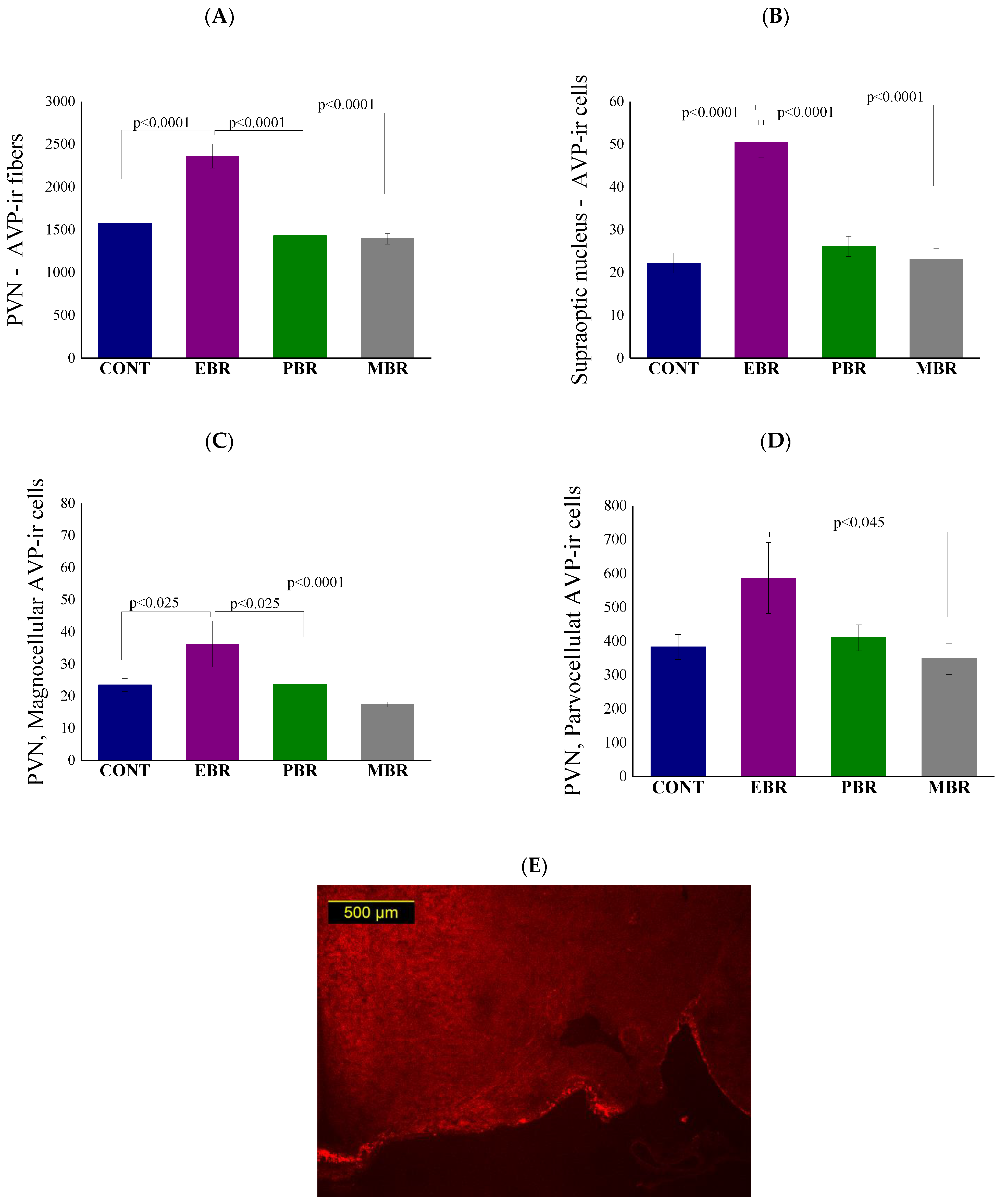
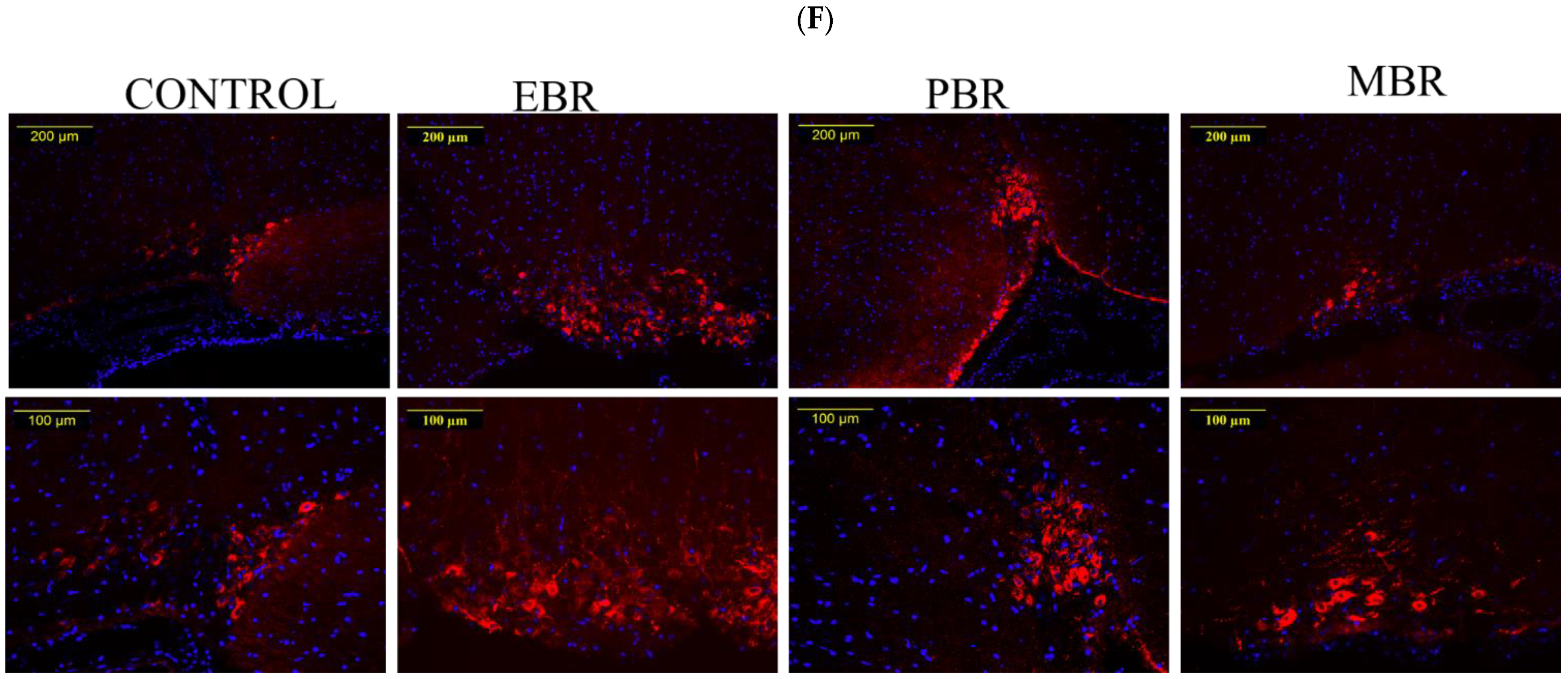
2.8. Brain AVP Levels 8 Days Post-PSS Exposure
3. Discussion
4. Materials and Methods
4.1. Animals
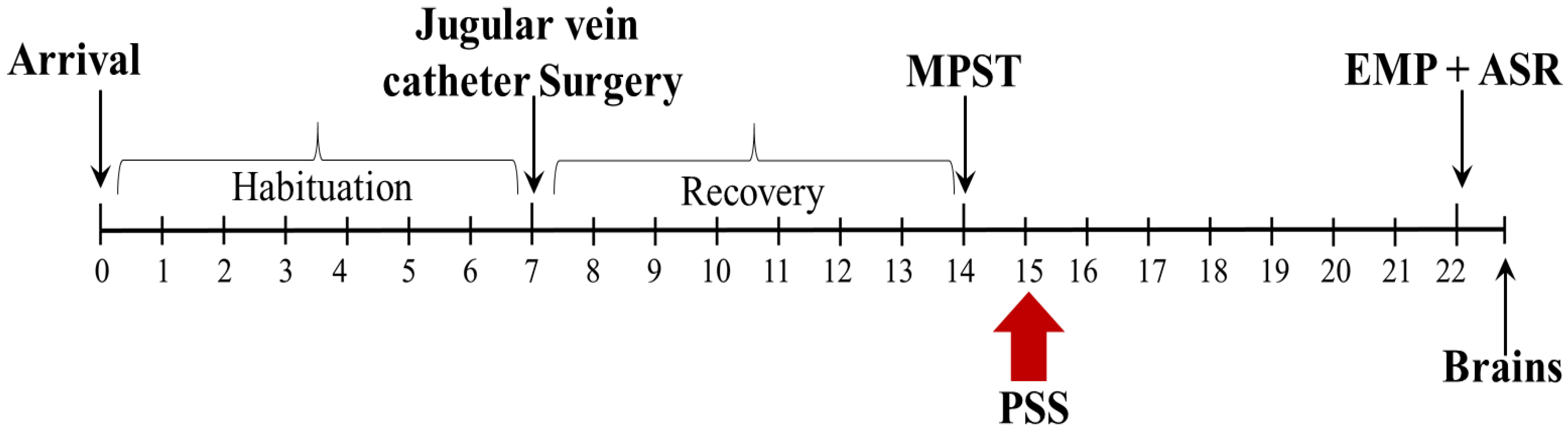
4.2. Experimental Design
4.3. Predator Scent Stress (PSS)
4.4. Behavioral Measurements
4.5. Cut-Off Behavioral Criteria (CBC) Model
4.6. Mesh Barrier Housing
4.7. Jugular Vein Catheter Surgery and Blood Sampling
4.8. Measurement of Serum Corticosterone
4.9. Methylprednisolone Suppression Test
4.10. Immunohistochemistry
4.11. Quantification of MR and GR Translocation Patterns in Hippocampal Subregions
4.12. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| ASR | Acoustic startle response |
| AVP | Arginine vasopressin |
| DG | Dentate gyrus |
| EBR | Extreme behavioral response |
| EPM | Elevated plus-maze |
| FKBP5 | FK506-binding protein 51 |
| GC | Glucocorticoid |
| GR | Glucocorticoid receptor |
| HPA | Hypothalamus–pituitary–adrenal |
| MBR | Minimal behavioral response |
| MPST | Methylprednisolone suppression test |
| MR | Mineralocorticoid receptor |
| PBR | Partial behavioral response |
| PSS | Predator scent stress |
| PTSD | Posttraumatic stress disorder |
| PVN | Paraventricular nucleus of the hypothalamus |
| RM | Repeated measure |
| SCN | Suprachiasmatic nucleus |
References
- McEwen, B.S. The neurobiology and neuroendocrinology of stress. Psychiatr. Clin. N. Am. 2002, 25, 469–494. [Google Scholar] [CrossRef]
- Yehuda, R. Psychoneuroendocrinology of Post-Traumatic Stress Disorder. Psychiatr. Clin. N. Am. 1998, 21, 359–379. [Google Scholar] [CrossRef]
- Danan, D.; Matar, M.A.; Kaplan, Z.; Zohar, J.; Cohen, H. Blunted basal corticosterone pulsatility predicts post-exposure susceptibility to PTSD phenotype in rats. Psychoneuroendocrinology 2018, 87, 35–42. [Google Scholar] [CrossRef]
- McGaugh, J.L.; Roozendaal, B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr. Opin. Neurobiol. 2002, 12, 205–210. [Google Scholar] [CrossRef]
- Goldstein, D.S.; McEwen, B. Allostasis, Homeostats, and the Nature of Stress. Stress 2002, 5, 55–58. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; (DSM-IV); American Psychiatric Association: Washington, DC, USA, 1995; Volume 152. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 0-89042-555-8. [Google Scholar]
- Delahanty, D.; Raimonde, A.; Spoonster, E. Initial posttraumatic urinary cortisol levels predict subsequent PTSD symptoms in motor vehicle accident victims. Biol. Psychiatry 2000, 48, 940–947. [Google Scholar] [CrossRef]
- McFarlane, A.C.; Atchison, M.; Yehuda, R. The acute stress response following motor vehicle accidents and its relation to PTSD. Ann. N. Y. Acad. Sci. 1997, 821, 437–441. [Google Scholar] [CrossRef]
- McFarlane, A.C.; Barton, C.A.; Yehuda, R.; Wittert, G. Cortisol response to acute trauma and risk of posttraumatic stress disorder. Psychoneuroendocrinology 2011, 36, 720–727. [Google Scholar] [CrossRef]
- McFarlane, S. Dendritic Morphogenesis. Mol. Neurobiol. 2000, 22, 001–010. [Google Scholar] [CrossRef]
- Witteveen, A.B.; Huizink, A.C.; Slottje, P.; Bramsen, I.; Smid, T.; Van Der Ploeg, H.M. Associations of cortisol with posttraumatic stress symptoms and negative life events: A study of police officers and firefighters. Psychoneuroendocrinology 2010, 35, 1113–1118. [Google Scholar] [CrossRef]
- Windle, R.J.; Wood, S.A.; Shanks, N.; Lightman, S.L.; Ingram, C. Ultradian Rhythm of Basal Corticosterone Release in the Female Rat: Dynamic Interaction with the Response to Acute Stress. Endocrinology 1998, 139, 443–450. [Google Scholar] [CrossRef]
- Sarabdjitsingh, R.A.; Isenia, S.; Polman, A.; Mijalkovic, J.; Lachize, S.; Datson, N.; De Kloet, E.R.; Meijer, O.C. Disrupted Corticosterone Pulsatile Patterns Attenuate Responsiveness to Glucocorticoid Signaling in Rat Brain. Endocrinology 2010, 151, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.J.; Spiga, F.; Gupta, R.; Zhao, Z.; Lightman, S.L.; Terry, J.R. Rapid intra-adrenal feedback regulation of glucocorticoid synthesis. J. R. Soc. Interface 2015, 12, 20140875. [Google Scholar] [CrossRef]
- George, C.L.; Birnie, M.; Flynn, B.P.; Kershaw, Y.M.; Lightman, S.L.; Conway-Campbell, B.L. Ultradian glucocorticoid exposure directs gene-dependent and tissue-specific mRNA expression patterns in vivo. Mol. Cell. Endocrinol. 2017, 439, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiga, F.; Zavala, E.; Walker, J.J.; Zhao, Z.; Terry, J.R.; Lightman, S.L. Dynamic responses of the adrenal steroidogenic regulatory network. Proc. Natl. Acad. Sci. USA 2017, 114, E6466–E6474. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.J.; Spiga, F.; Waite, E.; Zhao, Z.; Kershaw, Y.; Terry, J.R.; Lightman, S.L. The Origin of Glucocorticoid Hormone Oscillations. PLoS Biol. 2012, 10, e1001341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henley, D.E.; Leendertz, J.A.; Russell, G.; Wood, S.A.; Taheri, S.; Woltersdorf, W.W.; Lightman, S.L. Development of an automated blood sampling system for use in humans. J. Med. Eng. Technol. 2009, 33, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Matar, M.A.; Joseph, Z. Animal Models of Post-Traumatic Stress Disorder. Curr. Protoc. Neurosci. 2013, 64, 9–45. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Zohar, J.; Matar, M. The relevance of differential response to trauma in an animal model of posttraumatic stress disorder. Biol. Psychiatry 2003, 53, 463–473. [Google Scholar] [CrossRef]
- Cohen, H.; Zohar, J.; Gidron, Y.; Matar, M.A.; Belkind, D.; Loewenthal, U.; Kozlovsky, N.; Kaplan, Z. Blunted HPA Axis Response to Stress Influences Susceptibility to Posttraumatic Stress Response in Rats. Biol. Psychiatry 2006, 59, 1208–1218. [Google Scholar] [CrossRef]
- Cohen, H.; Matar, M.A.; Buskila, D.; Kaplan, Z.; Zohar, J. Early Post-Stressor Intervention with High-Dose Corticosterone Attenuates Posttraumatic Stress Response in an Animal Model of Posttraumatic Stress Disorder. Biol. Psychiatry 2008, 64, 708–717. [Google Scholar] [CrossRef]
- Zohar, J.; Juven-Wetzler, A.; Sonnino, R.; Cwikel-Hamzany, S.; Balaban, E.; Cohen, H. New insights into secondary prevention in post-traumatic stress disorder. Dialog. Clin. Neurosci. 2011, 13, 301–309. [Google Scholar]
- Ii, D.A.D.; Adler, G.K.; Nguyen, D.P.; Klerman, E.B. Biological Time Series Analysis Using a Context Free Language: Applicability to Pulsatile Hormone Data. PLoS ONE 2014, 9, e104087. [Google Scholar] [CrossRef] [Green Version]
- Andrews, M.H.; Wood, S.A.; Windle, R.J.; Lightman, S.L.; Ingram, C.D. Acute Glucocorticoid Administration Rapidly Suppresses Basal and Stress-Induced Hypothalamo-Pituitary-Adrenal Axis Activity. Endocrinology 2012, 153, 200–211. [Google Scholar] [CrossRef]
- McEwen, B.S. The neurobiology of stress: From serendipity to clinical relevance11Published on the World Wide Web on 22 November. Brain Res. 2000, 886, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialog. Clin. Neurosci. 2006, 8, 383–395. [Google Scholar]
- Whitaker, A.M.; Farooq, M.A.; Edwards, S.; Gilpin, N.W. Post-traumatic stress avoidance is attenuated by corticosterone and associated with brain levels of steroid receptor co-activator-1 in rats. Stress 2016, 19, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, D.A.; Arbuckle, M.R.; Travis, M.J.; Dwyer, J.B.; Van Schalkwyk, G.I.; Ressler, K.J. An Integrated Neuroscience Perspective on Formulation and Treatment Planning for Posttraumatic Stress Disorder. JAMA Psychiatry 2017, 74, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehuda, R.; Southwick, S.M.; Krystal, J.H.; Bremner, D.; Charney, D.S.; Mason, J.W. Enhanced suppression of cortisol following dexamethasone administration in posttraumatic stress disorder. Am. J. Psychiatry 1993, 150, 83–86. [Google Scholar] [CrossRef]
- de Kloet, C.; Vermetten, E.; Geuze, E.; Kavelaars, A.; Heijnen, C.; Westenberg, H. Assessment of HPA-axis function in posttraumatic stress disorder: Pharmacological and non-pharmacological challenge tests, a review. J. Psychiatr. Res. 2006, 40, 550–567. [Google Scholar] [CrossRef]
- Morris, M.C.; Compas, B.E.; Garber, J. Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: A systematic review and meta-analysis. Clin. Psychol. Rev. 2012, 32, 301–315. [Google Scholar]
- Yehuda, R.; Lowy, M.T.; Southwick, S.M.; Shaffer, D.; Giller, E.L. Lymphocyte glucocorticoid receptor number in posttraumatic stress disorder. Am. J. Psychiatry 1991, 148, 499–504. [Google Scholar] [CrossRef]
- Yehuda, R.; Boisoneau, D.; Lowy, M.T.; Giller, E.L. Dose-Response Changes in Plasma Cortisol and Lymphocyte Glucocorticoid Receptors Following Dexamethasone Administration in Combat Veterans with and without Posttraumatic Stress Disorder. Arch. Gen. Psychiatry 1995, 52, 583–593. [Google Scholar] [CrossRef]
- De Kloet, C.S.; Vermetten, E.; Bikker, A.; Meulman, E.; Geuze, E.; Kavelaars, A.; Westenberg, H.G.M.; Heijnen, C.J. Leukocyte glucocorticoid receptor expression and immunoregulation in veterans with and without post-traumatic stress disorder. Mol. Psychiatry 2007, 12, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Gotovac, K.; Sabioncello, A.; Rabatic, S.; Berki, T.; Dekaris, D. Flow cytometric determination of glucocorticoid receptor (GCR) expression in lymphocyte subpopulations: Lower quantity of GCR in patients with post-traumatic stress disorder (PTSD). Clin. Exp. Immunol. 2003, 131, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Matić, G.; Milutinović, D.V.; Nestorov, J.; Elaković, I.; Jovanović, S.M.; Perišić, T.; Dunđerski, J.; Damjanović, S.; Knežević, G.; Špirić, Ž.; et al. Lymphocyte glucocorticoid receptor expression level and hormone-binding properties differ between war trauma-exposed men with and without PTSD. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 43, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Gola, H.; Engler, A.; Morath, J.; Adenauer, H.; Elbert, T.; Kolassa, I.-T.; Engler, H. Reduced Peripheral Expression of the Glucocorticoid Receptor α Isoform in Individuals with Posttraumatic Stress Disorder: A Cumulative Effect of Trauma Burden. PLoS ONE 2014, 9, e86333. [Google Scholar] [CrossRef] [Green Version]
- Yehuda, R.; A Golier, J.; Yang, R.-K.; Tischler, L. Enhanced sensitivity to glucocorticoids in peripheral mononuclear leukocytes in posttraumatic stress disorder. Biol. Psychiatry 2004, 55, 1110–1116. [Google Scholar] [CrossRef]
- Golier, J.A.; Schmeidler, J.; Legge, J.; Yehuda, R. Enhanced cortisol suppression to dexamethasone associated with Gulf War deployment. Psychoneuroendocrinology 2006, 31, 1181–1189. [Google Scholar] [CrossRef]
- Shalev, A.Y.; Videlock, E.J.; Peleg, T.; Segman, R.; Pitman, R.K.; Yehuda, R. Stress hormones and post-traumatic stress disorder in civilian trauma victims: A longitudinal study. Part I: HPA axis responses. Int. J. Neuropsychopharmacol. 2008, 11, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savić, D.; Knezevic, G.; Damjanovic, S.; Spiric, Z.; Matić, G. The role of personality and traumatic events in cortisol levels – Where does PTSD fit in? Psychoneuroendocrinology 2012, 37, 937–947. [Google Scholar] [CrossRef]
- Dunlop, B.W.; Wong, A. The hypothalamic-pituitary-adrenal axis in PTSD: Pathophysiology and treatment interventions. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 89, 361–379. [Google Scholar] [CrossRef]
- Van Zuiden, M.; Heijnen, C.J.; Maas, M.; Amarouchi, K.; Vermetten, E.; Geuze, E.; Kavelaars, A. Glucocorticoid sensitivity of leukocytes predicts PTSD, depressive and fatigue symptoms after military deployment: A prospective study. Psychoneuroendocrinology 2012, 37, 1822–1836. [Google Scholar] [CrossRef] [PubMed]
- Galatzer-Levy, I.R.; Steenkamp, M.M.; Brown, A.D.; Qian, M.; Inslicht, S.; Henn-Haase, C.; Otte, C.; Yehuda, R.; Neylan, T.C.; Marmar, C.R. Cortisol response to an experimental stress paradigm prospectively predicts long-term distress and resilience trajectories in response to active police service. J. Psychiatr. Res. 2014, 56, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steudte-Schmiedgen, S.; Stalder, T.; Schönfeld, S.; Wittchen, H.-U.; Trautmann, S.; Alexander, N.; Miller, R.; Kirschbaum, C. Hair cortisol concentrations and cortisol stress reactivity predict PTSD symptom increase after trauma exposure during military deployment. Psychoneuroendocrinology 2015, 59, 123–133. [Google Scholar] [CrossRef]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Hubold, C.; Fischer, B.; Peters, A. Modeling the hypothalamus–pituitary–adrenal system: Homeostasis by interacting positive and negative feedback. J. Biol. Phys. 2009, 35, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Russell, G.; Lightman, S. The human stress response. Nat. Rev. Endocrinol. 2019, 15, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Russell, G.M.; Henley, D.E.; Leendertz, J.; Douthwaite, J.A.; Wood, S.A.; Stevens, A.; Woltersdorf, W.W.; Peeters, B.W.M.M.; Ruigt, G.S.F.; White, A.; et al. Rapid Glucocorticoid Receptor-Mediated Inhibition of Hypothalamic–Pituitary–Adrenal Ultradian Activity in Healthy Males. J. Neurosci. 2010, 30, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- Russell, G.M.; Kalafatakis, K.; Lightman, S.L. The Importance of Biological Oscillators for Hypothalamic-Pituitary-Adrenal Activity and Tissue Glucocorticoid Response: Coordinating Stress and Neurobehavioural Adaptation. J. Neuroendocr. 2015, 27, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Kalsbeek, A.; van der Spek, R.; Lei, J.; Endert, E.; Buijs, R.; Fliers, E. Circadian rhythms in the hypothalamo–pituitary–adrenal (HPA) axis. Mol. Cell. Endocrinol. 2012, 349, 20–29. [Google Scholar] [CrossRef]
- Oltmanns, K.M.; Fehm, H.L.; Peters, A. Chronic fentanyl application induces adrenocortical insufficiency. J. Intern. Med. 2005, 257, 478–480. [Google Scholar] [CrossRef]
- Wagner, U.; Degirmenci, M.; Drosopoulos, S.; Perras, B.; Born, J. Effects of Cortisol Suppression on Sleep-Associated Consolidation of Neutral and Emotional Memory. Biol. Psychiatry 2005, 58, 885–893. [Google Scholar] [CrossRef]
- Avital, A.; Segal, M.; Richter-Levin, G. Contrasting Roles of Corticosteroid Receptors in Hippocampal Plasticity. J. Neurosci. 2006, 26, 9130–9134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, A.; Conrad, M.; Hubold, C.; Schweiger, U.; Fischer, B.; Fehm, H.L. The principle of homeostasis in the hypothalamus-pituitary-adrenal system: New insight from positive feedback. Am. J. Physiol. Integr. Comp. Physiol. 2007, 293, R83–R98. [Google Scholar] [CrossRef] [PubMed]
- Ruoff, P.; Agafonov, O.; Tveit, D.M.; Thorsen, K.; Drengstig, T. Homeostatic controllers compensating for growth and perturbations. PLoS ONE 2019, 14, e0207831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, L.U.; D’Orsogna, M.R.; Chou, T. Perturbing the Hypothalamic–Pituitary–Adrenal Axis: A Mathematical Model for Interpreting PTSD Assessment Tests. Comput. Psychiatry 2018, 2, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Tasker, J.G.; Herman, J. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic–pituitary–adrenal axis. Stress 2011, 14, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, T.; Makino, S.; Matsumoto, R.; Nakayama, S.; Nishiyama, M.; Terada, Y.; Hashimoto, K. Regulation of Glucocorticoid Receptor Transcription and Nuclear Translocation during Single and Repeated Immobilization Stress. Endocrinology 2010, 151, 4344–4355. [Google Scholar] [CrossRef]
- Chen, A.C.; Etkin, A. Hippocampal Network Connectivity and Activation Differentiates Post-Traumatic Stress Disorder from Generalized Anxiety Disorder. Neuropsychopharmacology 2013, 38, 1889–1898. [Google Scholar] [CrossRef] [Green Version]
- Maggio, N.; Segal, M. Differential Corticosteroid Modulation of Inhibitory Synaptic Currents in the Dorsal and Ventral Hippocampus. J. Neurosci. 2009, 29, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Petrides, T.; Georgopoulos, P.; Kostopoulos, G.; Papatheodoropoulos, C. The GABAA receptor-mediated recurrent inhibition in ventral compared with dorsal CA1 hippocampal region is weaker, decays faster and lasts less. Exp. Brain Res. 2007, 177, 370–383. [Google Scholar] [CrossRef]
- Keralapurath, M.M.; Clark, J.K.; Hammond, S.; Wagner, J.J. Cocaine- or stress-induced metaplasticity of LTP in the dorsal and ventral hippocampus. Hippocampus 2014, 24, 577–590. [Google Scholar] [CrossRef]
- Maggio, N.; Segal, M. Differential Modulation of Long-Term Depression by Acute Stress in the Rat Dorsal and Ventral Hippocampus. J. Neurosci. 2009, 29, 8633–8638. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoropoulos, C. Striking differences in synaptic facilitation along the dorsoventral axis of the hippocampus. Neuroscience 2015, 301, 454–470. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Central effects of stress hormones in health and disease: Understanding the protective and damaging effects of stress and stress mediators. Eur. J. Pharmacol. 2008, 583, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Oitzl, M.S.; De Kloet, E.R. Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav. Neurosci. 1992, 106, 62–71. [Google Scholar] [CrossRef]
- Oitzl, M.S.; Fluttert, M.; De Kloet, E.R. The effect of corticosterone on reactivity to spatial novelty is mediated by central mineralocorticosteroid receptors. Eur. J. Neurosci. 1994, 6, 1072–1079. [Google Scholar] [CrossRef]
- Khaksari, M.; Rashidy-Pour, A.; Vafaei, A. Central mineralocorticoid receptors are indispensable for corticosterone-induced impairment of memory retrieval in rats. Neuroscience 2007, 149, 729–738. [Google Scholar] [CrossRef]
- Cornelisse, S.; Joëls, M.; Smeets, T. A Randomized Trial on Mineralocorticoid Receptor Blockade in Men: Effects on Stress Responses, Selective Attention, and Memory. Neuropsychopharmacology 2011, 36, 2720–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, A.J.; Mott, D.D. Functional neuroanatomy of amygdalohippocampal interconnections and their role in learning and memory. J. Neurosci. Res. 2017, 95, 797–820. [Google Scholar] [CrossRef] [Green Version]
- Bannerman, D.; Rawlins, J.; McHugh, S.; Deacon, R.; Yee, B.; Bast, T.; Zhang, W.-N.; Pothuizen, H.; Feldon, J. Regional dissociations within the hippocampus—memory and anxiety. Neurosci. Biobehav. Rev. 2004, 28, 273–283. [Google Scholar] [CrossRef]
- Fanselow, M.S.; Dong, H.-W. Are the Dorsal and Ventral Hippocampus Functionally Distinct Structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoryan, G.; Segal, M. Lasting Differential Effects on Plasticity Induced by Prenatal Stress in Dorsal and Ventral Hippocampus. Neural Plast. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjelstrup, K.G.; Tuvnes, F.A.; Steffenach, H.-A.; Murison, R.; Moser, E.I.; Moser, M.-B. Reduced fear expression after lesions of the ventral hippocampus. Proc. Natl. Acad. Sci. USA 2002, 99, 10825–10830. [Google Scholar] [CrossRef] [Green Version]
- McHugh, S.B.; Deacon, R.M.J.; Rawlins, J.N.P.; Bannerman, D.M. Amygdala and Ventral Hippocampus Contribute Differentially to Mechanisms of Fear and Anxiety. Behav. Neurosci. 2004, 118, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Risold, P.Y.; Swanson, L.W. Structural Evidence for Functional Domains in the Rat Hippocampus. Science 1996, 272, 1484–1486. [Google Scholar] [CrossRef]
- Wang, M.E.; Fraize, N.P.; Yin, L.; Yuan, R.K.; Petsagourakis, D.; Wann, E.; Muzzio, I.A. Differential roles of the dorsal and ventral hippocampus in predator odor contextual fear conditioning. Hippocampus 2013, 23, 451–466. [Google Scholar] [CrossRef]
- Herman, J.P.; Mueller, N.K. Role of the ventral subiculum in stress integration. Behav. Brain Res. 2006, 174, 215–224. [Google Scholar] [CrossRef]
- Herman, J.P.; Tasker, J.; Ziegler, D.R.; Cullinan, W.E. Local circuit regulation of paraventricular nucleus stress integration. Pharmacol. Biochem. Behav. 2002, 71, 457–468. [Google Scholar] [CrossRef]
- Sapolsky, R.M.; Krey, L.C.; McEwen, B.S. Glucocorticoid-sensitive hippocampal neurons are involved in terminating the adrenocortical stress response. Proc. Natl. Acad. Sci. USA 1984, 81, 6174–6177. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, L.; Sapolsky, R. The Role of the Hippocampus in Feedback Regulation of the Hypothalamic-Pituitary-Adrenocortical Axis. Endocr. Rev. 1991, 12, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Ratka, A.; Sutanto, W.; Bloemers, M.; De Kloet, R. On the Role of Brain Mineralocorticoid (Type I) and Glucocorticoid (Type II) Receptors in Neuroendocrine Regulation. Neuroendocrinology 1989, 50, 117–123. [Google Scholar] [CrossRef]
- Van Haarst, A.D.; Oitzl, M.S.; De Kloet, E.R. Facilitation of feedback inhibition through blockade of glucocorticoid receptors in the hippocampus. Neurochem. Res. 1997, 22, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Nasca, C.; Bigio, B.; Zelli, D.; Nicoletti, F.; McEwen, B.S. Mind the gap: Glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol. Psychiatry 2015, 20, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joëls, M.; Karst, H.; DeRijk, R.; de Kloet, E.R. The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 2008, 31, 1–7. [Google Scholar] [CrossRef]
- Groeneweg, F.L.; Karst, H.; De Kloet, E.R.; Joels, M. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J. Endocrinol. 2011, 209, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Joels, M.; De Kloet, E.R. 30 YEARS of the MINERALOCORTICOID RECEPTOR: The brain mineralocorticoid receptor: A saga in three episodes. J. Endocrinol. 2017, 234, T49–T66. [Google Scholar] [CrossRef]
- George, S.A.; Rodriguez-Santiago, M.; Riley, J.; Rodriguez, E.; Liberzon, I. The effect of chronic phenytoin administration on single prolonged stress induced extinction retention deficits and glucocorticoid upregulation in the rat medial prefrontal cortex. Psychopharmacology 2014, 232, 47–56. [Google Scholar] [CrossRef]
- Finsterwald, C.; Steinmetz, A.B.; Travaglia, A.; Alberini, C.M. From Memory Impairment to Posttraumatic Stress Disorder-Like Phenotypes: The Critical Role of an Unpredictable Second Traumatic Experience. J. Neurosci. 2015, 35, 15903–15915. [Google Scholar] [CrossRef] [Green Version]
- Perrine, S.A.; Eagle, A.L.; George, S.A.; Mulo, K.; Kohler, R.J.; Gerard, J.; Harutyunyan, A.; Hool, S.M.; Susick, L.; Schneider, B.L.; et al. Severe, multimodal stress exposure induces PTSD-like characteristics in a mouse model of single prolonged stress. Behav. Brain Res. 2016, 303, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulton, E.; Chamness, M.; Knox, D. Characterizing changes in glucocorticoid receptor internalization in the fear circuit in an animal model of post traumatic stress disorder. PLoS ONE 2018, 13, e0205144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagle, A.L.; Knox, D.; Roberts, M.M.; Mulo, K.; Liberzon, I.; Galloway, M.P.; Perrine, S.A. Single prolonged stress enhances hippocampal glucocorticoid receptor and phosphorylated protein kinase B levels. Neurosci. Res. 2013, 75, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Kozlovsky, N.; Matar, M.A.; Kaplan, Z.; Zohar, J.; Cohen, H. A distinct pattern of intracellular glucocorticoid-related responses is associated with extreme behavioral response to stress in an animal model of post-traumatic stress disorder. Eur. Neuropsychopharmacol. 2009, 19, 759–771. [Google Scholar] [CrossRef]
- Louvart, H.; Maccari, S.; Lesage, J.; Léonhardt, M.; Dickes-Coopman, A.; Darnaudéry, M. Effects of a single footshock followed by situational reminders on HPA axis and behaviour in the aversive context in male and female rats. Psychoneuroendocrinology 2006, 31, 92–99. [Google Scholar] [CrossRef]
- Van Zuiden, M.; Geuze, E.; Willemen, H.L.; Vermetten, E.; Maas, M.; Amarouchi, K.; Kavelaars, A.; Heijnen, C.J. Glucocorticoid Receptor Pathway Components Predict Posttraumatic Stress Disorder Symptom Development: A Prospective Study. Biol. Psychiatry 2012, 71, 309–316. [Google Scholar] [CrossRef]
- Malivoire, B.L.; Girard, T.A.; Patel, R.; Monson, C.M. Functional connectivity of hippocampal subregions in PTSD: Relations with symptoms. BMC Psychiatry 2018, 18, 129. [Google Scholar] [CrossRef] [PubMed]
- Kaouane, N.; Porte, Y.; Vallee, M.; Brayda-Bruno, L.; Mons, N.; Calandreau, L.; Marighetto, A.; Piazza, P.V.; Desmedt, A. Glucocorticoids Can Induce PTSD-Like Memory Impairments in Mice. Science 2012, 335, 1510–1513. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.L.; Zoladz, P.R.; Sweatt, J.D.; Diamond, D.M. Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J. Psychiatr. Res. 2011, 45, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009, 34, S186–S195. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Calabrese, F.; Anacker, C.; Racagni, G.; Pariante, C.M.; A Riva, M. Glucocorticoid Receptor and FKBP5 Expression Is Altered Following Exposure to Chronic Stress: Modulation by Antidepressant Treatment. Neuropsychopharmacology 2013, 38, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Gallo, L.I.; Ghini, A.A.; Pilipuk, G.P.; Galigniana, M.D. Differential Recruitment of Tetratricorpeptide Repeat Domain Immunophilins to the Mineralocorticoid Receptor Influences both Heat-Shock Protein 90-Dependent Retrotransport and Hormone-Dependent Transcriptional Activity. Biochemistry 2007, 46, 14044–14057. [Google Scholar] [CrossRef]
- Zannas, A.S.; Wiechmann, T.; Gassen, N.; Binder, E.B. Gene–Stress–Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacology 2016, 41, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criado-Marrero, M.; Gebru, N.T.; Gould, L.A.; Smith, T.M.; Kim, S.; Blackburn, R.J.; Dickey, C.A.; Blair, L.J. Early Life Stress and High FKBP5 Interact to Increase Anxiety-Like Symptoms through Altered AKT Signaling in the Dorsal Hippocampus. Int. J. Mol. Sci. 2019, 20, 2738. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.; Wagner, K.V.; Gaali, S.; Kirschner, A.; Kozany, C.; Rühter, G.; Dedic, N.; Häusl, A.S.; Hoeijmakers, L.; Westerholz, S.; et al. Pharmacological Inhibition of the Psychiatric Risk Factor FKBP51 Has Anxiolytic Properties. J. Neurosci. 2015, 35, 9007–9016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Duncan, L.; Ratanatharathorn, A.; E Aiello, A.; Almli, L.; Amstadter, A.B.; E Ashley-Koch, A.; Baker, D.G.; Beckham, J.C.; Bierut, L.J.; Bisson, J.; et al. Largest GWAS of PTSD (N=20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol. Psychiatry 2018, 23, 666–673. [Google Scholar] [CrossRef]
- Stein, M.B.; Chen, C.-Y.; Ursano, R.; Cai, T.; Gelernter, J.; Heeringa, S.G.; Jain, S.; Jensen, K.P.; Maihofer, A.X.; Mitchell, C.; et al. Genome-wide Association Studies of Posttraumatic Stress Disorder in 2 Cohorts of US Army Soldiers. JAMA Psychiatry 2016, 73, 695–704. [Google Scholar] [CrossRef]
- Binder, E.B. Association of FKBP5 Polymorphisms and Childhood Abuse With Risk of Posttraumatic Stress Disorder Symptoms in Adults. JAMA 2008, 299, 1291–1305. [Google Scholar] [CrossRef] [Green Version]
- Daskalakis, N.P.; Rijal, C.M.; King, C.; Huckins, L.M.; Ressler, K.J. Recent Genetics and Epigenetics Approaches to PTSD. Curr. Psychiatry Rep. 2018, 20, 1–12. [Google Scholar] [CrossRef]
- Kang, J.I.; Kim, T.Y.; Choi, J.H.; So, H.S.; Kim, S.J. Allele-specific DNA methylation level of FKBP5 is associated with post-traumatic stress disorder. Psychoneuroendocrinology 2019, 103, 1–7. [Google Scholar] [CrossRef]
- Klengel, T.; Mehta, D.; Anacker, C.; Rex-Haffner, M.; Pruessner, J.C.; Pariante, C.M.; Pace, T.W.W.; Mercer, K.B.; Mayberg, H.S.; Bradley, B.; et al. Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions. Nat. Neurosci. 2013, 16, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Sheerin, C.M.; Lind, M.J.; Bountress, K.E.; Marraccini, M.E.; Amstadter, A.B.; Bacanu, S.; Nugent, N.R. Meta-Analysis of Associations Between Hypothalamic-Pituitary-Adrenal Axis Genes and Risk of Posttraumatic Stress Disorder. J. Trauma. Stress 2020, 33, 688–698. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, L.; Li, G.; Cao, C.; Fang, R.; Liu, P.; Luo, S.; Zhang, X. Correlation between hypothalamic-pituitary-adrenal axis gene polymorphisms and posttraumatic stress disorder symptoms. Horm. Behav. 2020, 117, 104604. [Google Scholar] [CrossRef]
- Szmydynger-Chodobska, J.; Fox, L.M.; Lynch, K.M.; Zink, B.J.; Chodobski, A. Vasopressin Amplifies the Production of Proinflammatory Mediators in Traumatic Brain Injury. J. Neurotrauma 2010, 27, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabold, R.; Krieg, S.; Schöller, K.; Plesnila, N. Role of Vasopressin V1a and V2 Receptors for the Development of Secondary Brain Damage after Traumatic Brain Injury in Mice. J. Neurotrauma 2008, 25, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Vakili, A.; Kataoka, H.; Plesnila, N. Role of Arginine Vasopressin V1 and V2 Receptors for Brain Damage after Transient Focal Cerebral Ischemia. Br. J. Pharmacol. 2005, 25, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, G.; Rabadan-Diehl, C. Vasopressinergic regulation of the hypothalamic–pituitary–adrenal axis: Implications for stress adaptation. Regul. Pept. 2000, 96, 23–29. [Google Scholar] [CrossRef]
- Rabadán-Diehl, C.; Aguilera, G. Glucocorticoids Increase Vasopressin V1b Receptor Coupling to Phospholipase C. Endocrinology 1998, 139, 3220–3226. [Google Scholar] [CrossRef]
- Volpi, S.; Rabadan-Diehl, C.; Aguilera, G. Vasopressinergic Regulation of the Hypothalamic Pituitary Adrenal Axis and Stress Adaptation. Stress 2004, 7, 75–83. [Google Scholar] [CrossRef]
- Rivier, C.; Vale, W. Interaction of Corticotropin-Releasing Factor and Arginine Vasopressin on Adrenocorticotropin Secretion in Vivo. Endocrinology 1983, 113, 939–942. [Google Scholar] [CrossRef]
- Scaccianoce, S.; Muscolo, L.A.A.; Cigliana, G.; Navarra, D.; Nicolai, R.; Angelucci, L. Evidence for a Specific Role of Vasopressin in Sustaining Pituitary-Adrenocortical Stress Response in the Rat. Endocrinology 1991, 128, 3138–3143. [Google Scholar] [CrossRef]
- Windle, R.J.; Wood, S.A.; Kershaw, Y.M.; Lightman, S.L.; Ingram, C.; Harbuz, M.S. Increased Corticosterone Pulse Frequency During Adjuvant-Induced Arthritis and its Relationship to Alterations in Stress Responsiveness. J. Neuroendocr. 2001, 13, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Whitnall, M.H. Regulation of the hypothalamic corticotropin-releasing hormone neurosecretory system. Prog. Neurobiol. 1993, 40, 573–629. [Google Scholar] [CrossRef]
- Makara, G.B.; Mergl, Z.; Zelena, D. The Role of Vasopressin in Hypothalamo-Pituitary-Adrenal Axis Activation during Stress: An Assessment of the Evidence. Ann. New York Acad. Sci. USA 2004, 1018, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; O’Carroll, A.-M.; Young, W.S.; Lolait, S. The vasopressin Avpr1b receptor: Molecular and pharmacological studies. Stress 2010, 14, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Spiga, F.; Harrison, L.R.; Wood, S.; Knight, D.M.; MacSweeney, C.P.; Thomson, F.; Craighead, M.; Lightman, S.L. Blockade of the V1b receptor reduces ACTH, but not corticosterone secretion induced by stress without affecting basal hypothalamic–pituitary–adrenal axis activity. J. Endocrinol. 2008, 200, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, L.G.; Arentzen, R.; Reid, J.M.; Manning, R.W.; Wolfson, B.; Lawrence, K.L.; Baldino, F. Glucocorticoid sensitivity of vasopressin mRNA levels in the paraventricular nucleus of the rat. Proc. Natl. Acad. Sci. USA 1986, 83, 1145–1149. [Google Scholar] [CrossRef] [Green Version]
- Kiss, J.; Van Eekelen, J.; Reul, J.; Westphal, H.; De Kloet, E. Glucocorticoid Receptor in Magnocellular Neurosecretory Cells. Endocrinology 1988, 122, 444–449. [Google Scholar] [CrossRef]
- Patchev, V.K.; Mastorakos, G.; Brady, L.S.; Redwine, J.; Wilder, R.L.; Chrousos, G.P. Increased Arginine Vasopressin Secretion May Participate in the Enhanced Susceptibility of Lewis Rats to Inflammatory Disease. Neuroendocrinology 1993, 58, 106–110. [Google Scholar] [CrossRef]
- De Kloet, C.S.; Vermetten, E.; Geuze, E.; Wiegant, V.M.; Westenberg, H.G.M. Elevated plasma arginine vasopressin levels in veterans with posttraumatic stress disorder. J. Psychiatr. Res. 2008, 42, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.; Vengrober, A.; Ebstein, R.P. Affiliation buffers stress: Cumulative genetic risk in oxytocin–vasopressin genes combines with early caregiving to predict PTSD in war-exposed young children. Transl. Psychiatry 2014, 4, e370. [Google Scholar] [CrossRef] [Green Version]
- Keck, M. Corticotropin-releasing factor, vasopressin and receptor systems in depression and anxiety. Amino Acids 2006, 31, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Frijling, J.L.; Van Zuiden, M.; Nawijn, L.; Koch, S.B.J.; Neumann, I.D.; Veltman, D.J.; Olff, M. Salivary Oxytocin and Vasopressin Levels in Police Officers With and Without Post-Traumatic Stress Disorder. J. Neuroendocr. 2015, 27, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Reijnen, A.; Geuze, E.; Vermetten, E. Individual variation in plasma oxytocin and vasopressin levels in relation to the development of combat-related PTSD in a large military cohort. J. Psychiatr. Res. 2017, 94, 88–95. [Google Scholar] [CrossRef]
- Qin, C.; Li, J.; Tang, K. The Paraventricular Nucleus of the Hypothalamus: Development, Function, and Human Diseases. Endocrinology 2018, 159, 3458–3472. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.E. Electrophysiological and morphological properties of pre-autonomic neurones in the rat hypothalamic paraventricular nucleus. J. Physiol. 2001, 537, 161–177. [Google Scholar] [CrossRef]
- Abdallah, K.; Artola, A.; Monconduit, L.; Dallel, R.; Luccarini, P. Bilateral Descending Hypothalamic Projections to the Spinal Trigeminal Nucleus Caudalis in Rats. PLoS ONE 2013, 8, e73022. [Google Scholar] [CrossRef]
- Rasmusson, A.M.; Pinna, G.; Paliwal, P.; Weisman, D.; Gottschalk, C.; Charney, D.; Krystal, J.; Guidotti, A. Decreased Cerebrospinal Fluid Allopregnanolone Levels in Women with Posttraumatic Stress Disorder. Biol. Psychiatry 2006, 60, 704–713. [Google Scholar] [CrossRef]
- Almeida, F.; Barros, H.; Pinna, G. Neurosteroids and Neurotrophic Factors: What Is Their Promise as Biomarkers for Major Depression and PTSD? Int. J. Mol. Sci. 2021, 22, 1758. [Google Scholar] [CrossRef]
- Kil Kim, B.; Fonda, J.R.; Hauger, R.L.; Pinna, G.; Anderson, G.M.; Valovski, I.T.; Rasmusson, A.M. Composite contributions of cerebrospinal fluid GABAergic neurosteroids, neuropeptide Y and interleukin-6 to PTSD symptom severity in men with PTSD. Neurobiol. Stress 2020, 12, 100220. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Maayan, R.; Touati-Werner, D.; Kaplan, Z.; Matar, M.A.; Loewenthal, U.; Kozlovsky, N.; Weizman, R. Decreased circulatory levels of neuroactive steroids in behaviourally more extremely affected rats subsequent to exposure to a potentially traumatic experience. Int. J. Neuropsychopharmacol. 2006, 10, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Dong, E.; Matsumoto, K.; Uzunova, V.; Sugaya, I.; Takahata, H.; Nomura, H.; Watanabe, H.; Costa, E.; Guidotti, A. Brain 5 -dihydroprogesterone and allopregnanolone synthesis in a mouse model of protracted social isolation. Proc. Natl. Acad. Sci. USA 2001, 98, 2849–2854. [Google Scholar] [CrossRef] [Green Version]
- Pibiri, F.; Nelson, M.; Guidotti, A.; Costa, E.; Pinna, G. Decreased corticolimbic allopregnanolone expression during social isolation enhances contextual fear: A model relevant for posttraumatic stress disorder. Proc. Natl. Acad. Sci. USA 2008, 105, 5567–5572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinna, G. Animal Models of PTSD: The Socially Isolated Mouse and the Biomarker Role of Allopregnanolone. Front. Behav. Neurosci. 2019, 13, 114. [Google Scholar] [CrossRef]
- Pinna, G.; Costa, E.; Guidotti, A. Changes in brain testosterone and allopregnanolone biosynthesis elicit aggressive behavior. Proc. Natl. Acad. Sci. USA 2005, 102, 2135–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.-M.; Qiu, Z.-K.; Zhao, N.; Chen, H.-X.; Liu, Y.-Q.; Xu, J.-P.; Zhang, Y.-Z.; Yang, R.-F.; Li, Y.-F. Anxiolytic-like effects of YL-IPA08, a potent ligand for the translocator protein (18 kDa) in animal models of post-traumatic stress disorder. Int. J. Neuropsychopharmacol. 2014, 17, 1659–1669. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.P.; Ostrander, M.M.; Mueller, N.K.; Figueiredo, H. Limbic system mechanisms of stress regulation: Hypothalamo-pituitary-adrenocortical axis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 1201–1213. [Google Scholar] [CrossRef]
- Ito, R.; Lee, A.C. The role of the hippocampus in approach-avoidance conflict decision-making: Evidence from rodent and human studies. Behav. Brain Res. 2016, 313, 345–357. [Google Scholar] [CrossRef]
- Chase, H.W.; Clos, M.; Dibble, S.; Fox, P.; Grace, A.A.; Phillips, M.L.; Eickhoff, S.B. Evidence for an anterior–posterior differentiation in the human hippocampal formation revealed by meta-analytic parcellation of fMRI coordinate maps: Focus on the subiculum. NeuroImage 2015, 113, 44–60. [Google Scholar] [CrossRef] [Green Version]
- Szeszko, P.R.; Lehrner, A.; Yehuda, R. Glucocorticoids and Hippocampal Structure and Function in PTSD. Harv. Rev. Psychiatry 2018, 26, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.L.; Shin, L.M.; Wright, C.I. Neuroimaging Studies of Amygdala Function in Anxiety Disorders. Ann. N. Y. Acad. Sci. 2006, 985, 389–410. [Google Scholar] [CrossRef]
- Shin, L.M.; Liberzon, I. The Neurocircuitry of Fear, Stress, and Anxiety Disorders. Neuropsychopharmacology 2009, 35, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Amaral, O.; Luft, T.; Cammarota, M.; Izquierdo, I.; Roesler, R. Temporary inactivation of the dorsal hippocampus induces a transient impairment in retrieval of aversive memory. Behav. Brain Res. 2007, 180, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Dorey, R.; Piérard, C.; Chauveau, F.; David, V.; Béracochéa, D. Stress-Induced Memory Retrieval Impairments: Different Time-Course Involvement of Corticosterone and Glucocorticoid Receptors in Dorsal and Ventral Hippocampus. Neuropsychopharmacology 2012, 37, 2870–2880. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.-B.; Moser, E.I. Functional differentiation in the hippocampus. Hippocampus 1998, 8, 608–619. [Google Scholar] [CrossRef]
- Segal, M.; Richter-Levin, G.; Maggio, N. Stress-induced dynamic routing of hippocampal connectivity: A hypothesis. Hippocampus 2010, 20, 1332–1338. [Google Scholar] [CrossRef]
- Cohen, H.; Zohar, J.; Matar, M.A.; Kaplan, Z.; Geva, A.B. Unsupervised Fuzzy Clustering Analysis Supports Behavioral Cutoff Criteria in an Animal Model of Posttraumatic Stress Disorder. Biol. Psychiatry 2005, 58, 640–650. [Google Scholar] [CrossRef]
- Klapper-Goldstein, H.; Murninkas, M.; Gillis, R.; Mulla, W.; Levanon, E.; Elyagon, S.; Schuster, R.; Danan, D.; Cohen, H.; Etzion, Y. An implantable system for long-term assessment of atrial fibrillation substrate in unanesthetized rats exposed to underlying pathological conditions. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Ayyar, V.S.; Song, D.; Dubois, D.C.; Almon, R.R.; Jusko, W.J. Modeling Corticosteroid Pharmacokinetics and Pharmacodynamics, Part I: Determination and Prediction of Dexamethasone and Methylprednisolone Tissue Binding in the Rat. J. Pharmacol. Exp. Ther. 2019, 370, 318–326. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Pavlaki, A.N.; Alexandra, M.A.M.; Chrousos, G.P. Glucocorticoid therapy and adrenal suppression. In Endotext [Internet]; MDText. com, Inc.: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Knobloch, H.S.; Egrinevich, V. Evolution of oxytocin pathways in the brain of vertebrates. Front. Behav. Neurosci. 2014, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Swanson, L.W.; E Sawchenko, P. Hypothalamic Integration: Organization of the Paraventricular and Supraoptic Nuclei. Annu. Rev. Neurosci. 1983, 6, 269–324. [Google Scholar] [CrossRef] [PubMed]
- Morsink, M.C.; Joels, M.; Sarabdjitsingh, R.A.; Meijer, O.C.; De Kloet, E.R.; Datson, N. The dynamic pattern of glucocorticoid receptor-mediated transcriptional responses in neuronal PC12 cells. J. Neurochem. 2006, 99, 1282–1298. [Google Scholar] [CrossRef]
- Sarabdjitsingh, R.A.; Meijer, O.C.; Schaaf, M.J.; de Kloet, E.R. Subregion-specific differences in translocation patterns of mineralocorticoid and glucocorticoid receptors in rat hippocampus. Brain Res. 2009, 1249, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Van Der Laan, S.; Lachize, S.; Schouten, T.; Vreugdenhil, E.; De Kloet, E.; Meijer, O. Neuroanatomical distribution and colocalisation of nuclear receptor corepressor (N-CoR) and silencing mediator of retinoid and thyroid receptors (SMRT) in rat brain. Brain Res. 2005, 1059, 113–121. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Post Methylprednisolone Administration | EBR n = 4 | PBR n = 9 | MBR n = 8 | Statistics |
|---|---|---|---|---|
| 20 min | ||||
| 0–50% Suppression, (n) | 0 | 100%, (9/9) | 87.5%, (7/8) | Pearson χ2 = 12.35, df = 2, p < 0.0025 |
| 51–89% Suppression, (n) | 100%, (4/4) | 0 | 12.5%, (1/8) | |
| 90%+ Suppression, (n) | 0 | 0 | 0 | |
| 40 min | ||||
| 0–50% Suppression, (n) | 0 | 22%, (2/9) | 75%, (6/8) | Pearson χ2 = 11.23, df = 4, p < 0.025 |
| 51–89% Suppression, (n) | 25%, (1/4) | 67%, (6/9) | 12.5%, (1/8) | |
| 90%+ Suppression, (n) | 75%, (3/4) | 11%, (1/9) | 12.5%, (1/8) | |
| 70 min | ||||
| 0–50% Suppression, (n) | 0 | 11%, (1/9) | 62.5%, (5/8) | Pearson χ2 = 11.12, df = 4, p < 0.03 |
| 51–89% Suppression, (n) | 0 | 33%, (3/9) | 25%, (2/8) | |
| 90%+ Suppression, (n) | 100%, (4/4) | 56%, (5.9) | 12.5%, (1/8) | |
| Average Pulsatility during MPST | EBR n = 4 (I) | PBR n = 9 (II) | MBR n = 8 (III) | Statistics |
|---|---|---|---|---|
| Amplitudes average suppression 1 | 0.16 ± 0.1 | 0.036 ± 0.02 | 0 | I ≠ III (Bonferroni) I ≠ II, III (Fisher LSD) |
| Amplitudes average recovery 2 | 0.64 ± 0.15 | 0.53 ± 0.13 | 0.24 ± 0.09 | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danan, D.; Todder, D.; Zohar, J.; Cohen, H. Is PTSD-Phenotype Associated with HPA-Axis Sensitivity? Feedback Inhibition and Other Modulating Factors of Glucocorticoid Signaling Dynamics. Int. J. Mol. Sci. 2021, 22, 6050. https://doi.org/10.3390/ijms22116050
Danan D, Todder D, Zohar J, Cohen H. Is PTSD-Phenotype Associated with HPA-Axis Sensitivity? Feedback Inhibition and Other Modulating Factors of Glucocorticoid Signaling Dynamics. International Journal of Molecular Sciences. 2021; 22(11):6050. https://doi.org/10.3390/ijms22116050
Chicago/Turabian StyleDanan, Dor, Doron Todder, Joseph Zohar, and Hagit Cohen. 2021. "Is PTSD-Phenotype Associated with HPA-Axis Sensitivity? Feedback Inhibition and Other Modulating Factors of Glucocorticoid Signaling Dynamics" International Journal of Molecular Sciences 22, no. 11: 6050. https://doi.org/10.3390/ijms22116050