Figure 1.
GSK3β phosphorylation sites. (A) GSK3β phosphorylation consensus sequence. (B) GSK3β phosphorylation sites of β-catenin. (C) Three putative GSK3β phosphorylation sites of the human AHR (amino acid 1-848) which show best match to the consensus sequence. The numbers indicate the amino acid locations of the primary sequence. Orange font represents the serine and threonine sites for GSK3β phosphorylation. S45 (bold) of β-catenin represents the priming site of casein kinase 1. Phosphorylation of the priming site is necessary for GSK phosphorylation.
Figure 1.
GSK3β phosphorylation sites. (A) GSK3β phosphorylation consensus sequence. (B) GSK3β phosphorylation sites of β-catenin. (C) Three putative GSK3β phosphorylation sites of the human AHR (amino acid 1-848) which show best match to the consensus sequence. The numbers indicate the amino acid locations of the primary sequence. Orange font represents the serine and threonine sites for GSK3β phosphorylation. S45 (bold) of β-catenin represents the priming site of casein kinase 1. Phosphorylation of the priming site is necessary for GSK phosphorylation.
Figure 2.
AHR protein levels are regulated by GSK3β in HeLa cells. (A) HeLa cells were treated with 10 μM, 20 μM or 40 μM TDG for 8 h. Western blot analysis showed that AHR protein levels increased after TDG treatment. The images are representative of the replicate data (means ± SD, n = 3, *** p < 0.001). DMSO group (treated for 8 h) was arbitrarily set as one (with no error bar) for data normalization. One-way ANOVA with Dunnett multiple comparisons test was performed to determine statistical significance. (B) AHR protein levels increased after 5 mM LiCl treatment for 6 h. The images are representative of the replicate data (means ± SD, n = 3, * p < 0.05). No LiCl (no treatment) group was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (C) Transient knockdown of GSK3β (after 72 h) increased AHR protein levels. The images are representative of the replicate data (means ± SD, n = 3, * p < 0.05). Cells transfected with scramble shRNA were used as the negative control and was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (D) Transient expression of HA-GSK3β (after 72 h) decreased AHR protein levels. The images are representative of the replicate data (means ± SD, n = 3, ** p < 0.01) in one experiment and was repeated two more times with similar results. No plasmid group (NP) represents cells undergoing the same transient transfection without a plasmid. Unpaired t-test was used to determine the statistical significance. For A to D, each Western lane contained 30 μg of whole-cell lysate. Data in A for 10-20 μM treatment groups were normalized by β-actin (as shown) whereas the rest of the data were normalized by total protein stain.
Figure 2.
AHR protein levels are regulated by GSK3β in HeLa cells. (A) HeLa cells were treated with 10 μM, 20 μM or 40 μM TDG for 8 h. Western blot analysis showed that AHR protein levels increased after TDG treatment. The images are representative of the replicate data (means ± SD, n = 3, *** p < 0.001). DMSO group (treated for 8 h) was arbitrarily set as one (with no error bar) for data normalization. One-way ANOVA with Dunnett multiple comparisons test was performed to determine statistical significance. (B) AHR protein levels increased after 5 mM LiCl treatment for 6 h. The images are representative of the replicate data (means ± SD, n = 3, * p < 0.05). No LiCl (no treatment) group was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (C) Transient knockdown of GSK3β (after 72 h) increased AHR protein levels. The images are representative of the replicate data (means ± SD, n = 3, * p < 0.05). Cells transfected with scramble shRNA were used as the negative control and was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (D) Transient expression of HA-GSK3β (after 72 h) decreased AHR protein levels. The images are representative of the replicate data (means ± SD, n = 3, ** p < 0.01) in one experiment and was repeated two more times with similar results. No plasmid group (NP) represents cells undergoing the same transient transfection without a plasmid. Unpaired t-test was used to determine the statistical significance. For A to D, each Western lane contained 30 μg of whole-cell lysate. Data in A for 10-20 μM treatment groups were normalized by β-actin (as shown) whereas the rest of the data were normalized by total protein stain.

Figure 3.
AHR undergoes the LC-3-mediated autophagy after GSK3β phosphorylation. (A) AHR protein levels ± HA-GSK3β transient expression (72 h) were increased by CQ (40 μM for 6 h, last 6 h of the 72-h period of transient transfection) but not by MG132 (10 μM for 6 h, last 6 h of the 72-h period of transient transfection). The time and dose of CQ and MG132 were optimized for inhibition of degradation. The images are representative of the replicate data (means ± SD, n = 3, ** p < 0.01, **** p < 0.0001, ns represents not significant) in one experiment and was repeated once with similar results. DMSO treatment group (treated for 6 h) was arbitrarily set as one for data normalization. No plasmid (NP) group represents cells undergoing the same transient transfection as HA-GSK3β but without plasmid. Two-way ANOVA with Dunnett multiple comparisons test was performed to determine statistical significance. (B) β-catenin protein levels decreased after HA-GSK3β transient expression (72 h) and were increased ± HA-GSK3β by MG132 (10 μM for 6 h, last 6 h of the 72-h period of transient transfection). The images are representative of the replicate data (means ± SD, n = 5, ** p < 0.01, *** p < 0.001, **** p < 0.0001). DMSO treatment group was arbitrarily set as one for data normalization. No plasmid (NP) group represents cells undergoing the same transient transfection as HA-GSK3β but without plasmid. Two-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. (C) LC3B-II levels with HA-GSK3β transient expression (72 h) were higher than that in the no plasmid group (NP). No plasmid (NP) group represents cells undergoing the same transient transfection as HA-GSK3β but without plasmid. The fold change of LC3B-II increase after 6-h, 40 μM CQ treatment was also higher in the HA-GSK3β group. The images are representative of the replicate data (means ± SD, n = 3, **** p < 0.0001) in one experiment and was repeated once with similar results. No treatment (NT) group (treated for 6 h) was arbitrarily set as one for data normalization. Two-way ANOVA with Tukey multiple comparisons test was performed to determine statistical significance. (D) Immunoprecipitation of AHR from HeLa whole cell lysates ± GSK3β knockdown using anti-AHR SA210 polyclonal antibody, followed by K63-TUBE Far-western analysis showing less K63-ubiquitinated AHR in GSK3β knockdown group while more AHR (104 kDa) being precipitated when compared to the scramble shRNA-transfected group. The images are representative of the replicate data (means ± SD, n = 3, * p < 0.05) with scramble shRNA group set as one for normalization of three separate experiment (thus no error bar). For A to C, each Western lane contained 30 μg of whole-cell lysate. Data were normalized by total protein stain.
Figure 3.
AHR undergoes the LC-3-mediated autophagy after GSK3β phosphorylation. (A) AHR protein levels ± HA-GSK3β transient expression (72 h) were increased by CQ (40 μM for 6 h, last 6 h of the 72-h period of transient transfection) but not by MG132 (10 μM for 6 h, last 6 h of the 72-h period of transient transfection). The time and dose of CQ and MG132 were optimized for inhibition of degradation. The images are representative of the replicate data (means ± SD, n = 3, ** p < 0.01, **** p < 0.0001, ns represents not significant) in one experiment and was repeated once with similar results. DMSO treatment group (treated for 6 h) was arbitrarily set as one for data normalization. No plasmid (NP) group represents cells undergoing the same transient transfection as HA-GSK3β but without plasmid. Two-way ANOVA with Dunnett multiple comparisons test was performed to determine statistical significance. (B) β-catenin protein levels decreased after HA-GSK3β transient expression (72 h) and were increased ± HA-GSK3β by MG132 (10 μM for 6 h, last 6 h of the 72-h period of transient transfection). The images are representative of the replicate data (means ± SD, n = 5, ** p < 0.01, *** p < 0.001, **** p < 0.0001). DMSO treatment group was arbitrarily set as one for data normalization. No plasmid (NP) group represents cells undergoing the same transient transfection as HA-GSK3β but without plasmid. Two-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. (C) LC3B-II levels with HA-GSK3β transient expression (72 h) were higher than that in the no plasmid group (NP). No plasmid (NP) group represents cells undergoing the same transient transfection as HA-GSK3β but without plasmid. The fold change of LC3B-II increase after 6-h, 40 μM CQ treatment was also higher in the HA-GSK3β group. The images are representative of the replicate data (means ± SD, n = 3, **** p < 0.0001) in one experiment and was repeated once with similar results. No treatment (NT) group (treated for 6 h) was arbitrarily set as one for data normalization. Two-way ANOVA with Tukey multiple comparisons test was performed to determine statistical significance. (D) Immunoprecipitation of AHR from HeLa whole cell lysates ± GSK3β knockdown using anti-AHR SA210 polyclonal antibody, followed by K63-TUBE Far-western analysis showing less K63-ubiquitinated AHR in GSK3β knockdown group while more AHR (104 kDa) being precipitated when compared to the scramble shRNA-transfected group. The images are representative of the replicate data (means ± SD, n = 3, * p < 0.05) with scramble shRNA group set as one for normalization of three separate experiment (thus no error bar). For A to C, each Western lane contained 30 μg of whole-cell lysate. Data were normalized by total protein stain.


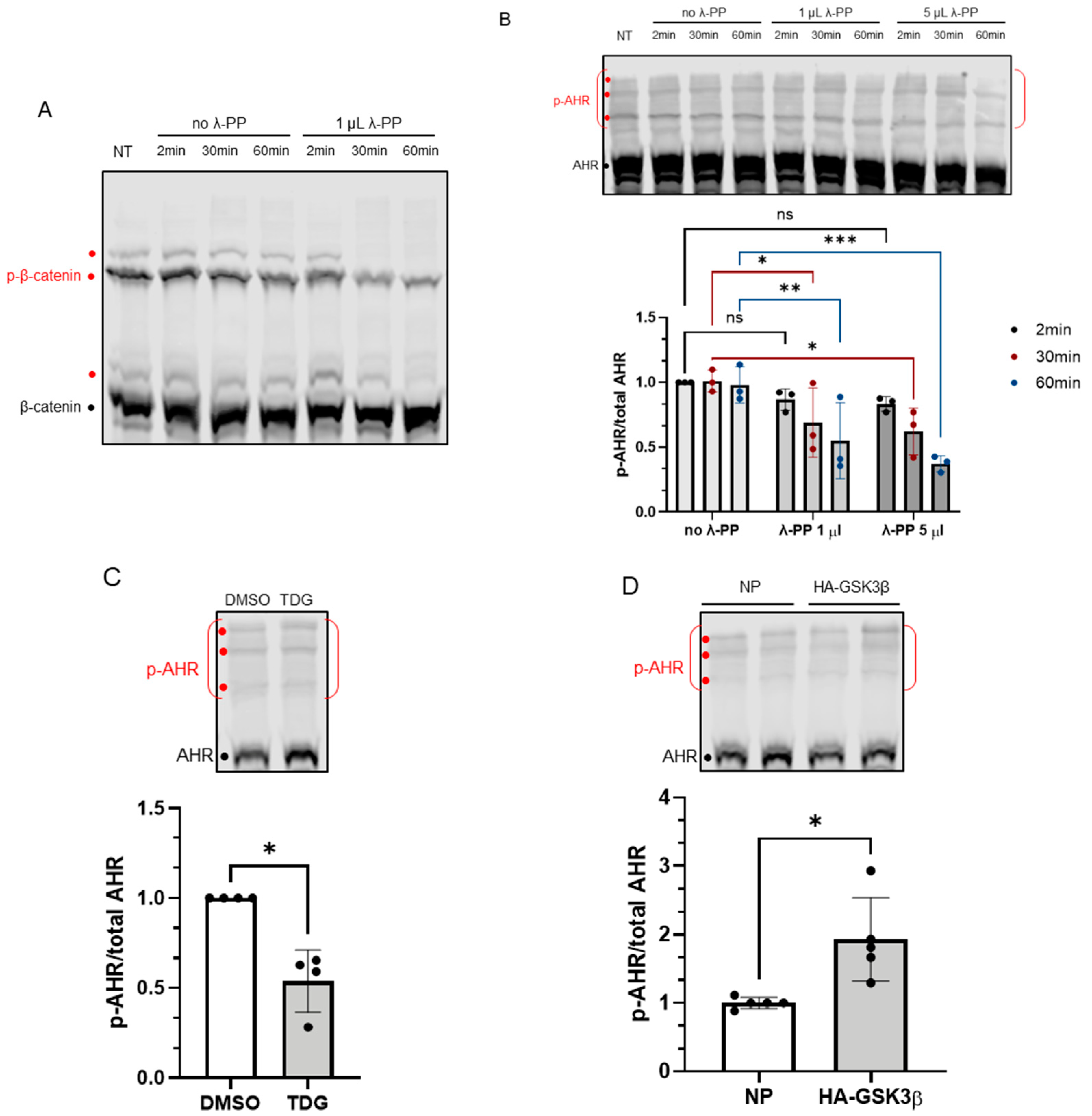
Figure 4.
Phosphorylation of AHR is affected by GSK3β activity. 7.5% acrylamide bis-Tris gel embedded with 20 μM phos-tag reagent was used to confirm the phosphorylation of AHR. (A) Phosphorylated (p-β-catenin) and unphosphorylated β-catenin can be separated, and the intensities of three phosphorylated bands (red dots) were noticeably decreased by 1 μL (80 units) of λ-PP treatment for 2, 30 or 60 min at 30 °C. NT represents no treatment. The top region of AHR blot in B-D shows three prominent phosphorylated AHR bands (p-AHR, red dots). The whole top region indicated by the red bracket is defined as the phosphorylated region and unphosphorylated AHR is indicated with the black dot. The y-axis (p-AHR/total AHR) represents intensity of the phosphorylated bracket region divided by the sum of the intensities of the bracket region plus unphosphorylated AHR. (B) Time- and dose-dependent decrease of the p-AHR/total AHR value after 1 μL (80 units) or 5 μL (400 units) of λ-PP treatment for 2, 30 or 60 min for 30 °C. The images are representative of the replicate data (means ± SD, n = 3, * p < 0.05, ** p < 0.01, *** p < 0.001, ns represents not significant). Two-minute of no λ-PP treatment group was arbitrarily set as one (with no error bar) for data normalization. Two-way ANOVA with Dunnett multiple comparisons test was performed to determine statistical significance. (C) After 40 μM TDG treatment for 8 h, the p-AHR/total AHR values decreased. The images are representative of the replicate data (means ± SD, n = 4, * p < 0.05). DMSO group (treated for 8 h) was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (D) Transient expression of HA-GSK3β increased the p-AHR/total AHR value. The images are representative of the replicate data (means ± SD, n = 5, * p < 0.05). No plasmid group (NP), which represents cells undergone same transient transfection protocol without the plasmid carrying HA-GSK3β cDNA, was arbitrarily set as one for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. For A to D, each Western lane contained 40 μg of whole-cell lysate. Data were normalized by total protein stain.
Figure 4.
Phosphorylation of AHR is affected by GSK3β activity. 7.5% acrylamide bis-Tris gel embedded with 20 μM phos-tag reagent was used to confirm the phosphorylation of AHR. (A) Phosphorylated (p-β-catenin) and unphosphorylated β-catenin can be separated, and the intensities of three phosphorylated bands (red dots) were noticeably decreased by 1 μL (80 units) of λ-PP treatment for 2, 30 or 60 min at 30 °C. NT represents no treatment. The top region of AHR blot in B-D shows three prominent phosphorylated AHR bands (p-AHR, red dots). The whole top region indicated by the red bracket is defined as the phosphorylated region and unphosphorylated AHR is indicated with the black dot. The y-axis (p-AHR/total AHR) represents intensity of the phosphorylated bracket region divided by the sum of the intensities of the bracket region plus unphosphorylated AHR. (B) Time- and dose-dependent decrease of the p-AHR/total AHR value after 1 μL (80 units) or 5 μL (400 units) of λ-PP treatment for 2, 30 or 60 min for 30 °C. The images are representative of the replicate data (means ± SD, n = 3, * p < 0.05, ** p < 0.01, *** p < 0.001, ns represents not significant). Two-minute of no λ-PP treatment group was arbitrarily set as one (with no error bar) for data normalization. Two-way ANOVA with Dunnett multiple comparisons test was performed to determine statistical significance. (C) After 40 μM TDG treatment for 8 h, the p-AHR/total AHR values decreased. The images are representative of the replicate data (means ± SD, n = 4, * p < 0.05). DMSO group (treated for 8 h) was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (D) Transient expression of HA-GSK3β increased the p-AHR/total AHR value. The images are representative of the replicate data (means ± SD, n = 5, * p < 0.05). No plasmid group (NP), which represents cells undergone same transient transfection protocol without the plasmid carrying HA-GSK3β cDNA, was arbitrarily set as one for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. For A to D, each Western lane contained 40 μg of whole-cell lysate. Data were normalized by total protein stain.

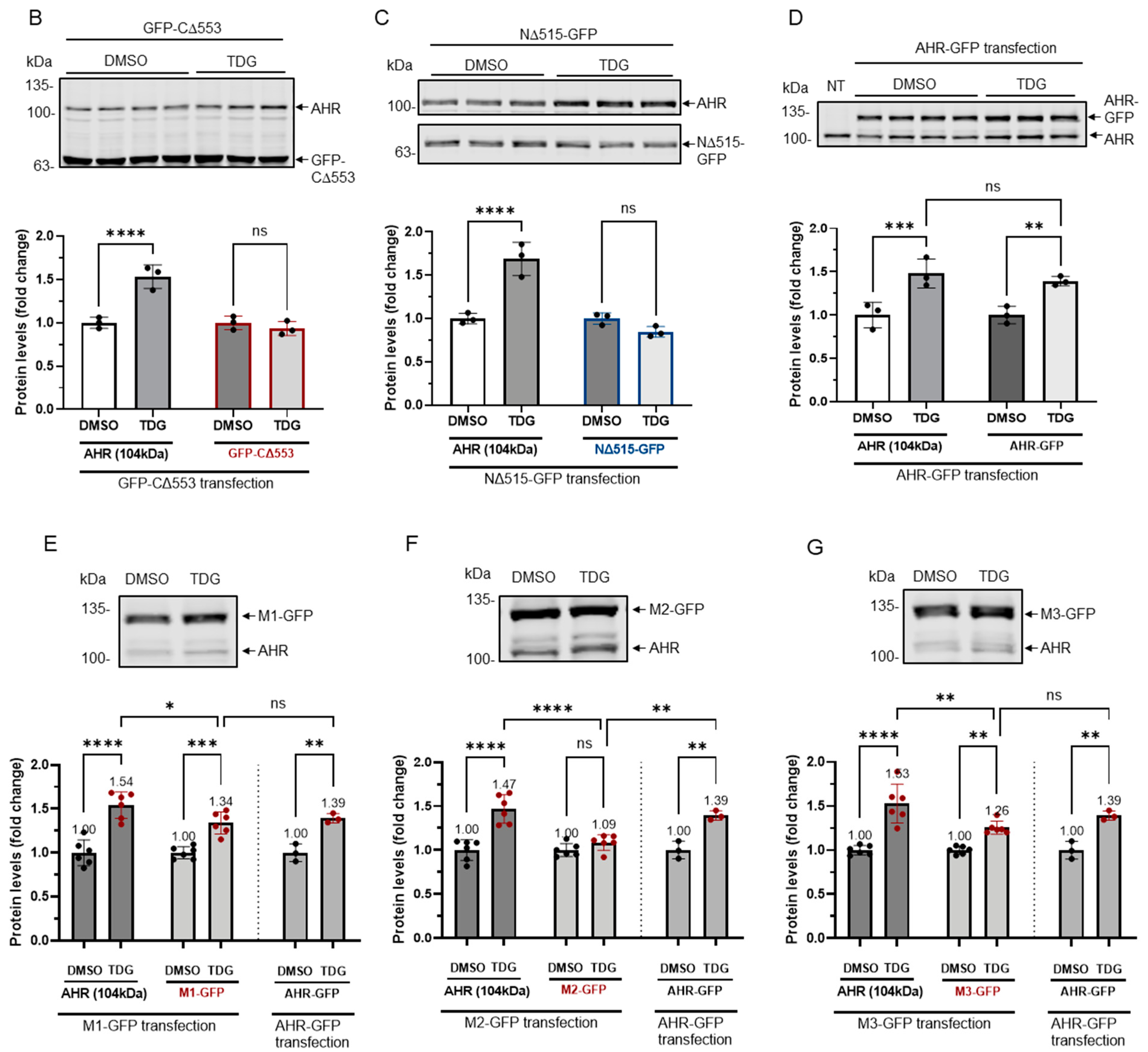
Figure 5.
Site-directed mutagenesis reveals three putative GSK3β phosphorylation sites of AHR. (A) A map showing GFP fusion expressing plasmids of full length human AHR and two truncated derivatives (pGFP2-N2-AHR, pGFP2-C2-CΔ553, and pGFP2-N2-NΔ515) and three AHR mutant GFP plasmids (S436A-S440A-S444A, M1; S689A-S693A-T697A, M2; S723A-S727A-T731A, M3). The names of the plasmid expressing the corresponding protein are listed on the left. Both GFP fusions of CΔ553 (B) and NΔ515 (C), 56 h after transient transfection, did not respond to TDG while the full-length AHR-GFP after transiently transfecting pGFP2-N2-AHR plasmid for 56 h (D) showed similar increase as the endogenous AHR proteins after TDG (40 μM, 8 h) treatment. The images are representative of the replicate data (means ± SD, n = 3, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns represents not significant) in one experiment, which was repeated once with similar results. DMSO treatment group was arbitrarily set as one for data normalization. NT represents no transfection. One-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. (E) M1-GFP mutant showed smaller increase (by 20%) when compared to endogenous AHR but similar increase as AHR-GFP (data from 5D) after TDG (40 μM, 8 h) treatment. (F) For M2-GFP mutant, its increase was minimal after treatment of 40 μM TDG for 8 h. The increased level was noticeably less when compared to AHR-GFP (data from 5D). (G) M3-GFP mutant showed 27% less increase when compared to the endogenous AHR and 13% less to AHR-GFP (data from 5D) after TDG (40 μM, 8 h) treatment. The images in (E-G) are representative of the replicate data (means ± SD, n = 6, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns represents not significant). DMSO treatment groups were arbitrarily set as one for data normalization. Two experiments of n = 3 each were performed so that error bars are present on the DMSO groups. The dotted lines separate the AHR-GFP transfection data that are the same data set as in 5D for easy reference. One-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. For B to G, each Western lane contained 30 μg of whole-cell lysate. Data were normalized by total protein stain. NΔ515-GFP (5C) was detected by the GFP antibody since this construct cannot be detected by SA210 AHR antibody while all other GFP fusions of AHR and derivatives were detected by SA210 AHR antibody.
Figure 5.
Site-directed mutagenesis reveals three putative GSK3β phosphorylation sites of AHR. (A) A map showing GFP fusion expressing plasmids of full length human AHR and two truncated derivatives (pGFP2-N2-AHR, pGFP2-C2-CΔ553, and pGFP2-N2-NΔ515) and three AHR mutant GFP plasmids (S436A-S440A-S444A, M1; S689A-S693A-T697A, M2; S723A-S727A-T731A, M3). The names of the plasmid expressing the corresponding protein are listed on the left. Both GFP fusions of CΔ553 (B) and NΔ515 (C), 56 h after transient transfection, did not respond to TDG while the full-length AHR-GFP after transiently transfecting pGFP2-N2-AHR plasmid for 56 h (D) showed similar increase as the endogenous AHR proteins after TDG (40 μM, 8 h) treatment. The images are representative of the replicate data (means ± SD, n = 3, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns represents not significant) in one experiment, which was repeated once with similar results. DMSO treatment group was arbitrarily set as one for data normalization. NT represents no transfection. One-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. (E) M1-GFP mutant showed smaller increase (by 20%) when compared to endogenous AHR but similar increase as AHR-GFP (data from 5D) after TDG (40 μM, 8 h) treatment. (F) For M2-GFP mutant, its increase was minimal after treatment of 40 μM TDG for 8 h. The increased level was noticeably less when compared to AHR-GFP (data from 5D). (G) M3-GFP mutant showed 27% less increase when compared to the endogenous AHR and 13% less to AHR-GFP (data from 5D) after TDG (40 μM, 8 h) treatment. The images in (E-G) are representative of the replicate data (means ± SD, n = 6, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns represents not significant). DMSO treatment groups were arbitrarily set as one for data normalization. Two experiments of n = 3 each were performed so that error bars are present on the DMSO groups. The dotted lines separate the AHR-GFP transfection data that are the same data set as in 5D for easy reference. One-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. For B to G, each Western lane contained 30 μg of whole-cell lysate. Data were normalized by total protein stain. NΔ515-GFP (5C) was detected by the GFP antibody since this construct cannot be detected by SA210 AHR antibody while all other GFP fusions of AHR and derivatives were detected by SA210 AHR antibody.


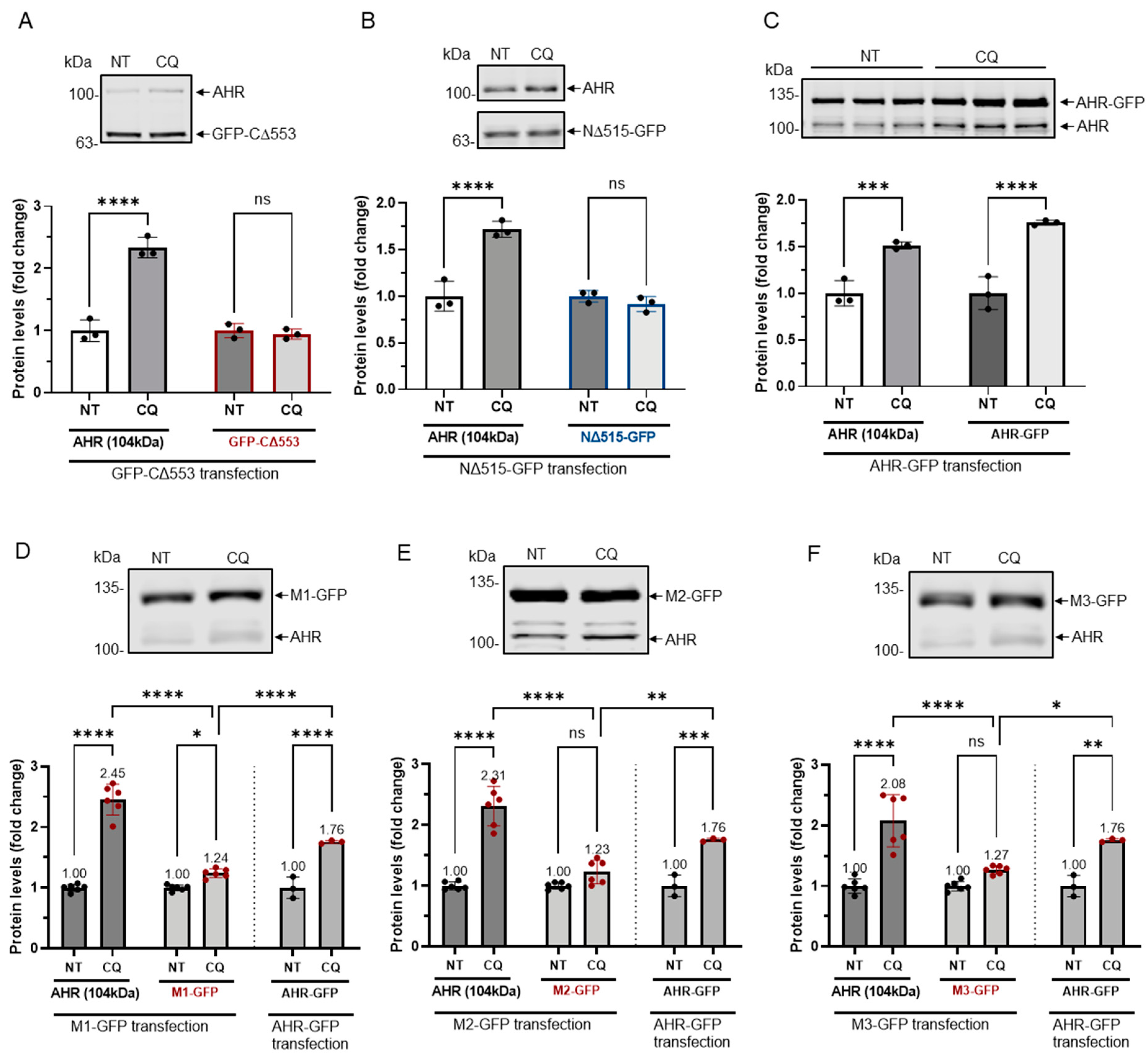
Figure 6.
GFP fusions of two truncated AHR and three mutants were less sensitive to lysosomal degradation. HeLa cells that were transiently transfected (for 54 h) with the plasmid expressing GFP-CΔ553 (A), NΔ515-GFP (B) or full length AHR-GFP (C) were treated with either no treatment (NT) or 40 μM CQ for 6 h. Only AHR-GFP showed similar increase as the endogenous AHR upon TDG treatment. The images are representative of the replicate data (means ± SD, n = 3, *** p < 0.001, **** p < 0.0001, ns represents not significant) in one experiment, and was repeated once with similar results. No treatment group (NT) was arbitrarily set as one for data normalization. One-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. HeLa cells that were transiently transfected (for 54 h) with the plasmid expressing M1-GFP (D), M2-GFP (E) or M3-GFP (F) showed a significant lower increase of 1.2- to 1.3-fold when compared to 2.0- to 2.5-fold increase of endogenous AHR upon treatment of 40 μM CQ for 6 h. These increases were significantly less when compared to AHR-GFP (data from 6C). The images in (D-F) are representative of the replicate data (means ± SD, n = 6, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). No treatment groups (NT) were arbitrarily set as one for data normalization. Two experiments of n = 3 each were performed so that error bars are present on the NT groups. The dotted lines separate the AHR-GFP transfection data that are presented in 6C for easy reference. One-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. For A to F, each Western lane contained 30 μg of whole-cell lysate. Data were normalized by total protein stain. NΔ515-GFP (6B) was detected by the GFP antibody since this construct could not be detected by SA210 AHR antibody while all other GFP fusions of AHR and derivatives were detected by SA210 AHR antibody.
Figure 6.
GFP fusions of two truncated AHR and three mutants were less sensitive to lysosomal degradation. HeLa cells that were transiently transfected (for 54 h) with the plasmid expressing GFP-CΔ553 (A), NΔ515-GFP (B) or full length AHR-GFP (C) were treated with either no treatment (NT) or 40 μM CQ for 6 h. Only AHR-GFP showed similar increase as the endogenous AHR upon TDG treatment. The images are representative of the replicate data (means ± SD, n = 3, *** p < 0.001, **** p < 0.0001, ns represents not significant) in one experiment, and was repeated once with similar results. No treatment group (NT) was arbitrarily set as one for data normalization. One-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. HeLa cells that were transiently transfected (for 54 h) with the plasmid expressing M1-GFP (D), M2-GFP (E) or M3-GFP (F) showed a significant lower increase of 1.2- to 1.3-fold when compared to 2.0- to 2.5-fold increase of endogenous AHR upon treatment of 40 μM CQ for 6 h. These increases were significantly less when compared to AHR-GFP (data from 6C). The images in (D-F) are representative of the replicate data (means ± SD, n = 6, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). No treatment groups (NT) were arbitrarily set as one for data normalization. Two experiments of n = 3 each were performed so that error bars are present on the NT groups. The dotted lines separate the AHR-GFP transfection data that are presented in 6C for easy reference. One-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. For A to F, each Western lane contained 30 μg of whole-cell lysate. Data were normalized by total protein stain. NΔ515-GFP (6B) was detected by the GFP antibody since this construct could not be detected by SA210 AHR antibody while all other GFP fusions of AHR and derivatives were detected by SA210 AHR antibody.

Figure 7.
Down-regulation of p23 in HeLa cells causes AHR not responsive to GSK3β regulation. (A) AHR protein levels were not altered by 8-h treatment of 10 μM or 20μM TDG in p23 stable knockdown (p23KD) HeLa cells. Treatment of 40 μM TDG increased AHR protein levels to 1.3-fold. The images are representative of the replicate data (means ± SD, n = 3, ** p < 0.01, ns represents not significant). DMSO group (treated for 8 h) was arbitrarily set as one (with no error bar) for data normalization. One-way ANOVA with Dunnett multiple comparisons test was performed to determine statistical significance. (B) Treatment of 5 mM LiCl for 6 h did not alter AHR protein levels in p23KD HeLa cells. The images are representative of the replicate data (means ± SD, n = 3, ns represents not significant). No LiCl (no treatment) group was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (C) β-catenin protein levels decreased while AHR protein levels showed no significant difference after transient expression of HA-GSK3β (72 h after transient transfection) in p23KD HeLa cells. The images are representative of the replicate data (means ± SD, n = 3, *p < 0.05, ns represents not significant). No plasmid group (NP) represents cells undergoing the same transient transfection protocol without the HA-GSK3β expressing plasmid and was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (D) β-catenin protein levels increased in p23KD HeLa cells after treatment of 5 mM LiCl for 6 h. The images are representative of the replicate data (means ± SD, n = 3 in one experiment, which was repeated once with similar results, *** p < 0.001). No LiCl (no treatment) group was arbitrarily set as one for data normalization. Unpaired t-test was used to determine the statistical significance. (E) p23KD HeLa cells were transiently transfected with the plasmid expressing GFP or GFP-p23. Transient expression of GFP-p23 caused AHR protein levels to increase when cells were treated 20 μM TDG for 8 h. The images are representative of the replicate data (means ± SD, n = 3 in one experiment, which was repeated once with similar results, * p < 0.05, *** p < 0.001, ns represents not significant). DMSO treatment group of GFP-transfected cells was arbitrarily set as one for data normalization. Two-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. For A to E, each Western lane contained 30 μg of whole-cell lysate. Data in A were normalized by β-actin (as shown) whereas the rest of the data were normalized by total protein stain.
Figure 7.
Down-regulation of p23 in HeLa cells causes AHR not responsive to GSK3β regulation. (A) AHR protein levels were not altered by 8-h treatment of 10 μM or 20μM TDG in p23 stable knockdown (p23KD) HeLa cells. Treatment of 40 μM TDG increased AHR protein levels to 1.3-fold. The images are representative of the replicate data (means ± SD, n = 3, ** p < 0.01, ns represents not significant). DMSO group (treated for 8 h) was arbitrarily set as one (with no error bar) for data normalization. One-way ANOVA with Dunnett multiple comparisons test was performed to determine statistical significance. (B) Treatment of 5 mM LiCl for 6 h did not alter AHR protein levels in p23KD HeLa cells. The images are representative of the replicate data (means ± SD, n = 3, ns represents not significant). No LiCl (no treatment) group was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (C) β-catenin protein levels decreased while AHR protein levels showed no significant difference after transient expression of HA-GSK3β (72 h after transient transfection) in p23KD HeLa cells. The images are representative of the replicate data (means ± SD, n = 3, *p < 0.05, ns represents not significant). No plasmid group (NP) represents cells undergoing the same transient transfection protocol without the HA-GSK3β expressing plasmid and was arbitrarily set as one (with no error bar) for data normalization. Unpaired t-test with Welch’s correction was used to determine the statistical significance. (D) β-catenin protein levels increased in p23KD HeLa cells after treatment of 5 mM LiCl for 6 h. The images are representative of the replicate data (means ± SD, n = 3 in one experiment, which was repeated once with similar results, *** p < 0.001). No LiCl (no treatment) group was arbitrarily set as one for data normalization. Unpaired t-test was used to determine the statistical significance. (E) p23KD HeLa cells were transiently transfected with the plasmid expressing GFP or GFP-p23. Transient expression of GFP-p23 caused AHR protein levels to increase when cells were treated 20 μM TDG for 8 h. The images are representative of the replicate data (means ± SD, n = 3 in one experiment, which was repeated once with similar results, * p < 0.05, *** p < 0.001, ns represents not significant). DMSO treatment group of GFP-transfected cells was arbitrarily set as one for data normalization. Two-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance. For A to E, each Western lane contained 30 μg of whole-cell lysate. Data in A were normalized by β-actin (as shown) whereas the rest of the data were normalized by total protein stain.


Figure 8.
Reduction of GSK3β activity suppresses the ligand-dependent activation of AHR target gene transcription. For A and B, HeLa cells were treated with 40 μM TDG for 9 h. At 5-h post-TDG treatment, cells were treated with either (A) 10 μM βNF, 10 μM CH223191 or both or (B) 1 μM 3MC, 10 μM CH223191 or both. TDG were able to partially block the increase of the cyp1a1 message levels triggered by βNF and 3MC. Co-treatment of TDG with CH223191 further decreased the cyp1a1 message levels. For A and B, the graphs represent replicate data (means ± SD, n = 3 except that n = 4 for DMSO, TDG + 3MC and TDG + 3MC + CH223191 groups in 8B, * p < 0.05, *** p < 0.001, **** p < 0.0001) and DMSO group was arbitrarily set as one (with no error bar) for data normalization. HeLa cells were transiently transfected for 70 h with the plasmid carrying scramble shRNA or GSK3β specific shRNA. Cells were then treated with (C) DMSO or 10 μM βNF, (D) DMSO or 5 μM BaP, (E) DMSO or 1 μM 3MC for 4 h. Cells were harvested at 74-h post transfection/4-h post ligand treatment. The increase level of the cyp1a1 message after ligands treatment were significantly lower in GSK3β knockdown groups. For C to E, graphs represent replicate data (means ± SD, n = 3, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001) with DMSO group arbitrarily set as one (with no error bar) for data normalization. (F) 7.5-h treatment of TDG (40 μM, treated at 70-77.5 h post-transfection) could not suppress the 3MC (1 μM, 3.5 h, treated at 74-77.5 h post-transfection) induced cyp1a1 messages in GSK3β shRNA transfected HeLa cells as observed in scramble shRNA transfected HeLa cells. The graph represents the replicate data (means ± SD, n = 3, * p < 0.05, *** p < 0.001, ns represents not significant) with 3MC groups minus TDG were arbitrarily set as one (with no error bar) for data normalization. Hep3B (G) and MCF-7 (H) cells were treated 40 μM TDG for 9 h. At 5-h post-TDG treatment, cells were treated with 1 μM 3MC for 4 h. The induced cyp1a1 message levels were suppressed by TDG in both cell lines. The graphs in G and H represent replicate data (means ± SD, n = 3, ** p < 0.01, **** p < 0.0001, ns represents not significant) with DMSO group arbitrarily set as one (with no error bar) for data normalization. For A to H, one-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance.
Figure 8.
Reduction of GSK3β activity suppresses the ligand-dependent activation of AHR target gene transcription. For A and B, HeLa cells were treated with 40 μM TDG for 9 h. At 5-h post-TDG treatment, cells were treated with either (A) 10 μM βNF, 10 μM CH223191 or both or (B) 1 μM 3MC, 10 μM CH223191 or both. TDG were able to partially block the increase of the cyp1a1 message levels triggered by βNF and 3MC. Co-treatment of TDG with CH223191 further decreased the cyp1a1 message levels. For A and B, the graphs represent replicate data (means ± SD, n = 3 except that n = 4 for DMSO, TDG + 3MC and TDG + 3MC + CH223191 groups in 8B, * p < 0.05, *** p < 0.001, **** p < 0.0001) and DMSO group was arbitrarily set as one (with no error bar) for data normalization. HeLa cells were transiently transfected for 70 h with the plasmid carrying scramble shRNA or GSK3β specific shRNA. Cells were then treated with (C) DMSO or 10 μM βNF, (D) DMSO or 5 μM BaP, (E) DMSO or 1 μM 3MC for 4 h. Cells were harvested at 74-h post transfection/4-h post ligand treatment. The increase level of the cyp1a1 message after ligands treatment were significantly lower in GSK3β knockdown groups. For C to E, graphs represent replicate data (means ± SD, n = 3, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001) with DMSO group arbitrarily set as one (with no error bar) for data normalization. (F) 7.5-h treatment of TDG (40 μM, treated at 70-77.5 h post-transfection) could not suppress the 3MC (1 μM, 3.5 h, treated at 74-77.5 h post-transfection) induced cyp1a1 messages in GSK3β shRNA transfected HeLa cells as observed in scramble shRNA transfected HeLa cells. The graph represents the replicate data (means ± SD, n = 3, * p < 0.05, *** p < 0.001, ns represents not significant) with 3MC groups minus TDG were arbitrarily set as one (with no error bar) for data normalization. Hep3B (G) and MCF-7 (H) cells were treated 40 μM TDG for 9 h. At 5-h post-TDG treatment, cells were treated with 1 μM 3MC for 4 h. The induced cyp1a1 message levels were suppressed by TDG in both cell lines. The graphs in G and H represent replicate data (means ± SD, n = 3, ** p < 0.01, **** p < 0.0001, ns represents not significant) with DMSO group arbitrarily set as one (with no error bar) for data normalization. For A to H, one-way ANOVA with Sidak multiple comparisons test was performed to determine statistical significance.


Figure 9.
A proposed model of GSK3β role on AHR function and degradation. AHR is phosphorylated by GSK3β in a p23-dependent manner in HeLa cells. This phosphorylation is required for optimal activation of the ligand-dependent AHR target gene transcription. After phosphorylation, AHR is K63-ubiquitinated and is targeted for the LC3-mediated selective autophagy. When the p23 content is compromised in HeLa cells, AHR is more prone to degradation via autophagy, bypassing the GSK3β phosphorylation of AHR.
Figure 9.
A proposed model of GSK3β role on AHR function and degradation. AHR is phosphorylated by GSK3β in a p23-dependent manner in HeLa cells. This phosphorylation is required for optimal activation of the ligand-dependent AHR target gene transcription. After phosphorylation, AHR is K63-ubiquitinated and is targeted for the LC3-mediated selective autophagy. When the p23 content is compromised in HeLa cells, AHR is more prone to degradation via autophagy, bypassing the GSK3β phosphorylation of AHR.
Table 1.
M1, M2, and M3 using the pGFP2-N2-AHR plasmid as the template for QuikChange mutagenesis.
Table 1.
M1, M2, and M3 using the pGFP2-N2-AHR plasmid as the template for QuikChange mutagenesis.
| Target Sequence | Primer Sequence |
|---|
| S436A-S440A-S444A | Forward(OL904): 5′-a aat ggc act gct gga aaa gac gct gct acc aca gcc act cta agc aag g -3′ |
| | Reverse(OL905): 5′-c ctt gct tag agt ggc tgt ggt agc gtc ttt tcc agc agt gcc att t-3′ |
| S689A-S693A-T697A | Forward(OL906): 5′-gag ttc ccc tac aaa gct gaa atg gat gct atg cct tat gca cag aac ttt att tcc-3′ |
| | Reverse(OL907): 5′-gga aat aaa gtt ctg tgc ata agg cat agc atc cat ttc agc ttt gta ggg gaa ctc-3′ |
| S723A-S727A-T731A | Forward(OL908): 5′-c tac cct atg ggg gct ttt gaa cca gcc cca tac ccc gct act tct agt t-3′ |
| | Reverse(OL909): 5′-a act aga agt agc ggg gta tgg ggc tgg ttc aaa agc ccc cat agg gta g-3′ |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}