
Minor Intron Splicing from Basic Science to Disease



Abstract
:1. RNA and Splicing
2. The Other Side of the Splicing Coin—An Overview
3. mi-INTs Sequence Characteristics
4. Origin and Evolution: The U12 Splicing Pathway Is as Old as the U2 Pathway
5. The Functional Relevance of Minor Introns
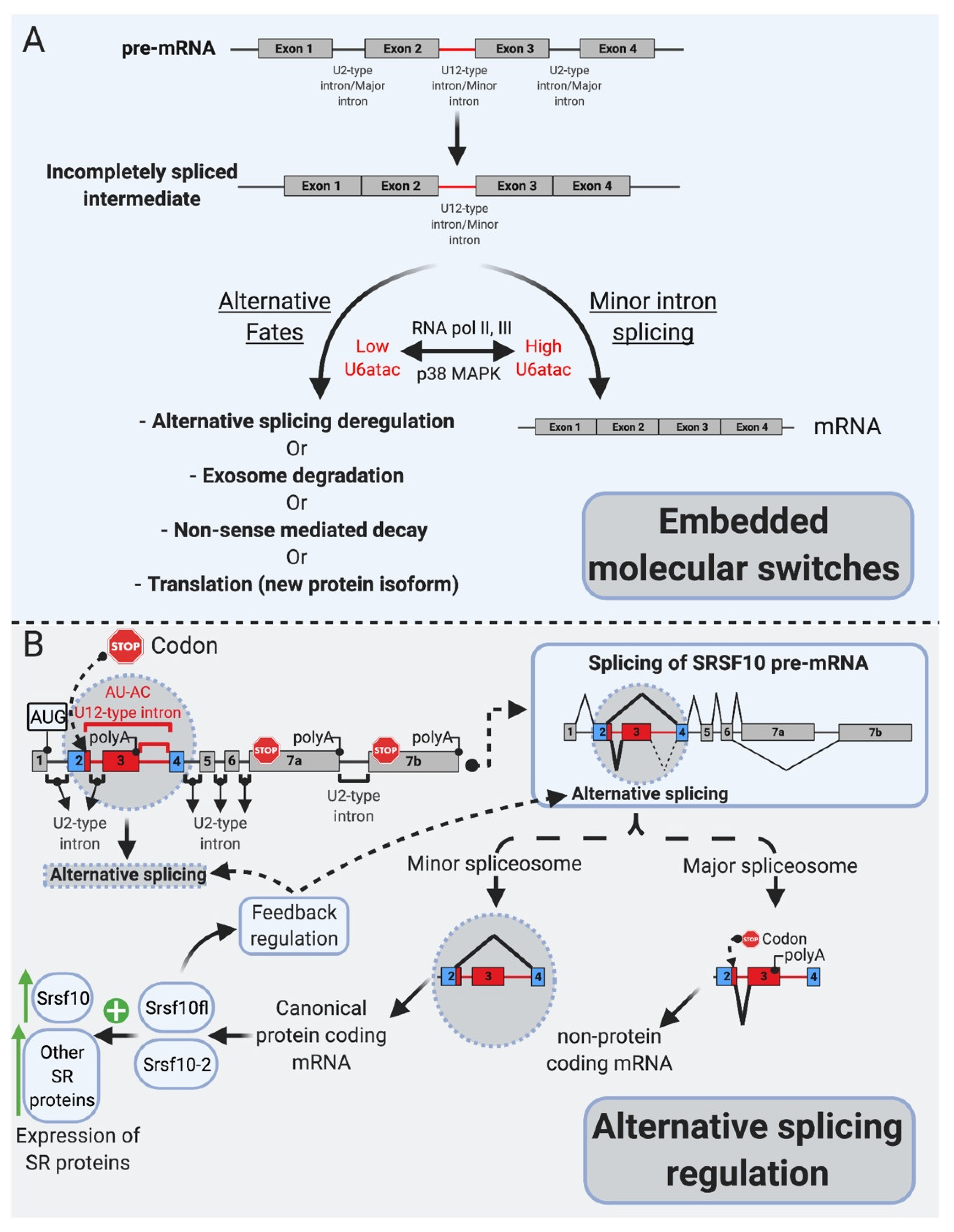
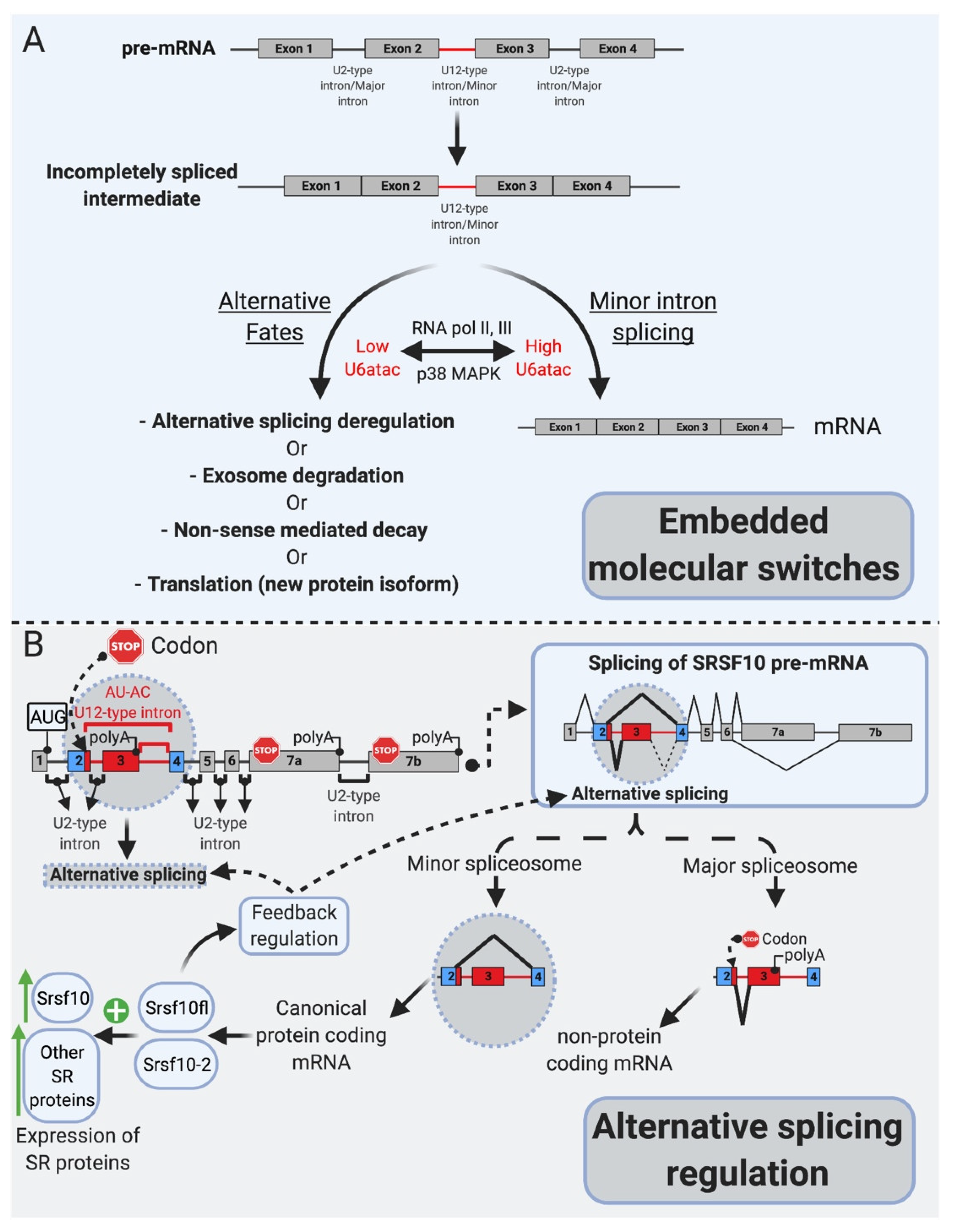
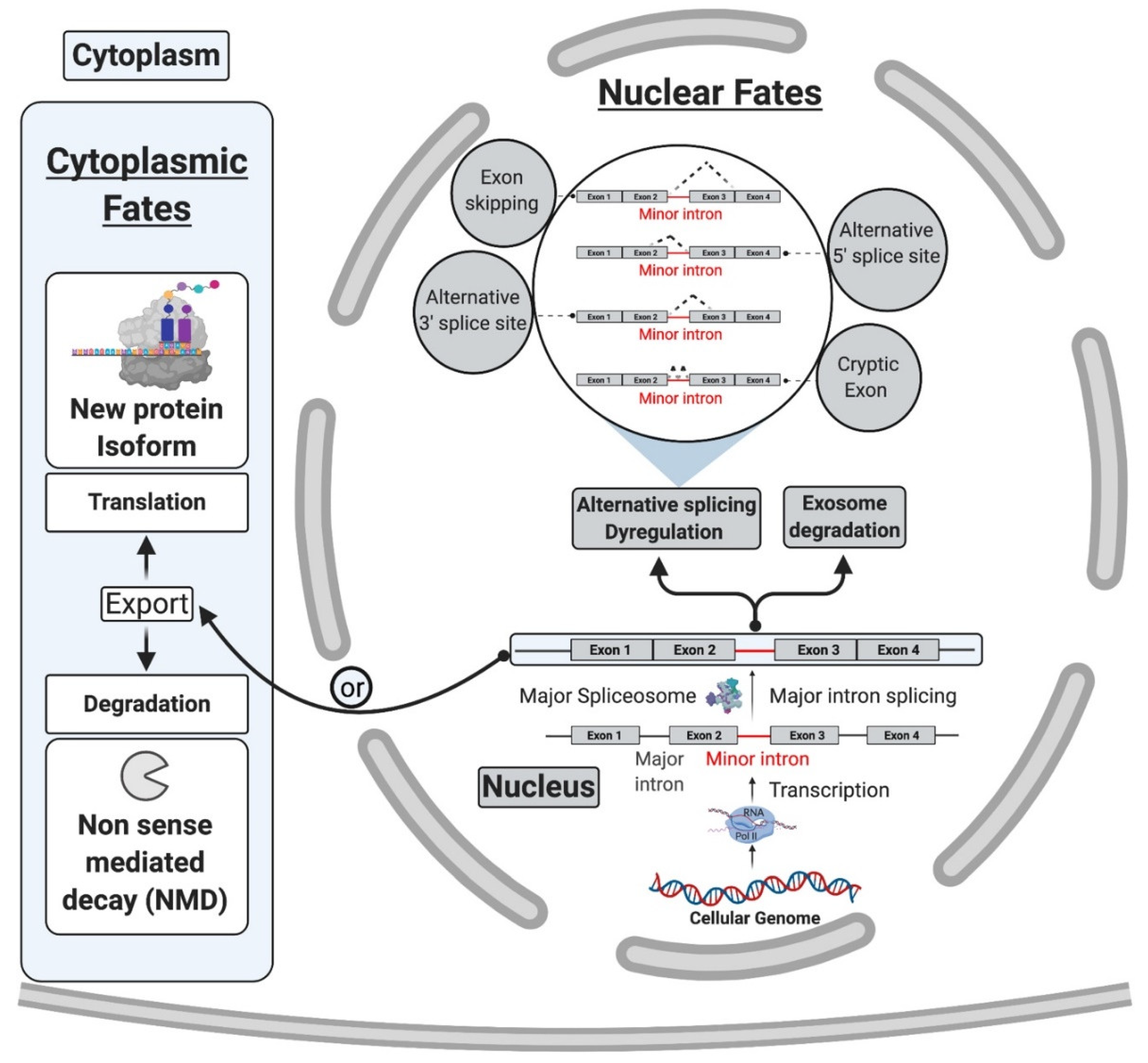
6. mi-INTs Splicing Is Regulated
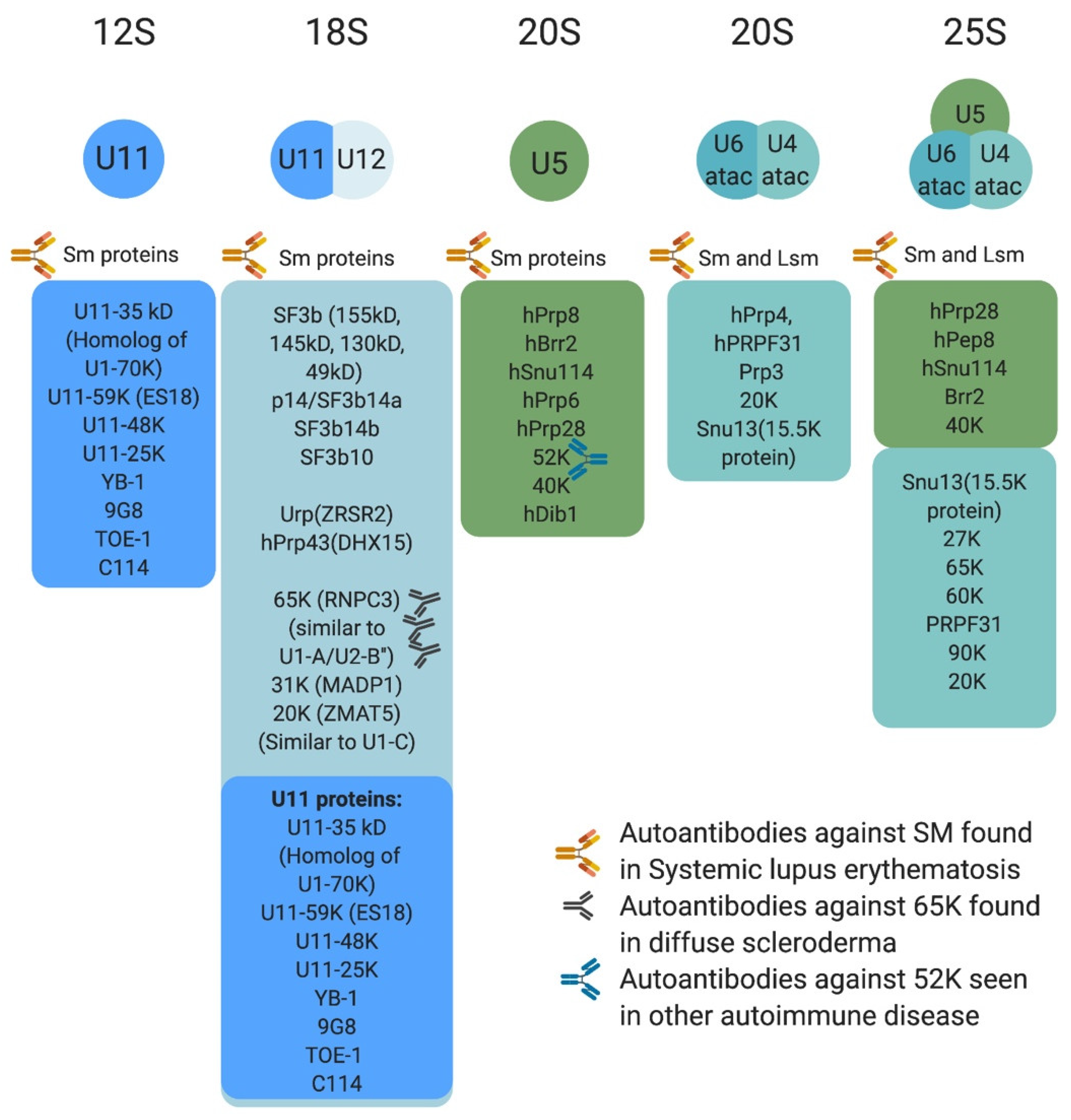
7. The Minor Spliceosome: Composition, Assembly, and Evolution
8. Splicing and Disease
9. Minor Intron Splicing and Diseases
10. Systemic vs. Tissue-Specific Pathologies
11. Minor Splicing and Disease Onset
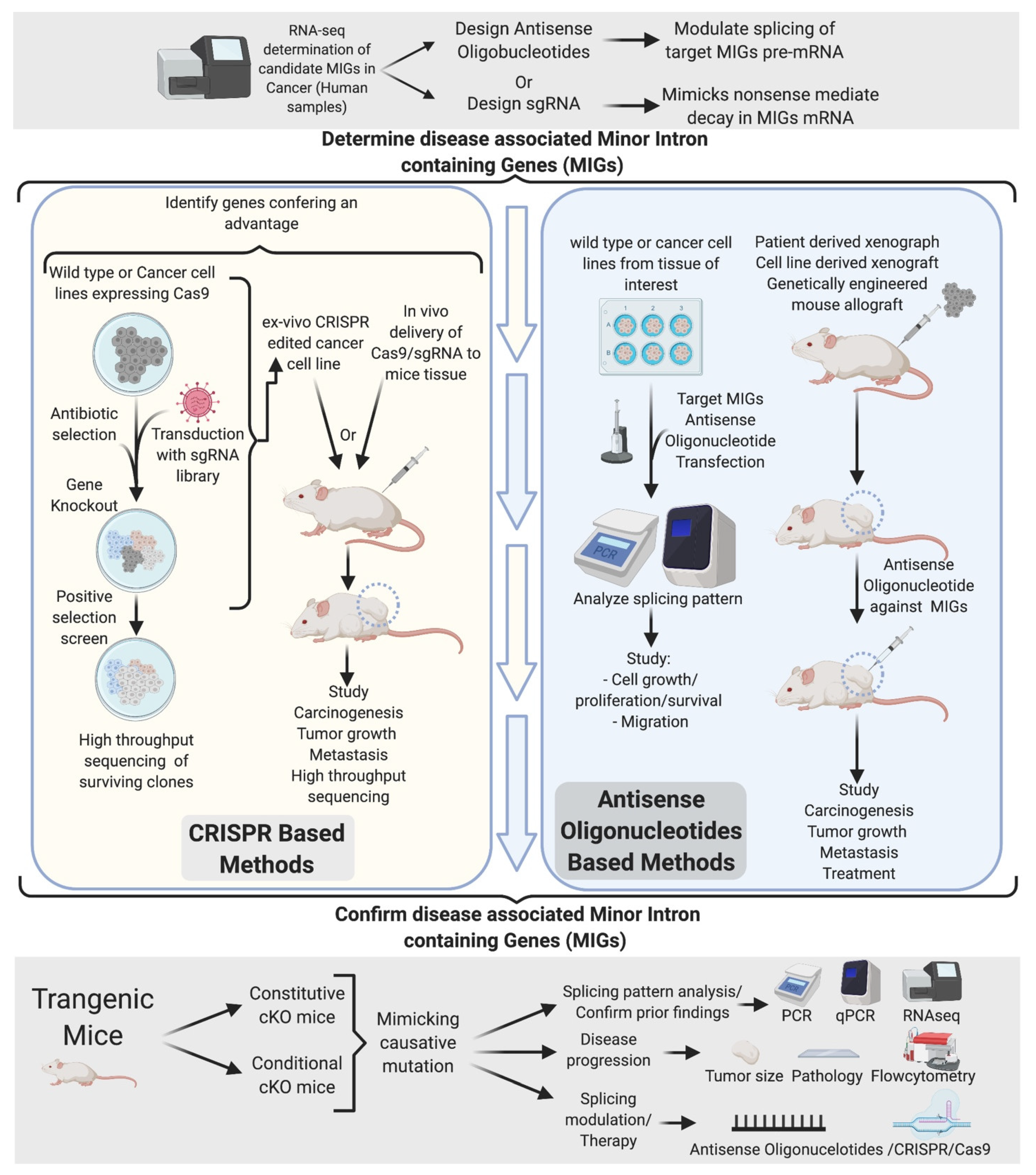
12. Identification of Key Disease-Associated miGs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Lévy, J.-P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytesin vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Abelson, J. RNA Processing and the Intervening Sequence Problem. Annu. Rev. Biochem. 1979, 48, 1035–1069. [Google Scholar] [CrossRef]
- Green, M.R. PRE-mRNA Splicing. Ann. Rev. Genet. 1986, 20, 671–708. [Google Scholar] [CrossRef]
- Padgett, R.A.; Grabowski, P.J.; Konarska, M.M.; Seiler, S.; Sharp, P.A. Splicing of Messenger RNA Precursors. Annu. Rev. Biochem. 1986, 55, 1119–1150. [Google Scholar] [CrossRef]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, S.L.; Padgett, R. Conserved Sequences in a Class of Rare Eukaryotic Nuclear Introns with Non-consensus Splice Sites. J. Mol. Biol. 1994, 239, 357–365. [Google Scholar] [CrossRef]
- Tarn, W.-Y.; Steitz, J.A. A Novel Spliceosome Containing U11, U12, and U5 snRNPs Excises a Minor Class (AT–AC) Intron In Vitro. Cell 1996, 84, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.A.; Steitz, J.A. Splicing double: Insights from the second spliceosome. Nat. Rev. Mol. Cell Biol. 2003, 4, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.L.; Padgett, R.A. Requirement of U12 snRNA for in Vivo Splicing of a Minor Class of Eukaryotic Nuclear Pre-mRNA Introns. Science 1996, 271, 1716–1718. [Google Scholar] [CrossRef]
- Jackson, L.J. A reappraisal of non-consensus mRNA splice sites. Nucleic Acids Res. 1991, 19, 3795–3798. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, R.C.; Incorvaia, R.A.; Padgett, R. Terminal Intron Dinucleotide Sequences Do Not Distinguish between U2- and U12-Dependent Introns. Mol. Cell 1997, 1, 151–160. [Google Scholar] [CrossRef]
- Sharp, P.A.; Burge, C.B. Classification of Introns: U2-Type or U12-Type. Cell 1997, 91, 875–879. [Google Scholar] [CrossRef] [Green Version]
- Burge, C.B.; Padgett, R.; Sharp, P.A. Evolutionary Fates and Origins of U12-Type Introns. Mol. Cell 1998, 2, 773–785. [Google Scholar] [CrossRef]
- Basu, M.K.; Makalowski, W.; Rogozin, I.B.; Koonin, E.V. U12 intron positions are more strongly conserved between animals and plants than U2 intron positions. Biol. Direct 2008, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turunen, J.J.; Niemelä, E.H.; Verma, B.; Frilander, M.J. The significant other: Splicing by the minor spliceosome. Wiley Interdiscip. Rev. RNA 2012, 4, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.W.; Van Nostrand, E.L.; Liang, T.Y. Discovery and Analysis of Evolutionarily Conserved Intronic Splicing Regulatory Elements. PLoS Genet. 2007, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Krainer, A.R. AT-AC Pre-mRNA Splicing Mechanisms and Conservation of Minor Introns in Voltage-Gated Ion Channel Genes. Mol. Cell. Biol. 1999, 19, 3225–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.A.; McCarthy, M.; Steitz, J.A. The splicing of U12-type introns can be a rate-limiting step in gene expression. EMBO J. 2002, 21, 3804–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, D.C.; LaRue, E.G.; Hershberger, C.E.; Roy, S.W.; Padgett, R.A. Comprehensive database and evolutionary dynamics of U12-type introns. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Montzka, K.A.; Steitz, J.A. Additional low-abundance human small nuclear ribonucleoproteins: U11, U12, etc. Proc. Natl. Acad. Sci. USA 1988, 85, 8885–8889. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.G.; Charette, J.M.; Spencer, D.F.; Gray, M.W. An early evolutionary origin for the minor spliceosome. Nat. Cell Biol. 2006, 443, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Carmel, L.; Csuros, M.; Koonin, E.V. Origin and evolution of spliceosomal introns. Biol. Direct 2012, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, W. The Exon Theory of Genes. Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 901–905. [Google Scholar] [CrossRef]
- Dibb, N.J.; Newman, A.J. Evidence that introns arose at proto-splice sites. EMBO J. 1989, 8, 2015–2021. [Google Scholar] [CrossRef]
- Dibb, N. Proto-splice site model of intron origin. J. Theor. Biol. 1991, 151, 405–416. [Google Scholar] [CrossRef]
- Sverdlov, A.V.; Rogozin, I.B.; Babenko, V.N.; Koonin, E.V. Reconstruction of Ancestral Protosplice Sites. Curr. Biol. 2004, 14, 1505–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, G.C.; Padgett, R. Conservation of functional features of U6atac and U12 snRNAs between vertebrates and higher plants. RNA 1999, 5, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younis, I.; Dittmar, K.; Wang, W.; Foley, S.W.; Berg, M.G.; Hu, K.Y.; Wei, Z.; Wan, L.; Dreyfuss, G. Minor introns are embedded molecular switches regulated by highly unstable U6atac snRNA. eLife 2013, 2, e00780. [Google Scholar] [CrossRef]
- Meinke, S.; Goldammer, G.; Weber, A.I.; Tarabykin, V.; Neumann, A.; Preussner, M.; Heyd, F. Srsf10 and the minor spliceosome control tissue-specific and dynamic SR protein expression. eLife 2020, 9, e56075. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [Green Version]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- López-Bigas, N.; Audit, B.; Ouzounis, C.; Parra, G.; Guigó, R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005, 579, 1900–1903. [Google Scholar] [CrossRef]
- Verma, B.; Akinyi, M.; Norppa, A.J.; Frilander, M.J. Minor spliceosome and disease. Semin. Cell Dev. Biol. 2018, 79, 103–112. [Google Scholar] [CrossRef]
- Shah, A.A.; Xu, G.; Rosen, A.; Hummers, L.K.; Wigley, F.M.; Elledge, S.J.; Casciola-Rosen, L. Brief Report: Anti–RNPC-3 Antibodies As a Marker of Cancer-Associated Scleroderma. Arthritis Rheumatol. 2017, 69, 1306–1312. [Google Scholar] [CrossRef]
- Fischer, D.; Wahlfors, T.; Mattila, H.; Oja, H.; Tammela, T.L.J.; Schleutker, J. MiRNA Profiles in Lymphoblastoid Cell Lines of Finnish Prostate Cancer Families. PLoS ONE 2015, 10, e0127427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Huang, Z.; Wang, Z.; Yin, C.; Zhou, L.; Zhang, L.; Huang, K.; Zhou, H.; Jiang, X.; Li, J.; et al. Identification of Novel Molecular Markers for Prognosis Estimation of Acute Myeloid Leukemia: Over-Expression of PDCD7, FIS1 and Ang2 May Indicate Poor Prognosis in Pretreatment Patients with Acute Myeloid Leukemia. PLoS ONE 2014, 9, e84150. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-F.; Mount, S.M.; Jarmołowski, A.; Makałowski, W. Evolutionary dynamics of U12-type spliceosomal introns. BMC Evol. Biol. 2010, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, R.C.; Peris, M.J.; Seyboldt, A.S.; Padgett, R.A. Role of the 3′ Splice Site in U12-Dependent Intron Splicing. Mol. Cell. Biol. 2001, 21, 1942–1952. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Gaubiercomella, P.; Delseny, M.; Grellet, F.; Van Montagu, M.; Rouze, P. Non–canonical introns are at least 109 years old. Nat. Genet. 1996, 14, 383–384. [Google Scholar] [CrossRef]
- Sharp, P.A. Five easy pieces. Science 1991, 254, 663–664. [Google Scholar] [CrossRef]
- Lynch, M. The evolution of spliceosomal introns. Curr. Opin. Genet. Dev. 2002, 12, 701–710. [Google Scholar] [CrossRef]
- Tarn, W.-Y.; Steitz, J.A. Pre-mRNA splicing: The discovery of a new spliceosome doubles the challenge. Trends Biochem. Sci. 1997, 22, 132–137. [Google Scholar] [CrossRef]
- Martin, W.; Koonin, E.V. Introns and the origin of nucleus–cytosol compartmentalization. Nat. Cell Biol. 2006, 440, 41–45. [Google Scholar] [CrossRef]
- Schneider, C.; Will, C.L.; Makarova, O.V.; Makarov, E.M.; Lührmann, R. Human U4/U6.U5 and U4atac/U6atac.U5 Tri-snRNPs Exhibit Similar Protein Compositions. Mol. Cell. Biol. 2002, 22, 3219–3229. [Google Scholar] [CrossRef] [Green Version]
- Basu, M.K.; Rogozin, I.B.; Koonin, E.V. Primordial spliceosomal introns were probably U2-type. Trends Genet. 2008, 24, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Scamborova, P.; Wong, A.; Steitz, J.A. An Intronic Enhancer Regulates Splicing of the Twintron of Drosophila melanogaster prospero Pre-mRNA by Two Different Spliceosomes. Mol. Cell. Biol. 2004, 24, 1855–1869. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Padgett, R. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 2009, 16, 1128–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Krainer, A.R. U1-Mediated Exon Definition Interactions between AT-AC and GT-AG Introns. Science 1996, 274, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Durbin, R. A computational scan for U12-dependent introns in the human genome sequence. Nucleic Acids Res. 2001, 29, 4006–4013. [Google Scholar] [CrossRef] [Green Version]
- Olthof, A.M.; Hyatt, K.C.; Kanadia, R.N. Minor intron splicing revisited: Identification of new minor intron-containing genes and tissue-dependent retention and alternative splicing of minor introns. BMC Genom. 2019, 20, 686. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Tarn, W.-Y.; Steitz, J.A. Highly Diverged U4 and U6 Small Nuclear RNAs Required for Splicing Rare AT-AC Introns. Science 1996, 273, 1824–1832. [Google Scholar] [CrossRef]
- Wassarman, K.M.; Steitz, J.A. The low-abundance U11 and U12 small nuclear ribonucleoproteins (snRNPs) interact to form a two-snRNP complex. Mol. Cell. Biol. 1992, 12, 1276–1285. [Google Scholar] [CrossRef] [Green Version]
- Frilander, M.J.; Steitz, J.A. Initial recognition of U12-dependent introns requires both U11/5′ splice-site and U12/branchpoint interactions. Genes Dev. 1999, 13, 851–863. [Google Scholar] [CrossRef] [Green Version]
- Will, C.L.; Schneider, C.; Reed, R.; Lührmann, R. Identification of Both Shared and Distinct Proteins in the Major and Minor Spliceosomes. Science 1999, 284, 2003–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golas, M.M.; Sander, B.; Will, C.L.; Lührmann, R.; Stark, H. Molecular Architecture of the Multiprotein Splicing Factor SF3b. Science 2003, 300, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Schneider, C.; Hossbach, M.; Urlaub, H.; Rauhut, R.; Elbashir, S.; Tuschl, T.; Lührmann, R. The human 18S U11/U12 snRNP contains a set of novel proteins not found in the U2-dependent spliceosome. RNA 2004, 10, 929–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, C.L.; Schneider, C.; Macmillan, A.M.; Katopodis, N.F.; Neubauer, C.; Wilms, M.; Lührmann, R.; Query, C.C. A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J. 2001, 20, 4536–4546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorkovic, Z.J.; Lehner, R.; Forstner, C.; Barta, A. Evolutionary conservation of minor U12-type spliceosome between plants and humans. RNA 2005, 11, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Jung, H.J.; Dinh, S.N.; Kang, H. Structural features important for the U12 snRNA binding and minor spliceosome assembly of Arabidopsis U11/U12-small nuclear ribonucleoproteins. RNA Biol. 2016, 13, 670–679. [Google Scholar] [CrossRef]
- Nottrott, S.; Hartmuth, K.; Fabrizio, P.; Urlaub, H.; Vidovic, I.; Ficner, R.; Luhrmann, R. Functional interaction of a novel 15.5 kD [U4/U6·U5] tri-snRNP protein with the 5’ stem-loop of U4 snRNA. EMBO J. 1999, 18, 6119–6133. [Google Scholar] [CrossRef] [Green Version]
- Frilander, M.J.; Steitz, J.A. Dynamic Exchanges of RNA Interactions Leading to Catalytic Core Formation in the U12-Dependent Spliceosome. Mol. Cell 2001, 7, 217–226. [Google Scholar] [CrossRef]
- Montes, M.; Sanford, B.L.; Comiskey, D.F.; Chandler, D.S. RNA Splicing and Disease: Animal Models to Therapies. Trends Genet. 2019, 35, 68–87. [Google Scholar] [CrossRef]
- Singh, R.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Madan, V.; Kanojia, D.; Jia, L.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A. ZRSR2 mutations cause dysregulated RNA splicing in MDS. Blood 2014, 124, 4609. [Google Scholar] [CrossRef]
- Jutzi, D.; Akinyi, M.; Mechtersheimer, J.; Frilander, M.J.; Ruepp, M.-D. The emerging role of minor intron splicing in neurological disorders. Cell Stress 2018, 2, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.D.; Lemoine, C.; Aquino, G.S.; Vaeth, A.M.; Kanadia, R.N. Minor spliceosome disruption causes limb growth defects without altering patterning. BioRxiv 2020. [Google Scholar] [CrossRef]
- Argente, J.; Flores, R.Z.; Gutiérrez-Arumí, A.; Verma, B.; Moreno, G.; Ángel, M.; Cuscó, I.; Oghabian, A.; Chowen, J.A.; Frilander, M.J.; et al. Defective minor spliceosome mRNA processing results in isolated familial growth hormone deficiency. EMBO Mol. Med. 2014, 6, 299–306. [Google Scholar] [CrossRef]
- Ng, B.; Yang, F.; Huston, D.P.; Yan, Y.; Yang, Y.; Xiong, Z.; Peterson, L.E.; Wang, H.; Yang, X.-F. Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J. Allergy Clin. Immunol. 2004, 114, 1463–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrank, B.; Götz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [Green Version]
- Otake, L.R.; Scamborova, P.; Hashimoto, C.; Steitz, J.A. The divergent U12-type spliceosome is required for pre-mRNA splicing and is essential for development in Drosophila. Mol. Cell 2002, 9, 439–446. [Google Scholar] [CrossRef]
- Doggett, K.; Williams, B.B.; Markmiller, S.; Geng, F.-S.; Coates, J.; Mieruszynski, S.; Ernst, M.; Thomas, T.; Heath, J.K. Early developmental arrest and impaired gastrointestinal homeostasis in U12-dependent splicing-defective Rnpc3-deficient mice. RNA 2018, 24, 1856–1870. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, M.; Olthof, A.M.; Aquino, G.S.; Hyatt, K.C.; Lemoine, C.; Drake, K.; Sturrock, N.; Nguyen, N.; al Seesi, S.; Kanadia, R.N. Minor spliceosome inactivation causes microcephaly, owing to cell cycle defects and death of self-amplifying radial glial cells. Development 2018, 145, 166322. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Salam, G.M.; Abdel-Hamid, M.S.; Issa, M.; Magdy, A.; El-Kotoury, A.; Amr, K. Expanding the phenotypic and mutational spectrum in microcephalic osteodysplastic primordial dwarfism type I. Am. J. Med. Genet. Part A 2012, 158A, 1455–1461. [Google Scholar] [CrossRef]
- Abdel-Salam, G.M.; Miyake, N.; Eid, M.M.; Abdel-Hamid, M.S.; Hassan, N.A.; Eid, O.M.; Effat, L.K.; El-Badry, T.H.; El-Kamah, G.Y.; El-Darouti, M.; et al. A homozygous mutation in RNU4ATAC as a cause of microcephalic osteodysplastic primordial dwarfism type I (MOPD I) with associated pigmentary disorder. Am. J. Med. Genet. Part A 2011, 155, 2885–2896. [Google Scholar] [CrossRef]
- Cologne, A.; Benoit-Pilven, C.; Besson, A.; Putoux, A.; Campan-Fournier, A.; Bober, M.B.; De Die-Smulders, C.E.; Paulussen, A.D.; Pinson, L.; Toutain, A.; et al. New insights into minor splicing-a transcriptomic analysis of cells derived from TALS patients. RNA 2019, 25, 1130–1149. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.J.; Morse, R.P. The neurologic findings in Taybi-Linder syndrome (MOPD I/III): Case report and review of the literature. Am. J. Med. Genet. Part A 2012, 158, 606–610. [Google Scholar] [CrossRef]
- Osman, E.Y.; Van Alstyne, M.; Yen, P.-F.; Lotti, F.; Feng, Z.; Ling, K.K.; Ko, C.-P.; Pellizzoni, L.; Lorson, C.L. Minor snRNA gene delivery improves the loss of proprioceptive synapses on SMA motor neurons. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Hastings, M.L.; Resta, N.; Traum, D.; Stella, A.; Guanti, G.; Krainer, A.R. An LKB1 AT-AC intron mutation causes Peutz-Jeghers syndrome via splicing at noncanonical cryptic splice sites. Nat. Struct. Mol. Biol. 2004, 12, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.A.; Brunetti-Pierre, N.; Kadasi, L.; Kovacova, V.; Van Maldergem, L.; De Brasi, D.; Salerno, M.; Gecz, J. Identification of three novel SEDL mutations, including mutation in the rare, non-canonical splice site of exon 4. Clin. Genet. 2003, 64, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, M.F.; Chalhoub, N.; Ben-Omran, T.; Kumar, P.; Kamel, H.; Ibrahim, K.; Mohamoud, Y.; Al-Dous, E.; Al-Azwani, I.; Malek, J.A.; et al. Mutation in noncoding RNA RNU12 causes early onset cerebellar ataxia. Ann. Neurol. 2017, 81, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Polaski, J.T.; Taylor, J.; Castel, P.; Chen, S.; Kobayashi, S.; Hogg, S.J.; Hayashi, Y.; Pineda, J.M.B.; El Marabti, E.; et al. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat. Genet. 2021, 53, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Edery, P.; Marcaillou, C.; Sahbatou, M.; Labalme, A.; Chastang, J.; Touraine, R.; Tubacher, E.; Senni, F.; Bober, M.B.; Nampoothiri, S.; et al. Association of TALS Developmental Disorder with Defect in Minor Splicing Component U4atac snRNA. Science 2011, 332, 240–243. [Google Scholar] [CrossRef]
- Farach, L.S.; Little, M.E.; Duker, A.L.; Logan, C.V.; Jackson, A.; Hecht, J.T.; Bober, M. The expanding phenotype of RNU4ATAC pathogenic variants to Lowry Wood syndrome. Am. J. Med. Genet. Part A 2018, 176, 465–469. [Google Scholar] [CrossRef]
- Merico, D.; Roifman, M.; Braunschweig, U.; Yuen, R.K.C.; Alexandrova, R.; Bates, A.; Reid, B.; Nalpathamkalam, T.; Wang, Z.; Thiruvahindrapuram, B.; et al. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat. Commun. 2015, 6, 8718. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, A.; Xie, J.; Liu, Y.F.; Poplawski, A.B.; Gomes, A.R.; Madanecki, P.; Fu, C.; Crowley, M.R.; Crossman, D.K.; Armstrong, L.; et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 2014, 46, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Reber, S.; Stettler, J.; Filosa, G.; Colombo, M.; Jutzi, D.; Lenzken, S.C.; Schweingruber, C.; Bruggmann, R.; Bachi, A.; Barabino, S.M.; et al. Minor intron splicing is regulated by FUS and affected by ALS -associated FUS mutants. EMBO J. 2016, 35, 1504–1521. [Google Scholar] [CrossRef]
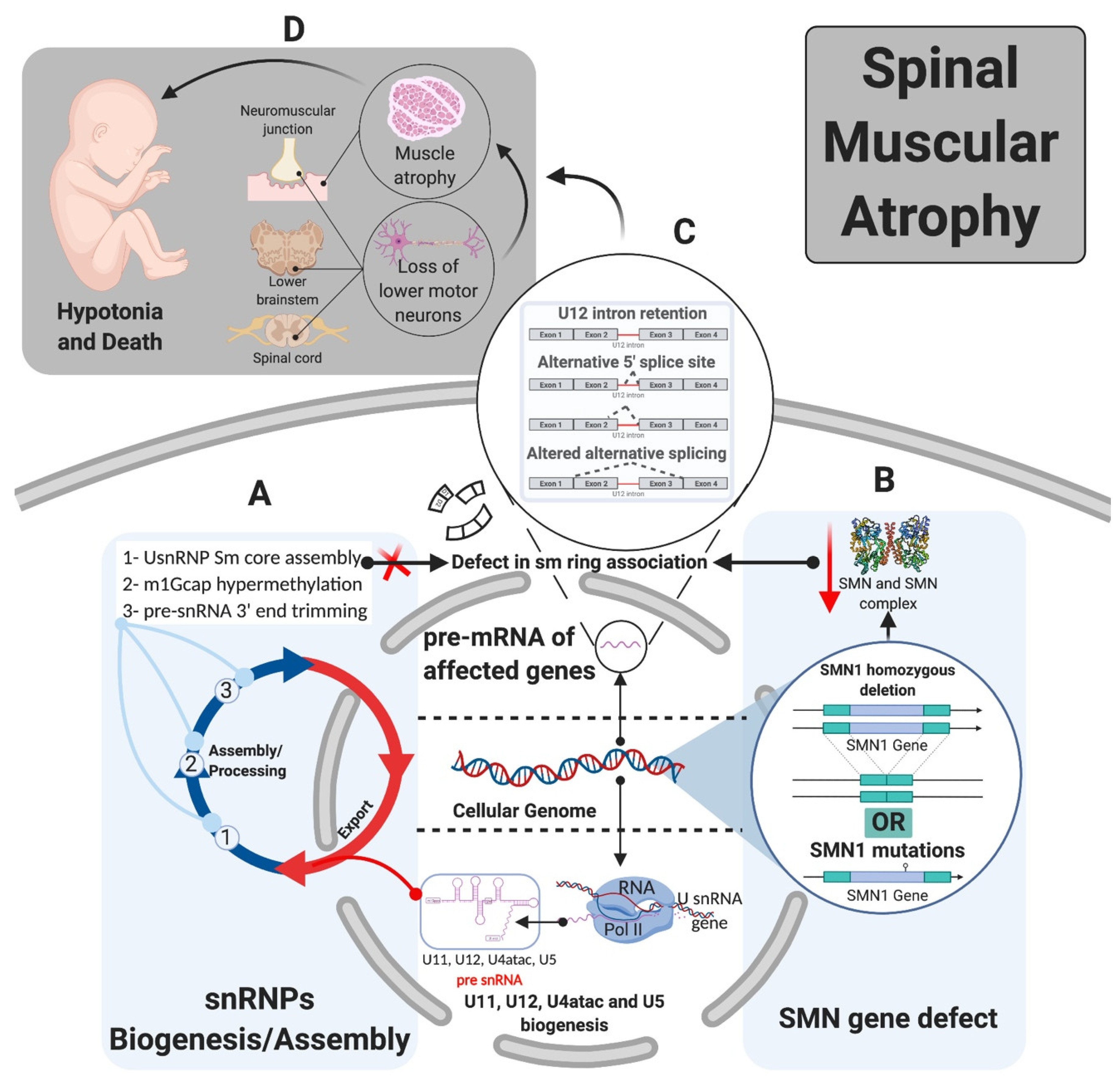
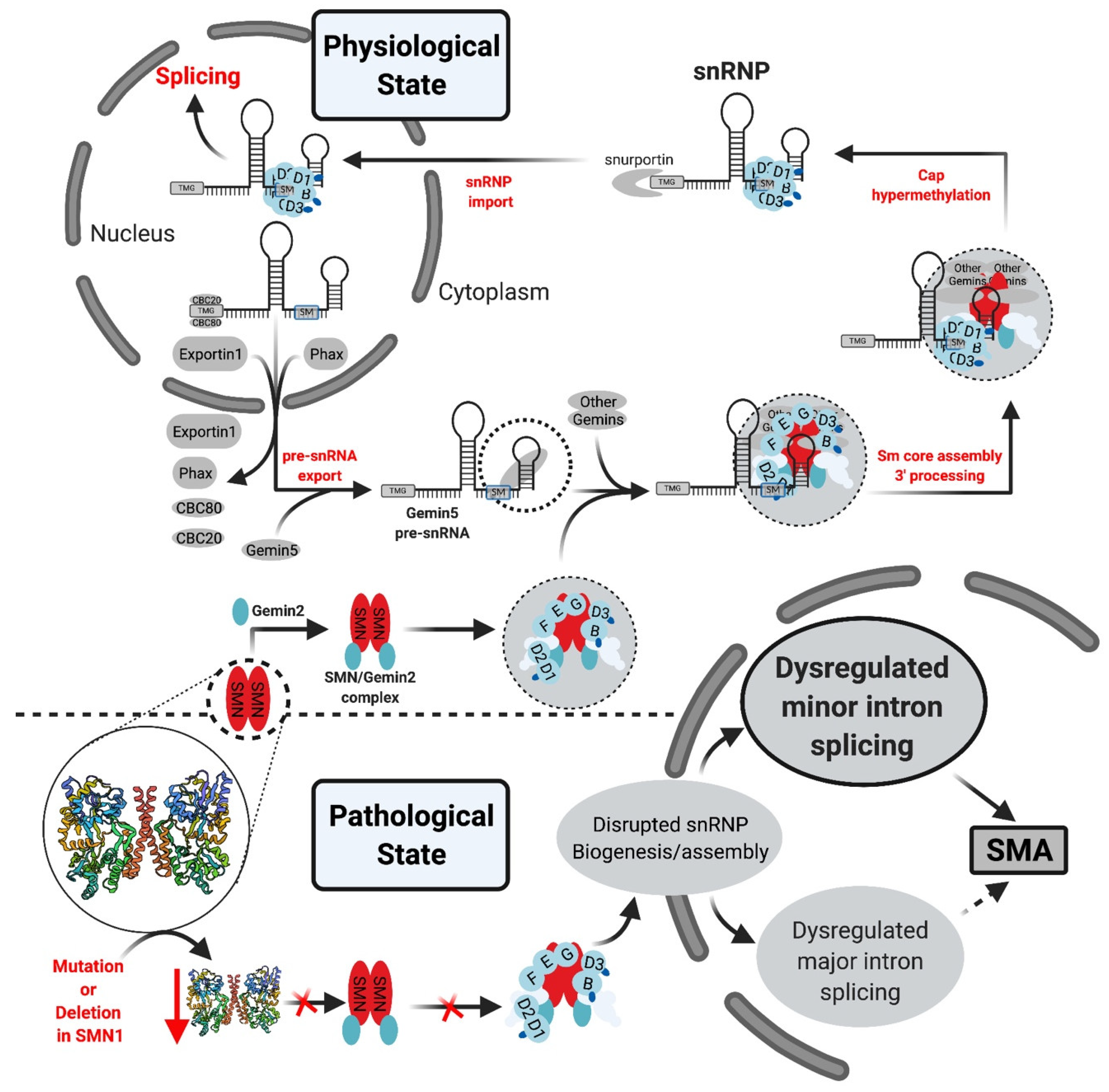
- Gabanella, F.; Butchbach, M.E.R.; Saieva, L.; Carissimi, C.; Burghes, A.H.M.; Pellizzoni, L. Ribonucleoprotein Assembly Defects Correlate with Spinal Muscular Atrophy Severity and Preferentially Affect a Subset of Spliceosomal snRNPs. PLoS ONE 2007, 2, e921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN Deficiency Causes Tissue-Specific Perturbations in the Repertoire of snRNAs and Widespread Defects in Splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, E.; Saieva, L.; Carrel, T.L.; Crawford, T.O.; Liu, D.; Lutz, C.; Beattie, C.E.; Pellizzoni, L.; Burghes, A.H. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum. Mol. Genet. 2009, 18, 2215–2229. [Google Scholar] [CrossRef]
- Boulisfane, N.; Choleza, M.; Rage, F.; Neel, H.; Soret, J.; Bordonné, R. Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum. Mol. Genet. 2010, 20, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Lotti, F.; Imlach, W.L.; Saieva, L.; Beck, E.S.; Hao, L.T.; Li, D.K.; Jiao, W.; Mentis, G.Z.; Beattie, C.E.; McCabe, B.D.; et al. An SMN-Dependent U12 Splicing Event Essential for Motor Circuit Function. Cell 2012, 151, 440–454. [Google Scholar] [CrossRef] [Green Version]
- Jangi, M.; Fleet, C.; Cullen, P.; Gupta, S.V.; Mekhoubad, S.; Chiao, E.; Allaire, N.; Bennett, C.F.; Rigo, F.; Krainer, A.R.; et al. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. USA 2017, 114, E2347–E2356. [Google Scholar] [CrossRef] [Green Version]
- Doktor, T.K.; Hua, Y.; Andersen, H.S.; Brøner, S.; Liu, Y.H.; Wieckowska, A.; Dembic, M.; Bruun, G.H.; Krainer, A.R.; Andresen, B.S. RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res. 2017, 45, 395–416. [Google Scholar] [CrossRef]
- Gilliam, A.C.; Steitz, J.A. Rare scleroderma autoantibodies to the U11 small nuclear ribonucleoprotein and to the trimethylguanosine cap of U small nuclear RNAs. Proc. Natl. Acad. Sci. USA 1993, 90, 6781–6785. [Google Scholar] [CrossRef] [Green Version]
- Zieve, G.W.; Sauterer, R.A.; Margolis, R.L. Cell Biology of the snRNP Particle. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Lehmeier, T.; Foulaki, K.; Lührmann, R. Evidence for three distinct D proteins, which react differentially with anti-Sm autoantibodies, in the cores of the major snRNPs U1, U2, U4/U6 and U5. Nucleic Acids Res. 1990, 18, 6475–6484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Liyanarachchi, S.; Akagi, K.; Nagy, R.; Li, J.; Dietrich, R.C.; Li, W.; Sebastian, N.; Wen, B.; Xin, B.; et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science 2011, 332, 238–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Lo, H.S.; Yang, H.; Gere, S.; Hu, Y.; Buetow, K.H.; Lee, M.P. Computational analysis and experimental validation of tumor-associated alternative RNA splicing in human cancer. Cancer Res. 2003, 63, 655–657. [Google Scholar] [PubMed]
- Markmiller, S.; Cloonan, N.; Lardelli, R.M.; Doggett, K.; Keightley, M.-C.; Boglev, Y.; Trotter, A.J.; Ng, A.Y.; Wilkins, S.J.; Verkade, H.; et al. Minor class splicing shapes the zebrafish transcriptome during development. Proc. Natl. Acad. Sci. USA 2014, 111, 3062–3067. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, R.; Kraus, W.L. The PARP Side of the Nucleus: Molecular Actions, Physiological Outcomes, and Clinical Targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [Green Version]
- Burghes, A.H.M.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, S.; Pellizzoni, L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015, 35, 8691–8700. [Google Scholar] [CrossRef] [Green Version]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef]
- Martinez-Montiel, N.; Rosas-Murrieta, N.H.; Ruiz, M.A.; Monjaraz-Guzman, E.; Martinez-Contreras, R. Alternative Splicing as a Target for Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Ma, Y.; Huang, T.; Chen, Y.; Peng, Y.; Li, B.; Li, J.; Zhang, Y.; Song, B.; Sun, X.; et al. Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol. Cell 2018, 72, 380–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
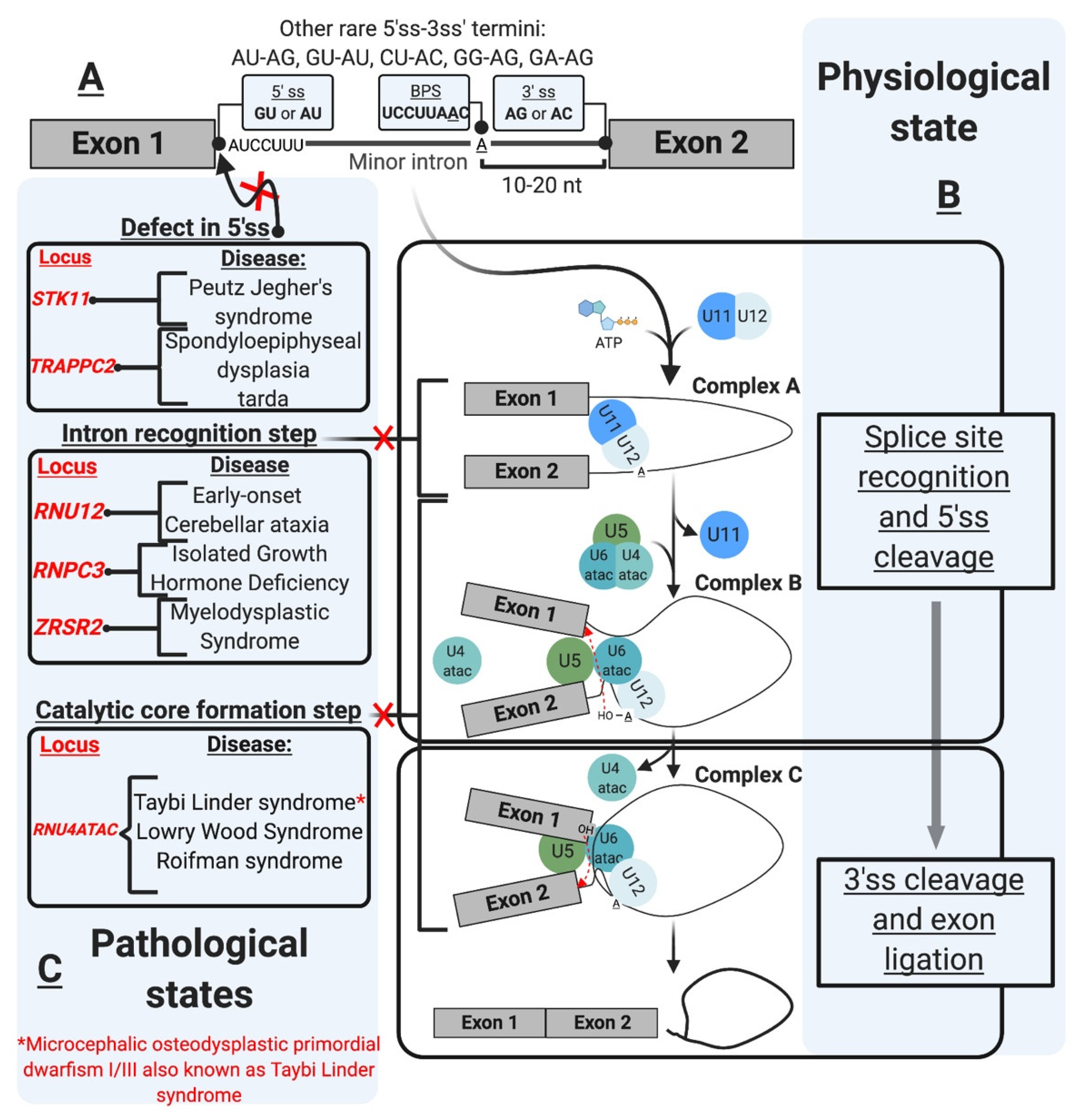
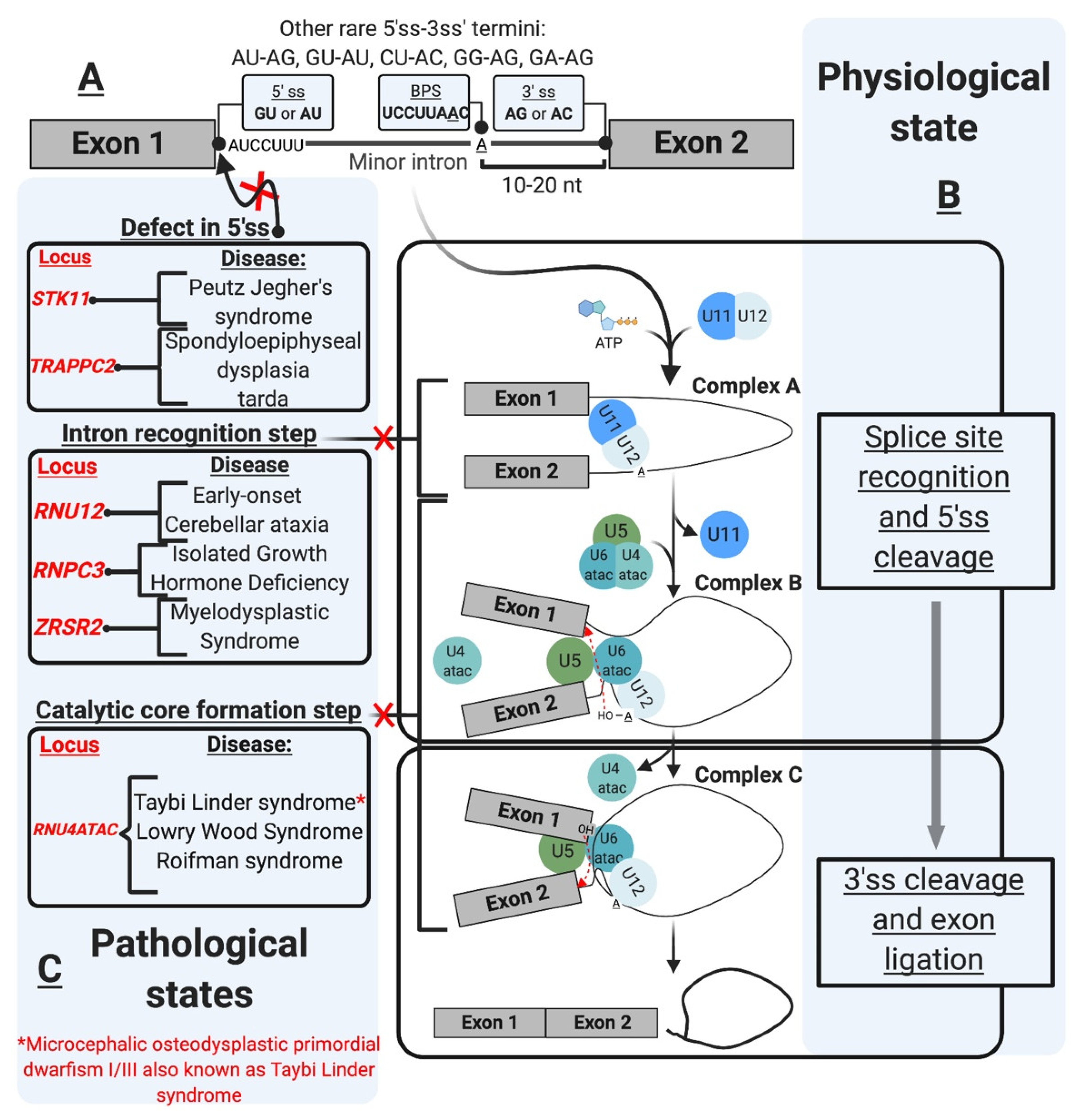
| Splicing Defect | Gene Affected | Disease | References |
|---|---|---|---|
| Aberrant 5’ splice site | STK11 | Peutz Jegher’s syndrome (PJS) | [85] |
| TRAPPC2 | Spondyloepiphyseal dysplasia tarda (SEDT) | [86] | |
| Intron recognition/mutant minor snRNP component | RNU12 | Early-onset cerebellar ataxia (EOCA) | [87] |
| RNPC3 | Isolated growth hormone deficiency (IGHD) | [74] | |
| ZRSR2 | Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) | [88] | |
| RNU4ATAC | Taybi-Linder syndrome (TLS) | [89] | |
| RNU4ATAC | Lowry Wood syndrome (LWS) | [90] | |
| RNU4ATAC | Roifman syndrome | [91] | |
| Branch point mutation | LZTR1 | Noonan syndrome (NS) | [88] |
| LZTR1 | Schwannomatosis | [92] | |
| Biogenesis/Assembly | FUS | Amyotrophic lateral sclerosis (ALS) | [93] |
| SMN1 | Spinal muscular atrophy (SMA) | [94,95,96,97,98,99,100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Marabti, E.; Malek, J.; Younis, I. Minor Intron Splicing from Basic Science to Disease. Int. J. Mol. Sci. 2021, 22, 6062. https://doi.org/10.3390/ijms22116062
El Marabti E, Malek J, Younis I. Minor Intron Splicing from Basic Science to Disease. International Journal of Molecular Sciences. 2021; 22(11):6062. https://doi.org/10.3390/ijms22116062
Chicago/Turabian StyleEl Marabti, Ettaib, Joel Malek, and Ihab Younis. 2021. "Minor Intron Splicing from Basic Science to Disease" International Journal of Molecular Sciences 22, no. 11: 6062. https://doi.org/10.3390/ijms22116062
APA StyleEl Marabti, E., Malek, J., & Younis, I. (2021). Minor Intron Splicing from Basic Science to Disease. International Journal of Molecular Sciences, 22(11), 6062. https://doi.org/10.3390/ijms22116062