
New Therapies to Correct the Cystic Fibrosis Basic Defect



Abstract
:1. Introduction
Classification of CFTR Defects: An Essential Step in CF Drug Discovery
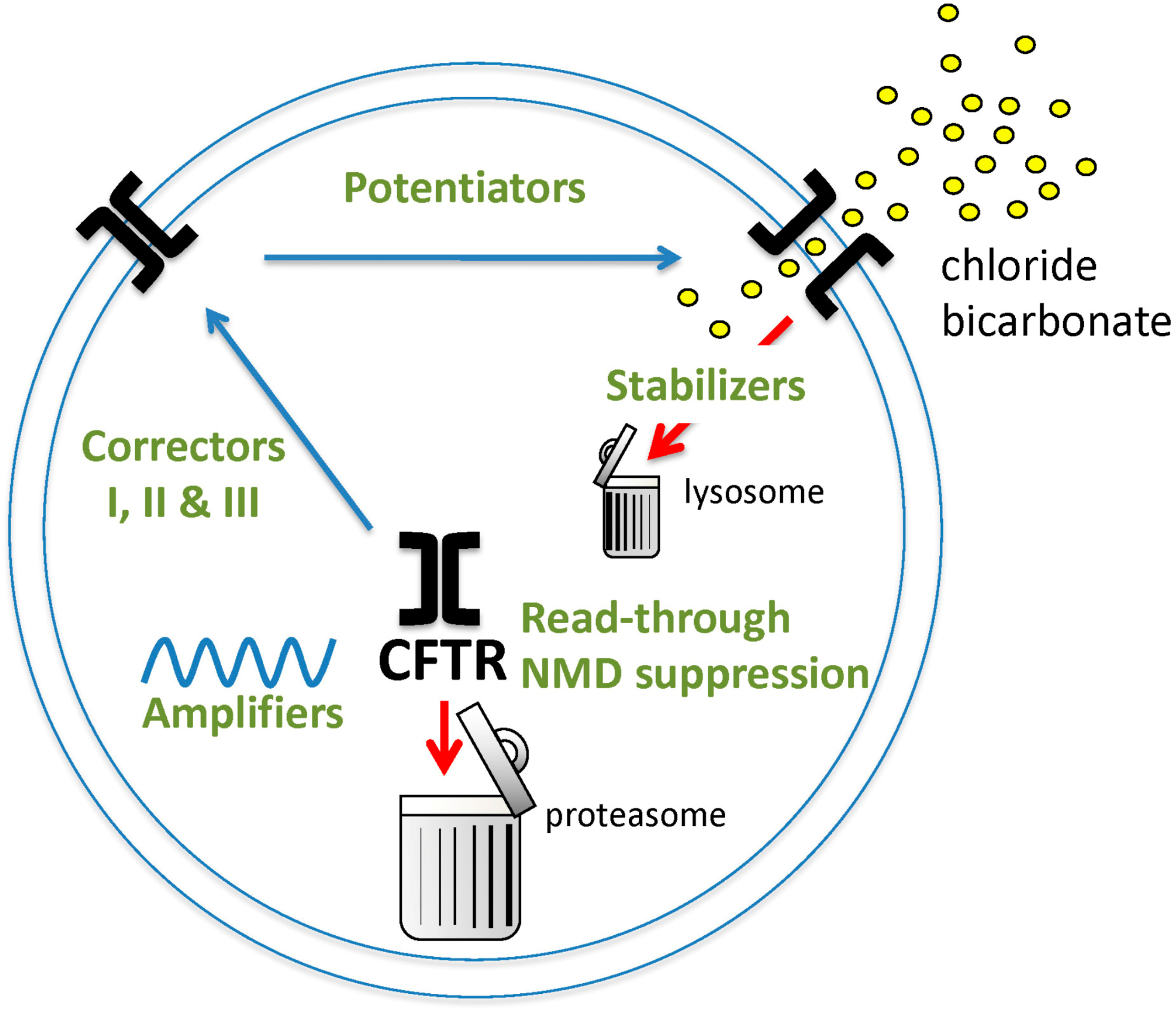
2. CFTR Modulators
2.1. First-Generation CFTR Modulators
2.1.1. Ivacaftor
2.1.2. Lumacaftor-Ivacaftor
2.1.3. Tezacaftor-Ivacaftor
2.2. Next-Generation CFTR Modulator Therapy
2.2.1. Elexacaftor–Tezacaftor–Ivacaftor
2.2.2. Other Modulators in the Pipeline
2.2.3. Personalized Medicine
2.3. CFTR Amplifiers
2.4. Pre-Termination Codon Agents
2.5. CFTR Stabilizers
2.6. Genetic Therapies
3. Alternate Ion Channel Modulation
3.1. ENaC Inhibitors
3.2. Calcium-Activated Chloride Secretion and TMEM16A Potentiator
3.3. Amphotericin
3.4. Anionophores
3.5. Antisense Oligonucleotides
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collins, F.S. Realizing the Dream of Molecularly Targeted Therapies for Cystic Fibrosis. N. Engl. J. Med. 2019, 381, 1863–1865. [Google Scholar] [CrossRef]
- Bergeron, C.; Cantin, A.M. Cystic Fibrosis: Pathophysiology of Lung Disease. Semin. Respir. Crit. Care Med. 2019, 40, 715–726. [Google Scholar] [CrossRef]
- Bardin, E.; Pastor, A.; Semeraro, M.; Golec, A.; Hayes, K.; Chevalier, B.; Berhal, F.; Prestat, G.; Hinzpeter, A.; Gravier-Pelletier, C.; et al. Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 2021, 213, 113195. [Google Scholar] [CrossRef]
- Somayaji, R.; Nichols, D.P.; Bell, S.C. Cystic fibrosis—Ten promising therapeutic approaches in the current era of care. Expert Opin. Investig. Drugs 2020, 29, 1107–1124. [Google Scholar] [CrossRef] [PubMed]
- Cftr2 Database. Available online: https://cftr2.org/ (accessed on 20 May 2021).
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Wilschanski, M.; Zielenski, J.; Markiewicz, D.; Tsui, L.-C.; Corey, M.; Levison, H.; Durie, P.R. Correlation of sweat chloride concentration with classes of the cystic fibrosis transmembrane conductance regulator gene mutations. J. Pediatr. 1995, 127, 705–710. [Google Scholar] [CrossRef]
- Stanke, F.; Tümmler, B. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e36. [Google Scholar] [CrossRef] [Green Version]
- Sugarman, A.E.; Rohlfs, E.M.; Silverman, L.M.; A Allitto, B. CFTR mutation distribution among U.S. Hispanic and African American individuals: Evaluation in cystic fibrosis patient and carrier screening populations. Genet. Med. 2004, 6, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.; Cuppens, H.; Rainisio, M.; Madessani, U.; Harms, H.; Hodson, M.; Mastella, G.; Navarro, J.; Strandvik, B.; McKenzie, S. Ercf Investigators of the. European Epidemiologic Registry of Cystic Fibrosis (Ercf): Comparison of Major Disease Manifestations between Patients with Different Classes of Mutations. Pediatr. Pulmonol. 2001, 31, 1–12. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linde, L.; Boelz, S.; Nissim-Rafinia, M.; Oren, Y.S.; Wilschanski, M.; Yaacov, Y.; Virgilis, D.; Neu-Yilik, G.; Kulozik, A.E.; Kerem, E.; et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J. Clin. Investig. 2007, 117, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A.; Awatade, N.T.; Felicio, V.M.; Silva, I.A.; Calucho, M.; Pereira, L.; Azevedo, P.; Cavaco, J.; Barreto, C.; Bertuzzo, C.; et al. The effect of premature termination codon mutations on CFTR mRNA abundance in human nasal epithelium and intestinal organoids: A basis for read-through therapies in cystic fibrosis. Hum. Mutat. 2018, 40, 326–334. [Google Scholar] [CrossRef]
- Estabrooks, S.; Brodsky, J.L. Regulation of Cftr Biogenesis by the Proteostatic Network and Pharmacological Modulators. Int. J. Mol. Sci. 2020, 21, 452. [Google Scholar] [CrossRef] [Green Version]
- Bobadilla, J.L.; Macek, M.; Fine, J.P.; Farrell, P.M. Cystic Fibrosis: A Worldwide Analysis of Cftr Mutations—Correlation with Incidence Data and Application to Screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Welsh, M.J. Regulation by Atp and Adp of Cftr Chloride Channels That Contain Mutant Nucleotide-Binding Domains. Science 1992, 257, 1701–1704. [Google Scholar] [CrossRef] [PubMed]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of Vx-770 in Persons with Cystic Fibrosis and the G551d-Cftr Mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, D.N.; Rich, D.P.; Ostedgaard, L.S.; Gregory, R.J.; Smith, A.E.; Welsh, M.J. Mutations in Cftr Associated with Mild-Disease-Form Cl- Channels with Altered Pore Properties. Nature 1993, 362, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B.; Flume, P.A.; Elborn, J.S.; Cooke, J.; Rowe, S.M.; McColley, S.A.; Rubenstein, R.C.; Higgins, M.; VX11-770-110 Study Group. Efficacy and Safety of Ivacaftor in Patients with Cystic Fibrosis Who Have an Arg117his-Cftr Mutation: A Double-Blind, Randomised Controlled Trial. Lancet Respir. Med. 2015, 3, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.-S.; Trapnell, B.C.; Curristin, S.; Cutting, G.R.; Crystal, R.G. Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat. Genet. 1993, 3, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Downey, D.G.; Beattie, D.; McCaughan, J.; Reid, A.; Stein, N.; Elborn, J.S. Lung function and disease severity in cystic fibrosis patients heterozygous for p.Arg117His. ERJ Open Res. 2017, 3, 00056–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haardt, M.; Benharouga, M.; Lechardeur, D.; Kartner, N.; Lukacs, G.L. C-terminal Truncations Destabilize the Cystic Fibrosis Transmembrane Conductance Regulator without Impairing Its Biogenesis. J. Biol. Chem. 1999, 274, 21873–21877. [Google Scholar] [CrossRef] [Green Version]
- Silvis, M.R.; Picciano, J.A.; Bertrand, C.; Weixel, K.; Bridges, R.J.; Bradbury, N.A. A Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Generates a Novel Internalization Sequence and Enhances Endocytic Rates. J. Biol. Chem. 2003, 278, 11554–11560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordoñez, C.L.; Geller, D.E. Ivacaftor in Subjects with Cystic Fibrosis Who Are Homozygous for the F508del-CFTR Mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A Cftr Potentiator in Patients with Cystic Fibrosis and the G551d Mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and Safety of Ivacaftor in Patients Aged 6 to 11 Years with Cystic Fibrosis with a G551d Mutation. Am. J. Respir Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical Mechanism of the Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor in G551D-mediated Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cftr Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Heltshe, S.L.; Mayer-Hamblett, N.; Burns, J.L.; Khan, U.; Baines, A.; Ramsey, B.W.; Rowe, S.M. Pseudomonas aeruginosa in Cystic Fibrosis Patients With G551D-CFTR Treated With Ivacaftor. Clin. Infect. Dis. 2015, 60, 703–712. [Google Scholar] [CrossRef]
- Frost, F.J.; Nazareth, D.S.; Charman, S.C.; Winstanley, C.; Walshaw, M.J. Ivacaftor Is Associated with Reduced Lung Infection by Key Cystic Fibrosis Pathogens. A Cohort Study Using National Registry Data. Ann. Am. Thorac. Soc. 2019, 16, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Guimbellot, J.; Baines, A.; Paynter, A.; Heltshe, S.; VanDalfsen, J.; Jain, M.; Rowe, S.; Sagel, S. Long term clinical effectiveness of ivacaftor in people with the G551D CFTR mutation. J. Cyst. Fibros. 2021, 20, 213–219. [Google Scholar] [CrossRef]
- Einarsson, G.G.; Ronan, N.J.; Mooney, D.; McGettigan, C.; Mullane, D.; NiChroinin, M.; Shanahan, F.; Murphy, D.M.; McCarthy, M.; McCarthy, Y.; et al. Extended-culture and culture-independent molecular analysis of the airway microbiota in cystic fibrosis following CFTR modulation with ivacaftor. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Higgins, M.; Konstan, M.W.; Sawicki, G.S.; Elbert, A.; et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J. Cyst. Fibros. 2020, 19, 68–79. [Google Scholar] [CrossRef]
- Kirwan, L.; Fletcher, G.; Harrington, M.; Jeleniewska, P.; Zhou, S.; Casserly, B.; Gallagher, C.G.; Greally, P.; Gunaratnam, C.; Herzig, M.; et al. Longitudinal Trends in Real-World Outcomes after Initiation of Ivacaftor. A Cohort Study from the Cystic Fibrosis Registry of Ireland. Ann. Am. Thorac. Soc. 2019, 16, 209–216. [Google Scholar] [CrossRef]
- Hebestreit, H.; Sauer-Heilborn, A.; Fischer, R.; Käding, M.; Mainz, J.G. Effects of ivacaftor on severely ill patients with cystic fibrosis carrying a G551D mutation. J. Cyst. Fibros. 2013, 12, 599–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, P.J.; Plant, B.J.; Nair, A.; Bicknell, S.; Simmonds, N.J.; Bell, N.J.; Shafi, N.T.; Daniels, T.; Shelmerdine, S.; Felton, I.; et al. Effects of Ivacaftor in Patients with Cystic Fibrosis Who Carry the G551d Mutation and Have Severe Lung Disease. Chest 2014, 146, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.; Niknian, M.; Gilmartin, G.; Pilewski, J.M.; VX11-770-901 Investigators. Effect of Ivacaftor in Patients with Advanced Cystic Fibrosis and a G551d-Cftr Mutation: Safety and Efficacy in an Expanded Access Program in the United States. J. Cyst. Fibros. 2016, 15, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Horsley, A.R.; Gustafsson, P.M.; A Macleod, K.; Saunders, C.; Greening, A.P.; Porteous, D.; Davies, J.; Cunningham, S.; Alton, E.W.; Innes, J. A Lung clearance index is a sensitive, repeatable and practical measure of airways disease in adults with cystic fibrosis. Thorax 2007, 63, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, P.M.; A De Jong, P.; Tiddens, H.A.W.M.; Lindblad, A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax 2007, 63, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Klingel, M.; Black, P.; Powers, M.R.; Grasemann, H.; Solomon, M.; Sagel, S.D.; Donaldson, S.H.; Rowe, S.M.; Rosenfeld, M. Changes in Lung Clearance Index in Preschool-aged Patients with Cystic Fibrosis Treated with Ivacaftor (GOAL): A Clinical Trial. Am. J. Respir. Crit. Care Med. 2018, 198, 526–528. [Google Scholar] [CrossRef]
- Hoare, S.; McEvoy, S.; McCarthy, C.J.; Kilcoyne, A.; Brady, D.; Gibney, B.; Gallagher, C.G.; McKone, E.F.; Dodd, J.D. Ivacaftor Imaging Response in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 189, 484. [Google Scholar] [CrossRef]
- Sheikh, S.I.; Long, F.R.; McCoy, K.S.; Johnson, T.; Ryan-Wenger, N.A.; Hayes, D. Computed tomography correlates with improvement with ivacaftor in cystic fibrosis patients with G551D mutation. J. Cyst. Fibros. 2015, 14, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassagnon, G.; Hubert, D.; Fajac, I.; Burgel, P.-R.; Revel, M.-P. Long-term computed tomographic changes in cystic fibrosis patients treated with ivacaftor. Eur. Respir. J. 2016, 48, 249–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, D.; McCoy, K.S.; I Sheikh, S. Improvement of sinus disease in cystic fibrosis with ivacaftor therapy. Am. J. Respir. Crit. Care Med. 2014, 190, 468. [Google Scholar] [CrossRef]
- Davies, J.C.; Cunningham, S.; Harris, W.T.; Lapey, A.; E Regelmann, W.; Sawicki, G.S.; Southern, K.W.; Robertson, S.; Green, Y.; Cooke, J.; et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016, 4, 107–115. [Google Scholar] [CrossRef]
- Smith, H.; Rayment, J.H. Sustained recovery of exocrine pancreatic function in a teenager with cystic fibrosis treated with ivacaftor. Pediatr. Pulmonol. 2020, 55, 2493–2494. [Google Scholar] [CrossRef]
- Megalaa, R.; Gopalareddy, V.; Champion, E.; Goralski, J.L. Time for a gut check: Pancreatic sufficiency resulting from CFTR modulator use. Pediatr. Pulmonol. 2019, 54, E16–E18. [Google Scholar] [CrossRef]
- Nichols, A.; Davies, J.; Jones, D.; Carr, S. Restoration of exocrine pancreatic function in older children with cystic fibrosis on ivacaftor. Paediatr. Respir. Rev. 2020, 35, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Munce, D.; Lim, M.; Akong, K. Persistent recovery of pancreatic function in patients with cystic fibrosis after ivacaftor. Pediatr. Pulmonol. 2020, 55, 3381–3383. [Google Scholar] [CrossRef] [PubMed]
- Kounis, I.; Lévy, P.; Rebours, V. Ivacaftor CFTR Potentiator Therapy is Efficient for Pancreatic Manifestations in Cystic Fibrosis. Am. J. Gastroenterol. 2018, 113, 1058–1059. [Google Scholar] [CrossRef] [PubMed]
- Franck-Thompson, E.; Watson, D.; Benoit, C.M.; Landvik, S.; McNamara, J. The Association of Pediatric Cystic Fibrosis-Related Diabetes Screening on Clinical Outcomes by Center: A Cf Patient Registry Study. J. Cyst. Fibros 2020, 19, 316–320. [Google Scholar] [CrossRef]
- Prinz, N.; Wosniok, J.; Staab, D.; Ballmann, M.; Dopfer, C.; Regenfuss, N.; Rosenecker, J.; Schramm, D.; Holl, R.W.; Nahrlich, L. Glucose Tolerance in Patients with Cystic Fibrosis—Results from the German Cystic Fibrosis Registry. Klin. Padiatr. 2020, 232, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.D.; Laguna, T.; Leschyshyn, J.; E Regelmann, W.; Dunitz, J.M.; Billings, J.; Moran, A. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: A small pilot study. Pediatr. Diabetes 2013, 14, 417–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, D.; McCoy, K.S.; Sheikh, S.I. Resolution of Cystic Fibrosis–related Diabetes with Ivacaftor Therapy. Am. J. Respir. Crit. Care Med. 2014, 190, 590–591. [Google Scholar] [CrossRef] [PubMed]
- Gaines, H.; Jones, K.R.; Lim, J.; Medhi, N.F.; Chen, S.; Scofield, R.H. Effect of Cftr Modulator Therapy on Cystic Fibrosis-Related Diabetes. J. Diabetes Complicat. 2021, 35, 107845. [Google Scholar] [CrossRef]
- Salvatore, D.; Terlizzi, V.; Francalanci, M.; Taccetti, G.; Messore, B.; Biglia, C.; Pisi, G.; Calderazzo, M.A.; Caloiero, M.; Pizzamiglio, G.; et al. Ivacaftor improves lung disease in patients with advanced CF carrying CFTR mutations that confer residual function. Respir. Med. 2020, 171, 106073. [Google Scholar] [CrossRef]
- Rosenfeld, M.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Chilvers, M.; Higgins, M.; Tian, S.; et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2–5 years (KLIMB). J. Cyst. Fibros. 2019, 18, 838–843. [Google Scholar] [CrossRef] [Green Version]
- Trimble, A.T.; Donaldson, S.H. Ivacaftor withdrawal syndrome in cystic fibrosis patients with the G551D mutation. J. Cyst. Fibros. 2018, 17, e13–e16. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Da Fonte, D.F.; Avramescu, R.G.; Premchandar, A.; Bagdany, M.; Xu, H.; Bensinger, D.; Stubba, D.; Schmidt, B.; Matouk, E.; et al. Mutation-specific dual potentiators maximize rescue of CFTR gating mutants. J. Cyst. Fibros. 2020, 19, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Durmowicz, A.G.; Lim, R.; Rogers, H.; Rosebraugh, C.J.; Chowdhury, B.A. The U.S. Food and Drug Administration’s Experience with Ivacaftor in Cystic Fibrosis. Establishing Efficacy Using In Vitro Data in Lieu of a Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. [Google Scholar] [CrossRef]
- Rowe, S.M.; Pyle, L.C.; Jurkevante, A.; Varga, K.; Collawn, J.; Sloane, P.A.; Woodworth, B.; Mazur, M.; Fulton, J.; Fan, Y.; et al. Deltaf508 Cftr Processing Correction and Activity in Polarized Airway and Non-Airway Cell Monolayers. Pulm. Pharmacol. Ther. 2010, 23, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeson, S.L.; Morgan, A.J.; Liu, J.F.; Aslanian, A.M.; Nguyen, S.; Bridson, G.W.; Brummel, C.L.; Wu, L.; Tung, R.D.; Pilja, L.; et al. Altering Metabolic Profiles of Drugs by Precision Deuteration 2: Discovery of a Deuterated Analog of Ivacaftor with Differentiated Pharmacokinetics for Clinical Development. J. Pharmacol. Exp. Ther. 2017, 362, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.J.; Loo, M.A.; Pind, S.; Williams, D.B.; Goldberg, A.L.; Riordan, J.R. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995, 83, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Strub, M.D.; McCray, P.B., Jr. Transcriptomic and Proteostasis Networks of Cftr and the Development of Small Molecule Modulators for the Treatment of Cystic Fibrosis Lung Disease. Genes 2020, 11, 546. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shelat, A.A.; Guy, R.K.; Gopinath, V.S.; Ma, T.; Du, K.; Lukacs, G.L.; Taddei, A.; Folli, C.; Pedemonte, N.; et al. Nanomolar Affinity Small Molecule Correctors of Defective Delta F508-Cftr Chloride Channel Gating. J. Biol. Chem. 2003, 278, 35079–36085. [Google Scholar] [CrossRef] [Green Version]
- Clancy, J.P.; Rowe, S.M.; Accurso, F.J.; Aitken, M.L.; Amin, R.S.; Ashlock, M.A.; Ballmann, M.; Boyle, M.P.; Bronsveld, I.; Campbell, P.W.; et al. Results of a Phase Iia Study of Vx-809, an Investigational Cftr Corrector Compound, in Subjects with Cystic Fibrosis Homozygous for the F508del-Cftr Mutation. Thorax 2012, 67, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Boyle, M.P.; Bell, S.C.; Konstan, M.W.; McColley, S.A.; Rowe, S.M.; Rietschel, E.; Huang, X.; Waltz, D.; Patel, N.R.; Rodman, D.; et al. A Cftr Corrector (Lumacaftor) and a Cftr Potentiator (Ivacaftor) for Treatment of Patients with Cystic Fibrosis Who Have a Phe508del Cftr Mutation: A Phase 2 Randomised Controlled Trial. Lancet Respir. Med. 2014, 2, 527–538. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; McKone, E.F.; Moss, R.B.; Marigowda, G.; Tian, S.; Waltz, D.; Huang, X.; Lubarsky, B.; Rubin, J.; Millar, S.J.; et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): A phase 3, extension study. Lancet Respir. Med. 2017, 5, 107–118. [Google Scholar] [CrossRef]
- Dagenais, R.V.E.; Su, V.C.H.; Quon, B.S. Real-World Safety of CFTR Modulators in the Treatment of Cystic Fibrosis: A Systematic Review. J. Clin. Med. 2020, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Guevara, M.T.; McColley, S. The safety of lumacaftor and ivacaftor for the treatment of cystic fibrosis. Expert Opin. Drug Saf. 2017, 16, 1305–1311. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.S.; Finkbeiner, W.E.; et al. Some Gating Potentiators, Including Vx-770, Diminish Deltaf508-Cftr Functional Expression. Sci. Transl. Med. 2014, 6, 246ra97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.; Lekstrom-Himes, J.A.; Wang, L.T. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef]
- Schneider, E.K. Cytochrome P450 3A4 Induction: Lumacaftor versus Ivacaftor Potentially Resulting in Significantly Reduced Plasma Concentration of Ivacaftor. Drug Metab. Lett. 2018, 12, 71–74. [Google Scholar] [CrossRef]
- Munck, A.; Kerem, E.; Ellemunter, H.; Campbell, D.; Wang, L.T.; Ahluwalia, N.; Owen, C.A.; Wainwright, C. Tezacaftor/Ivacaftor in People with Cystic Fibrosis Heterozygous for Minimal Function Cftr Mutations. J. Cyst. Fibros. 2020, 19, 962–968. [Google Scholar] [CrossRef]
- McKone, E.F.; DiMango, E.A.; Sutharsan, S.; Barto, T.L.; Campbell, D.; Ahluwalia, N.; Higgins, M.; Owen, C.A.; Tullis, E. A phase 3, randomized, double-blind, parallel-group study to evaluate tezacaftor/ivacaftor in people with cystic fibrosis heterozygous for F508del-CFTR and a gating mutation. J. Cyst. Fibros. 2021, 20, 234–242. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; Van Der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, C.; Sutharsan, S.; Epaud, R.; Klingsberg, R.C.; Fischer, R.; Rowe, S.M.; Audhya, P.K.; Ahluwalia, N.; You, X.; Ferro, T.J.; et al. Tezacaftor/ivacaftor in people with cystic fibrosis who stopped lumacaftor/ivacaftor due to respiratory adverse events. J. Cyst. Fibros. 2021, 20, 228–233. [Google Scholar] [CrossRef]
- Walker, S.; Flume, P.; McNamara, J.; Solomon, M.; Chilvers, M.; Chmiel, J.; Harris, R.S.; Haseltine, E.; Stiles, D.; Li, C.; et al. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11 years with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 708–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.C.; Sermet-Gaudelus, I.; Naehrlich, L.; Harris, R.S.; Campbell, D.; Ahluwalia, N.; Short, C.; Haseltine, E.; Panorchan, P.; Saunders, C.; et al. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. J. Cyst. Fibros. 2021, 20, 68–77. [Google Scholar] [PubMed]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M. Simon, A.; et al. Mechanism-Based Corrector Combination Restores Deltaf508-Cftr Folding and Function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D. False dawn for cystic fibrosis disease modifiers? Nat. Rev. Drug Discov. 2014, 13, 713–714. [Google Scholar] [CrossRef]
- Rowe, S.M.; McColley, S.; Rietschel, E.; Li, X.; Bell, S.C.; Konstan, M.W.; Marigowda, G.; Waltz, D.; Boyle, M.P. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann. Am. Thorac. Soc. 2016, 14, 213–219. [Google Scholar]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Safirstein, J.; Grant, J.J.; Clausen, E.; Savant, D.; Dezube, R.; Hong, G. Biliary disease and cholecystectomy after initiation of elexacaftor/ivacaftor/tezacaftor in adults with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 506–510. [Google Scholar] [CrossRef]
- Rotolo, S.M.; Duehlmeyer, S.; Slack, S.M.; Jacobs, H.R.; Heckman, B. Testicular Pain Following Initiation of Elexacaftor/Tezacaftor/Ivacaftor in Males with Cystic Fibrosis. J. Cyst. Fibros. 2020, 19, e39–e41. [Google Scholar] [CrossRef]
- O’Connor, K.E.; Goodwin, D.L.; Nesmith, A.; Garcia, B.; Mingora, C.; Ladores, S.L.; Rowe, S.M.; Krick, S.; Solomon, G.M. Elexacafator/tezacaftor/ivacaftor resolves subfertility in females with CF: A two center case series. J. Cyst. Fibros. 2021, 20, 399–401. [Google Scholar] [CrossRef]
- Song, Y.; Palacios, A.C.; Thiagalingam, A.; Middleton, P.G. Azithromycin and tezacaftor/ivacaftor is associated with first-degree heart block in an adult with cystic fibrosis. J. Cyst. Fibros. 2021, 20, e19–e21. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Taylor-Cousar, J.L.; Davies, J.; Gibson, R.L.; Mall, M.; McKone, E.F.; McNally, P.; Ramsey, B.W.; Rayment, J.H.; Rowe, S.M.; et al. A Phase 3 Open-Label Study of ELX/TEZ/IVA in Children 6 through 11 Years of Age with CF and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- O’Shea, K.M.; O’Carroll, O.M.; Carroll, C.; Grogan, B.; Connolly, A.; O’Shaughnessy, L.; Nicholson, T.T.; Gallagher, C.G.; McKone, E.F. Efficacy of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease. Eur. Respir. J. 2021, 57, 2003079. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Durieu, I.; Chiron, R.; Ramel, S.; Danner-Boucher, I.; Prevotat, A.; Grenet, D.; Marguet, C.; Reynaud-Gaubert, M.; Macey, J.; et al. Rapid Improvement after Starting Elexacaftor-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- DiMango, E.; Overdevest, J.; Keating, C.; Francis, S.F.; Dansky, D.; Gudis, D. Effect of highly effective modulator treatment on sinonasal symptoms in cystic fibrosis. J. Cyst. Fibros. 2021, 20, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Vaccarin, C.; Lukacs, G.L. Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- Cuyx, S.; De Boeck, K. Treating the Underlying Cystic Fibrosis Transmembrane Conductance Regulator Defect in Patients with Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Zemanick, E.T.; Konstan, M.W.; VanDevanter, D.R.; Rowe, S.M.; Clancy, J.; Odem-Davis, K.; Skalland, M.; Mayer-Hamblett, N. Measuring the impact of CFTR modulation on sweat chloride in cystic fibrosis: Rationale and design of the CHEC-SC study. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- Eckford, P.D.W.; McCormack, J.; Munsie, L.; He, G.; Stanojevic, S.; Pereira, S.L.; Ho, K.; Avolio, J.; Bartlett, C.; Yang, J.Y.; et al. The Cf Canada-Sick Kids Program in Individual Cf Therapy: A Resource for the Advancement of Personalized Medicine in Cf. J. Cyst. Fibros. 2019, 18, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, A.S.; Fürstová, E.; Vonk, A.M.; Ferrante, M.; Verfaillie, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekman, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; Groot, K.M.D.W.-D.; Brandsma, A.M.; de Jong, N.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; de Boeck, K.; Ecfs Strategic Planning Task Force on “Speeding up access to new drugs for CF”. Theranostics by Testing Cftr Modulators in Patient-Derived Materials: The Current Status and a Proposal for Subjects with Rare Cftr Mutations. J. Cyst. Fibros. 2019, 18, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; Groot, K.M.D.W.-D.; Arets, H.G.; der Wilt, R.E.M.-V.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef] [Green Version]
- Groot, K.M.D.W.-D.; Berkers, G.; van der Wilt, R.E.M.; van der Meer, R.; Vonk, A.; Dekkers, J.F.; Geerdink, M.; Michel, S.; Kruisselbrink, E.; Vries, R.; et al. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J. Cyst. Fibros. 2020, 19, 614–619. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Dousova, T.; Ramalho, S.; Centeio, R.; Clarke, L.A.; Railean, V.; Botelho, H.M.; Holubova, A.; Valaskova, I.; Yeh, J.T.; et al. Organoids as a Personalized Medicine Tool for Ultra-Rare Mutations in Cystic Fibrosis: The Case of S955p and 1717-2a>G. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165905. [Google Scholar] [CrossRef]
- Molinski, S.V.; Ahmadi, S.; Ip, W.; Ouyang, H.; Villella, A.; Miller, J.P.; Lee, P.S.; Kulleperuma, K.; Du, K.; Di Paola, M.; et al. Orkambi(R) and Amplifier Co-Therapy Improves Function from a Rare Cftr Mutation in Gene-Edited Cells and Patient Tissue. EMBO Mol. Med. 2017, 9, 1224–1243. [Google Scholar] [CrossRef]
- Flume, P.; Sawicki, G.; Pressler, T.; Schwarz, C.; Fajac, I.; Layish, D.; Bialek, P.; Wilson, S.; Kang, L.; McLaughlin, B.; et al. WS01.2 Phase 2 initial results evaluating PTI-428, a novel CFTR amplifier, in patients with cystic fibrosis. J. Cyst. Fibros. 2018, 17, S1–S2. [Google Scholar] [CrossRef]
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Aviram, M.; Bdolah-Abram, T.; Bebok, Z.; Shushi, L.; et al. Gentamicin-Induced Correction of Cftr Function in Patients with Cystic Fibrosis and Cftr Stop Mutations. N. Engl. J. Med. 2003, 349, 1433–1441. [Google Scholar] [CrossRef] [Green Version]
- Aksit, M.; Bowling, A.; Evans, T.; Joynt, A.; Osorio, D.; Patel, S.; West, N.; Merlo, C.; Sosnay, P.; Cutting, G.; et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J. Cyst. Fibros. 2019, 18, 606–613. [Google Scholar] [CrossRef]
- Keenan, M.M.; Huang, L.; Jordan, N.J.; Wong, E.; Cheng, Y.; Valley, H.C.; Mahiou, J.; Liang, F.; Bihler, H.; Mense, M.; et al. Nonsense-mediated RNA Decay Pathway Inhibition Restores Expression and Function of W1282X CFTR. Am. J. Respir. Cell Mol. Biol. 2019, 61, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Crawford, D.K.; Mullenders, J.; Pott, J.; Boj, S.F.; Landskroner-Eiger, S.; Goddeeris, M.M. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 2021, 20, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Alroy, I.; Shohat, M.; Eshkar-Oren, I.; Huertas, P. Translational Read-through of Ctns Nonsense Mutation and Attenuation F Ctns Nonsense-Mediated Mrna Decay by Elx-02 Treatment. Mol. Genet. Metab. 2018, 123, S18. [Google Scholar] [CrossRef]
- Mutyam, V.; Sharma, J.; Li, Y.; Peng, N.; Chen, J.; Tang, L.P.; Falk Libby, E.; Singh, A.K.; Conrath, K.; Rowe, S.M. Novel Correctors and Potentiators Enhance Functional Rescue of Cftr Nonsense Mutation Translational Readthrough. Am. J. Respir. Cell Mol. Biol. 2021, 64, 604–616. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Apaja, P.; Lukacs, G.L. Protein quality control at the plasma membrane. Curr. Opin. Cell Biol. 2011, 23, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moniz, S.; Sousa, M.; Moraes, B.J.; Mendes, A.I.; Palma, M.; Barreto, C.; Fragata, J.I.; Amaral, M.D.; Matos, P. HGF Stimulation of Rac1 Signaling Enhances Pharmacological Correction of the Most Prevalent Cystic Fibrosis Mutant F508del-CFTR. ACS Chem. Biol. 2012, 8, 432–442. [Google Scholar] [CrossRef]
- Alshafie, W.; Chappe, F.G.; Li, M.; Anini, Y.; Chappe, V.M. Vip Regulates Cftr Membrane Expression and Function in Calu-3 Cells by Increasing Its Interaction with Nherf1 and P-Erm in a Vpac1- and Pkcepsilon-Dependent Manner. Am. J. Physiol. Cell Physiol. 2014, 307, C107–C119. [Google Scholar] [CrossRef] [Green Version]
- Marozkina, N.V.; Yemen, S.; Borowitz, M.; Liu, L.; Plapp, M.; Sun, F.; Islam, R.; Erdmann-Gilmore, P.; Townsend, R.R.; Lichti, C.F.; et al. Hsp 70/Hsp 90 organizing protein as a nitrosylation target in cystic fibrosis therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 11393–11398. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, S.H.; Solomon, G.M.; Zeitlin, P.L.; Flume, P.A.; Casey, A.; McCoy, K.; Zemanick, E.T.; Mandagere, A.; Troha, J.M.; Shoemaker, S.A.; et al. Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. J. Cyst. Fibros. 2017, 16, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, A.L.; Thornell, I.M.; Singh, B.; Shah, V.S.; Stoltz, D.A.; Zabner, J.; Sinn, P.L. A Novel AAV-mediated Gene Delivery System Corrects CFTR Function in Pigs. Am. J. Respir. Cell Mol. Biol. 2019, 61, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Brommel, C.M.; Cooney, A.L.; Sinn, P.L. Adeno-Associated Virus-Based Gene Therapy for Lifelong Correction of Genetic Disease. Hum. Gene Ther. 2020, 31, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Breunig, C.T.; Durovic, T.; Neuner, A.M.; Baumann, V.; Wiesbeck, M.F.; Köferle, A.; Götz, M.; Ninkovic, J.; Stricker, S.H. One step generation of customizable gRNA vectors for multiplex CRISPR approaches through string assembly gRNA cloning (STAgR). PLoS ONE 2018, 13, e0196015. [Google Scholar] [CrossRef]
- Zhou, Z.P.; Yang, L.L.; Cao, H.; Chen, Z.R.; Zhang, Y.; Wen, X.-Y.; Hu, J.; Wen, X.-Y. In Vitro Validation of a CRISPR-Mediated CFTR Correction Strategy for Preclinical Translation in Pigs. Hum. Gene Ther. 2019, 30, 1101–1116. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Bleich, M.; Greger, R.; Schreiber, R.; Kunzelmann, K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J. Clin. Investig. 1998, 102, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Stutts, M.J.; Canessa, C.M.; Olsen, J.C.; Hamrick, M.; Cohn, J.A.; Rossier, B.C.; Boucher, R.C. Cftr as a Camp-Dependent Regulator of Sodium Channels. Science 1995, 269, 847–850. [Google Scholar] [CrossRef]
- Knowles, M.R.; Boucher, R.C. Mucus Clearance as a Primary Innate Defense Mechanism for Mammalian Airways. J. Clin. Invest. 2002, 109, 571–577. [Google Scholar] [CrossRef]
- Mall, M.; Grubb, B.R.; Harkema, J.R.; O’Neal, W.K.; Boucher, R.C. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493. [Google Scholar] [CrossRef]
- Mall, M.A. ENaC inhibition in cystic fibrosis: Potential role in the new era of CFTR modulator therapies. Eur. Respir. J. 2020, 56, 2000946. [Google Scholar] [CrossRef] [PubMed]
- Nickolaus, P.; Jung, B.; Sabater, J.; Constant, S.; Gupta, A. Preclinical evaluation of the epithelial sodium channel inhibitor BI 1265162 for treatment of cystic fibrosis. ERJ Open Res. 2020, 6. [Google Scholar] [CrossRef]
- Goss, C.H.; Jain, R.; Seibold, W.; Picard, A.C.; Hsu, M.C.; Gupta, A.; Fajac, I. An Innovative Phase Ii Trial to Establish Proof of Efficacy and Optimal Dose of a New Inhaled Epithelial Sodium Channel Inhibitor Bi 1265162 in Adults and Adolescents with Cystic Fibrosis: Balance-Cf™ 1. ERJ Open Res. 2020, 6. [Google Scholar] [CrossRef]
- Danahay, H.; Gosling, M. TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis? Int. J. Mol. Sci. 2020, 21, 2386. [Google Scholar] [CrossRef] [Green Version]
- Yerxa, B.R.; Sabater, J.R.; Davis, C.W.; Stutts, M.J.; Lang-Furr, M.; Picher, M.; Jones, A.C.; Cowlen, M.; Dougherty, R.; Boyer, J.; et al. Pharmacology of INS37217 [P1-(Uridine 5′)-P4- (2′-deoxycytidine 5′)tetraphosphate, Tetrasodium Salt], a Next-Generation P2Y2 Receptor Agonist for the Treatment of Cystic Fibrosis. J. Pharmacol. Exp. Ther. 2002, 302, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, P.L.; Boyle, M.P.; Guggino, W.B.; Molina, L. A Phase I Trial of Intranasal Moli1901 for Cystic Fibrosis. Chest 2004, 125, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Accurso, F.J.; Moss, R.B.; Wilmott, R.W.; Anbar, R.D.; Schaberg, A.E.; Durham, T.A.; Ramsey, B.W. Denufosol Tetrasodium in Patients with Cystic Fibrosis and Normal to Mildly Impaired Lung Function. Am. J. Respir. Crit. Care Med. 2011, 183, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Durham, T.; Navratil, T.; Schaberg, A.; Accurso, F.J.; Wainwright, C.; Barnes, M.; Moss, R.B. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J. Cyst. Fibros. 2012, 11, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Grasemann, H.; Stehling, F.; Brunar, H.; Widmann, R.; Laliberte, T.W.; Molina, L.; Döring, G.; Ratjen, F. Inhalation of Moli1901 in Patients With Cystic Fibrosis. Chest 2007, 131, 1461–1466. [Google Scholar] [CrossRef]
- Danahay, H.L.; Lilley, S.; Fox, R.; Charlton, H.; Sabater, J.; Button, B.; McCarthy, C.; Collingwood, S.P.; Gosling, M. TMEM16A Potentiation: A Novel Therapeutic Approach for the Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 946–954. [Google Scholar] [CrossRef] [Green Version]
- Danahay, H.L.; Morris, D.G.; Gosling, M. Reply to Olschewski et al.: TMEM16A Potentiation: Possible Drawbacks. Am. J. Respir. Crit. Care Med. 2020, 202, 905–906. [Google Scholar] [CrossRef] [PubMed]
- Muraglia, K.A.; Chorghade, R.; Kim, B.R.; Tang, X.X.; Shah, V.S.; Grillo, A.S.; Daniels, P.; Cioffi, A.G.; Karp, P.H.; Zhu, L.; et al. Small-molecule ion channels increase host defences in cystic fibrosis airway epithelia. Nat. Cell Biol. 2019, 567, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Chorghade, R.S.; Kim, B.R.; Launspach, J.L.; Karp, P.H.; Welsh, M.J.; Burke, M.D. Amphotericin B induces epithelial voltage responses in people with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 540–550. [Google Scholar] [CrossRef]
- Li, H.; Valkenier, H.; Judd, L.W.; Brotherhood, P.R.; Hussain, S.; Cooper, J.A.; Jurcek, O.; Sparkes, H.A.; Sheppard, D.N.; Davis, A.P. Efficient, Non-Toxic Anion Transport by Synthetic Carriers in Cells and Epithelia. Nat. Chem. 2016, 8, 24–32. [Google Scholar] [CrossRef]
- Hernando, E.; Capurro, V.; Cossu, C.; Fiore, M.; Garcia-Valverde, M.; Soto-Cerrato, V.; Perez-Tomas, R.; Moran, O.; Zegarra-Moran, O.; Quesada, R. Small Molecule Anionophores Promote Transmembrane Anion Permeation Matching Cftr Activity. Sci. Rep. 2018, 8, 2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossu, C.; Fiore, M.; Baroni, D.; Capurro, V.; Caci, E.; Garcia-Valverde, M.; Quesada, R.; Moran, O. Anion-Transport Mechanism of a Triazole-Bearing Derivative of Prodigiosine: A Candidate for Cystic Fibrosis Therapy. Front. Pharmacol. 2018, 9, 852. [Google Scholar] [CrossRef]
- Fiore, M.; Cossu, C.; Capurro, V.; Picco, C.; Ludovico, A.; Mielczarek, M.; Carreira-Barral, I.; Caci, E.; Baroni, D.; Quesada, R.; et al. Small molecule-facilitated anion transporters in cells for a novel therapeutic approach to cystic fibrosis. Br. J. Pharmacol. 2019, 176, 1764–1779. [Google Scholar] [CrossRef]
- Gianotti, A.; Capurro, V.; Delpiano, L.; Mielczarek, M.; García-Valverde, M.; Carreira-Barral, I.; Ludovico, A.; Fiore, M.; Baroni, D.; Moran, O.; et al. Small Molecule Anion Carriers Correct Abnormal Airway Surface Liquid Properties in Cystic Fibrosis Airway Epithelia. Int. J. Mol. Sci. 2020, 21, 1488. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef] [Green Version]
- Crosby, J.R.; Zhao, C.; Jiang, C.; Bai, D.; Katz, M.; Greenlee, S.; Kawabe, H.; McCaleb, M.; Rotin, D.; Guo, S.; et al. Inhaled ENaC antisense oligonucleotide ameliorates cystic fibrosis-like lung disease in mice. J. Cyst. Fibros. 2017, 16, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaels, W.; Bridges, R.J.; Hastings, M.L. Antisense oligonucleotide-mediated correction of CFTR splicing improves chloride secretion in cystic fibrosis patient-derived bronchial epithelial cells. Nucleic Acids Res. 2020, 48, 7454–7467. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Hamblett, N.; Nichols, D.P.; Odem-Davis, K.; A Riekert, K.; Sawicki, G.S.; Donaldson, S.H.; Ratjen, F.; Konstan, M.W.; Simon, N.; Rosenbluth, D.B.; et al. Evaluating the Impact of Stopping Chronic Therapies after Modulator Drug Therapy in Cystic Fibrosis: The SIMPLIFY Study Design. Ann. Am. Thorac. Soc. 2021. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drug (Trade Name) | Mode of Action | Age | Mutation Class | Alleles |
|---|---|---|---|---|
| Ivacaftor (Kalydeko) | Potentiator | 2 years and older | III, IV | G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R, R117H, other rare mutations (see text) |
| Ivacaftor and lumacaftor (Orkambi) | Potentiator and corrector | 12 years and older | II | F508del homozygous |
| Ivacaftor and tezacaftor (Symdeko) | Potentiator and second-generation corrector | 12 years and older | II/II, or II/RF * | F508del homozygous or F508del/RF * |
| Ivacaftor, tezacaftor and elexacaftor (Trikafta) | Potentiator and second-generation corrector and next-generation corrector | 12 years and older | II/II, II/other | A diagnosis of CF and at least one F508del OR another CFTR responsive mutation ** |
| Molecule ID | Company | Mechanism | Clinical Trial Number |
|---|---|---|---|
| VX-561 deutivacaftor | Vertex Pharmaceuticals | Potentiator | NCT03911713 |
| ABBV-974 | AbbVie | Potentiator | NCT02707562 NCT02690519 |
| ABBV-2451 | AbbVie | Potentiator | NCT03540524 |
| ABBC-3067 | AbbVie | Potentiator | NCT03969888 |
| QBW251 icenticaftor | Novartis | Potentiator | NCT02190604 |
| VX-121 | Vertex Pharmaceuticals | Corrector | NCT03912233 NCT03768089 |
| VX-440 olacaftor | Vertex Pharmaceuticals | Corrector | NCT02951182 |
| VX-659 bamocaftor | Vertex Pharmaceuticals | Corrector | NCT03447249 |
| ABBV-2222 galicaftor | AbbVie | Corrector | NCT03969888 |
| ELX-02 | Eloxx Pharmaceuticals | Read-through | NCT04135495 NCT04126473 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergeron, C.; Cantin, A.M. New Therapies to Correct the Cystic Fibrosis Basic Defect. Int. J. Mol. Sci. 2021, 22, 6193. https://doi.org/10.3390/ijms22126193
Bergeron C, Cantin AM. New Therapies to Correct the Cystic Fibrosis Basic Defect. International Journal of Molecular Sciences. 2021; 22(12):6193. https://doi.org/10.3390/ijms22126193
Chicago/Turabian StyleBergeron, Christelle, and André M. Cantin. 2021. "New Therapies to Correct the Cystic Fibrosis Basic Defect" International Journal of Molecular Sciences 22, no. 12: 6193. https://doi.org/10.3390/ijms22126193
APA StyleBergeron, C., & Cantin, A. M. (2021). New Therapies to Correct the Cystic Fibrosis Basic Defect. International Journal of Molecular Sciences, 22(12), 6193. https://doi.org/10.3390/ijms22126193