
Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms

Abstract
:1. Introduction
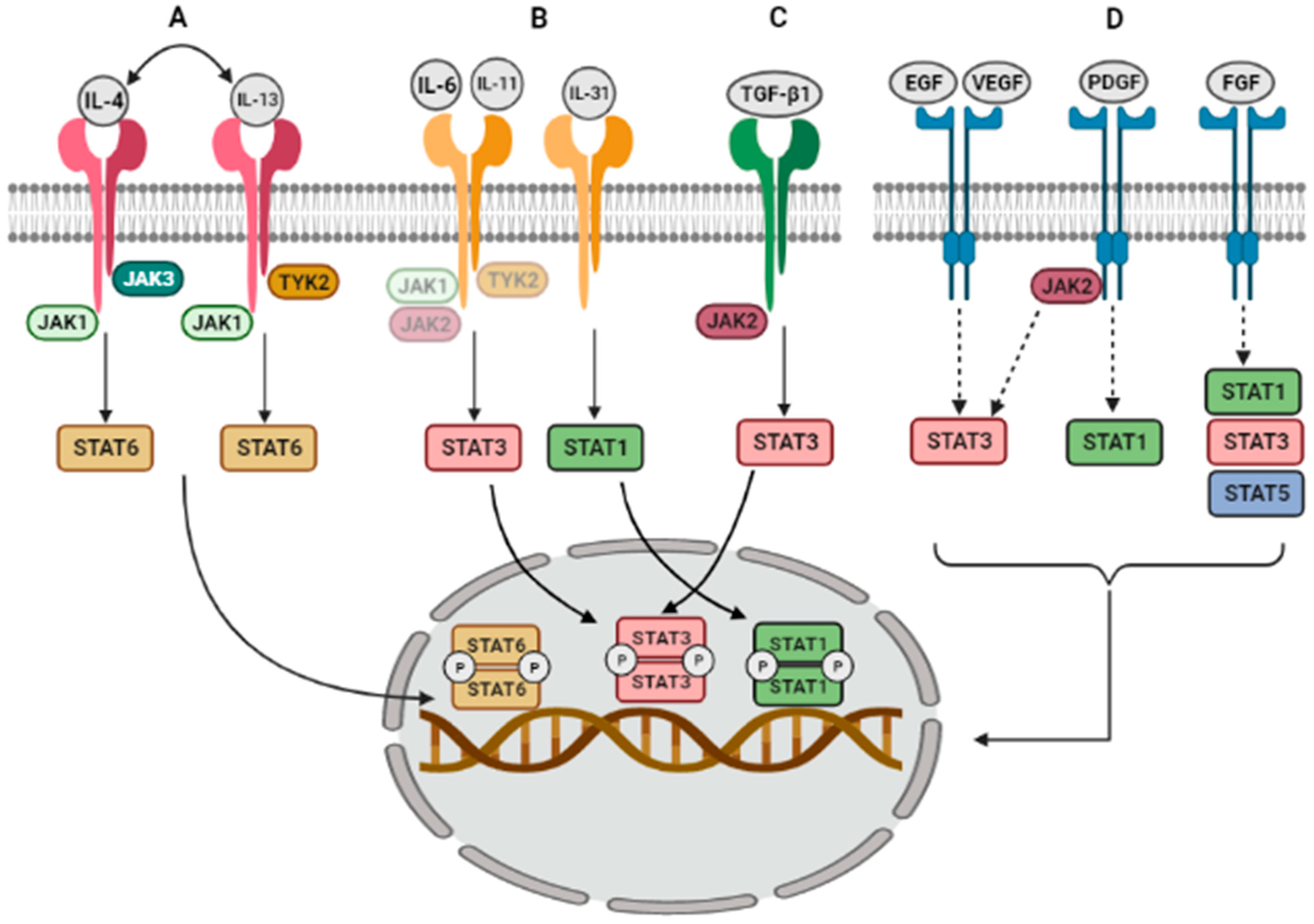
2. JAK/STAT Activation Mechanisms
2.1. Activation by Interleukins
2.2. Activation by Growth Factors
3. JAK/STAT Lung Expression and Distribution

{kind=link}
{kind=link}
{kind=link}
| JAK/STAT | Study Subject/ILD | JAK/STAT Lung Distribution | JAK/STAT mRNA and Protein Expression | Reference |
|---|---|---|---|---|
| JAK 1 | BLM-induced IPF mouse model | Inflammatory and epithelial cells | Increased mRNAexpression in lung tissue | [57] |
| p-JAK1 | BLM-induced IPF mouse model | Lung tissue | [58] | |
| JAK2 | BLM-induced IPF rat model | Fibrotic cells | [31] | |
| IPF patients | Hyperplastic alveolar cells and fibroblasts | Increased mRNA and protein expression in lung tissue | [31] | |
| IPF + pulmonary hypertension patients | Tunica intima and media of small pulmonary arteries | Increased mRNA and protein expression in pulmonary arteries | [59] | |
| p-JAK2 | BLM-induced IPF rat model | Nuclei of fibrotic cells | Increased protein expression in lung tissue | [31] |
| TYK2 | Progressive pulmonary sarcoidosis patients | Increased mRNA expression in bronchoalveolar lavage | [65] | |
| STAT1 | BLM-induced IPF mouse model | Inflammatory and epithelial cells | Increased mRNAexpression in lung tissue | [28,57,60] |
| BLM-induced IPF rat model | Alveolar macrophages | [61] | ||
| Sarcoidosis patients | Increased expression in blood and lung samples | [63,64] | ||
| p-STAT1 | BLM-induced IPF mouse model | Lung tissue | [58] | |
| STAT 3 | BLM-induced IPF mouse model | Alveolar macrophages, endothelial cells, and neutrophils | Increased mRNA and proteinexpression in lung tissue | [30,57] |
| BLM-induced IPF rat model | Fibrotic cells | Increased protein expression in lung tissue | [31] | |
| IPF patients | Hyperplastic alveolar cells and fibroblasts | Increased mRNA and protein expression in lung tissue | [31] | |
| IPF+ pulmonary hypertension patients | Tunica intima and media of small pulmonary arteries | Increased mRNA and protein expression in pulmonary arteries | [59] | |
| p-STAT3 | BLM-induced IPF mouse model | Myofibroblasts and alveolar macrophages | Increased protein expression in lung tissue | [30,62,67] |
| BLM-induced IPF rat model | Fibrotic cells and nuclei of fibrotic cells | Increased protein expression in lung tissue | [31] | |
| IPF patients | Areas of dense fibrosis, parenchymal cells adjacent to collagenous foci, (epithelial and hematopoietic origin), alveolar macrophages, myofibroblasts and nuclei of AECs | Increased protein expression in lung tissue and isolated IPF-lung-fibroblasts | [23,30,49,67] | |
| IPF+ pulmonary hypertension Patients | Nucleus of pulmonary artery cells, fibroblasts, and AECs | Increased protein expression in pulmonary arteries | [59] | |
| p-STAT5 | DIP mouse model | Increased p-STAT5 protein expression in alveolar macrophages | [66] | |
| STAT 6 | BLM-induced IPF mouse model | Increased mRNA expression | [20] |
4. Genetic Alterations
5. Cellular Processes Activated by JAK/STAT
5.1. Fibroblasts to Mesenchymal Transition
5.2. Epithelial to Mesenchymal Transition
5.3. Senescence
5.4. Apoptosis and Proliferation
5.5. Endoplasmic Reticulum Stress
5.6. Autophagy
6. Targeting JAK/STAT for ILDs Treatment
6.1. JAK Inhibitors
6.1.1. Tofacitinib
6.1.2. Ruxolitinib
6.1.3. Baricitinib
6.2. STAT Inhibitors
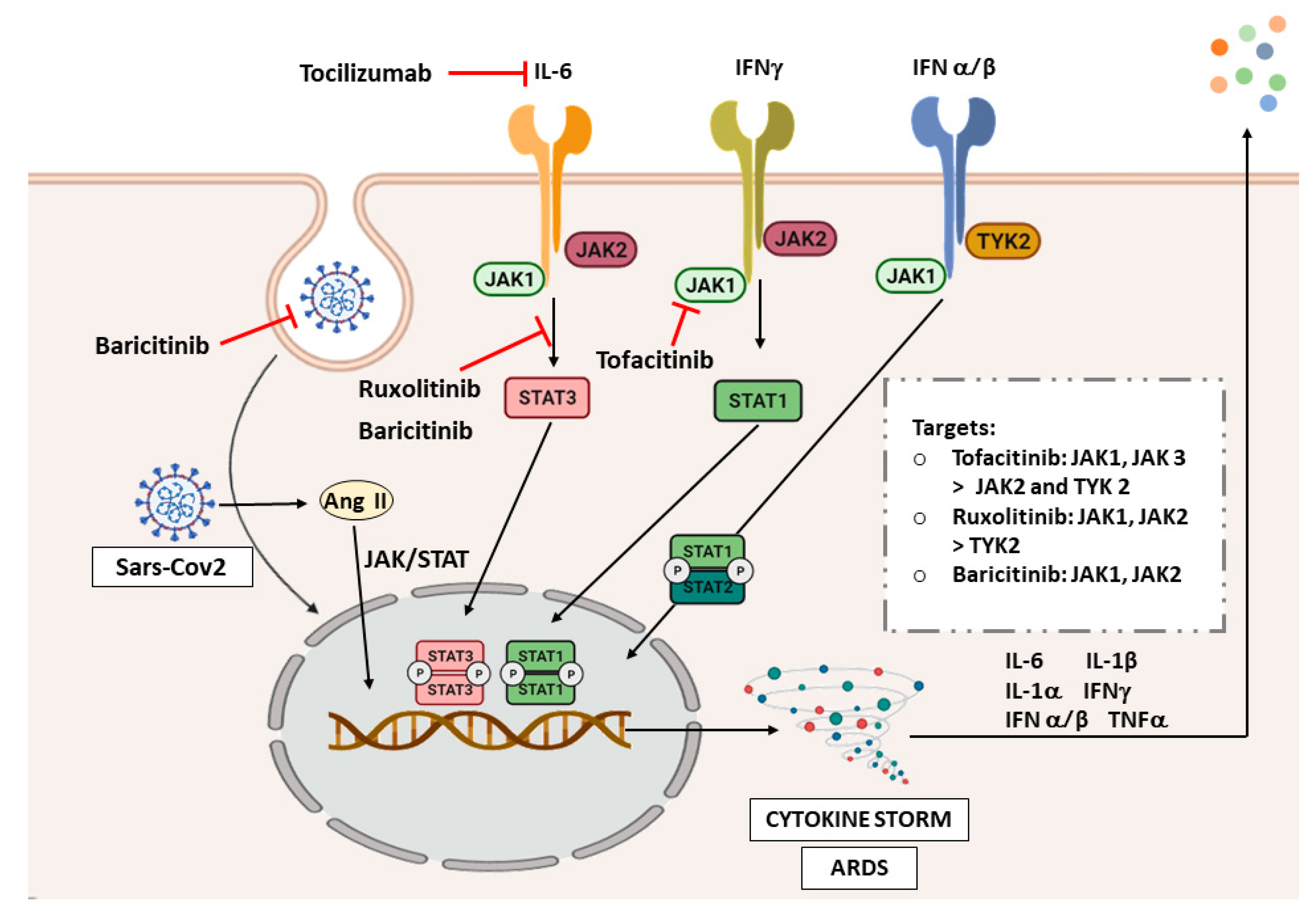
7. JAK/STAT and COVID-19
7.1. Interstitial Lung Disease and COVID-19
7.2. The JAK/STAT Pathway in COVID-19
7.3. JAK/STAT Inhibition as Therapeutic Strategy in COVID-19
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- American Thoracic Society. European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. Am. Thorac. Soc. AJRCCM 2002, 165, 277–304. [Google Scholar]
- Mueller-Mang, C.; Plank, C.; Ringl, H.; Dirisamer, A.; Herold, C.J. Interstitial Lung Diseases. Med Radiol. 2009, 333–355. [Google Scholar] [CrossRef]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, K.M.; Margaritopoulos, G.A.; Tomassetti, S.; Bonella, F.; Costabel, U.; Poletti, V. Interstitial lung disease. Eur. Respir. Rev. 2014, 23, 40–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller-Mang, C.; Grosse, C.; Schmid, K.; Stiebellehner, L.; Bankier, A.A. What Every Radiologist Should Know about Idiopathic Interstitial Pneumonias. Radiographics 2007, 27, 595–615. [Google Scholar] [CrossRef] [Green Version]
- Kligerman, S.J.; Groshong, S.; Brown, K.K.; Lynch, D.A. Nonspecific Interstitial Pneumonia: Radiologic, Clinical, and Pathologic Considerations. Radiographics 2009, 29, 73–87. [Google Scholar] [CrossRef]
- Siemińska, A.; Kuziemski, K. Respiratory bronchiolitis-interstitial lung disease. Orphanet J. Rare Dis. 2014, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Cordier, J.-F. Cryptogenic organizing pneumonia. Clin. Chest Med. 2004, 25, 727–738. [Google Scholar] [CrossRef]
- Bruminhent, J.; Yassir, S.; Pippim, J. Acute Interstitial Pneumonia (Hamman-Rich Syndrome) as a Cause of Idiopathic Acute Respiratory Distress Syndrome. Case Rep. Med. 2011, 2011, 1–4. [Google Scholar] [CrossRef] [Green Version]
- A Mikolasch, T.; Garthwaite, H.S.; Porter, J.C. Update in diagnosis and management of interstitial lung disease. Clin. Med. 2017, 17, 146–153. [Google Scholar] [CrossRef]
- Furini, F.; Carnevale, A.; Casoni, G.L.; Guerrini, G.; Cavagna, L.; Govoni, M.; Sciré, C.A. The Role of the Multidisciplinary Evaluation of Interstitial Lung Diseases: Systematic Literature Review of the Current Evidence and Future Perspectives. Front. Med. 2019, 6, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. [Internet] Frontiers; 2018. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2018.00287/full (accessed on 20 January 2021).
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef]
- Yan, Z.; Gibson, S.A.; Buckley, J.A.; Qin, H.; Benveniste, E.N. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin. Immunol. 2018, 189, 4–13. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Mohr, A.; Chatain, N.; Domoszlai, T.; Rinis, N.; Sommerauer, M.; Vogt, M.; Müller-Newen, G. Dynamics and non-canonical aspects of JAK/STAT signalling. Eur. J. Cell Biol. 2012, 91, 524–532. [Google Scholar] [CrossRef]
- Liu, T.; Jin, H.; Ullenbruch, M.; Hu, B.; Hashimoto, N.; Moore, B.; McKenzie, A.; Lukacs, N.W.; Phan, S. Regulation of Found in Inflammatory Zone 1 Expression in Bleomycin-Induced Lung Fibrosis: Role of IL-4/IL-13 and Mediation via STAT-6. J. Immunol. 2004, 173, 3425–3431. [Google Scholar] [CrossRef] [Green Version]
- Walford, H.H.; Doherty, T.A. STAT6 and Lung Inflammation. JAKSTAT [Internet]. 2013. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3876430/ (accessed on 1 February 2021).
- Moodley, Y.P.; Misso, N.L.A.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse Effects of Interleukin-6 on Apoptosis of Fibroblasts from Pulmonary Fibrosis and Normal Lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, D.V.; Prêle, C.M.; Wong, J.; Hogaboam, C.M.; McAnulty, R.; Laurent, G.J.; Zhang, S.S.-M.; Selman, M.; Mutsaers, S.E.; Knight, D.A. STAT3-Mediated Signaling Dysregulates Lung Fibroblast-Myofibroblast Activation and Differentiation in UIP/IPF. Am. J. Pathol. 2012, 180, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.P.; Scaffidi, A.K.; Misso, N.L.; Keerthisingam, C.; McAnulty, R.; Laurent, G.J.; Mutsaers, S.E.; Thompson, P.J.; Knight, D.A. Fibroblasts Isolated from Normal Lungs and Those with Idiopathic Pulmonary Fibrosis Differ in Interleukin-6/gp130-Mediated Cell Signaling and Proliferation. Am. J. Pathol. 2003, 163, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Ng, B.; Dong, J.; D’Agostino, G.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Ko, N.S.J.; Tan, J.; Chothani, S.P.; Huang, B.; et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaaw1237. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.; Dong, J.; Viswanathan, S.; Widjaja, A.A.; Paleja, B.; Adami, E.; Ko, N.S.J.; Wang, M.; Lim, S.; Tan, J.; et al. Fibroblast-specific IL11 signaling drives chronic inflammation in murine fibrotic lung disease. FASEB J. 2020, 34, 11802–11815. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.; Cook, S.A.; Schafer, S. Interleukin-11 signaling underlies fibrosis, parenchymal dysfunction, and chronic inflammation of the airway. Exp. Mol. Med. 2020, 52, 1871–1878. [Google Scholar] [CrossRef]
- Shi, K.; Jiang, J.; Ma, T.; Xie, J.; Duan, L.; Chen, R.; Song, P.; Yu, Z.; Liu, C.; Zhu, Q.; et al. Pathogenesis pathways of idiopathic pulmonary fibrosis in bleomycin-induced lung injury model in mice. Respir. Physiol. Neurobiol. 2014, 190, 113–117. [Google Scholar] [CrossRef]
- Zhang, Y.; Dees, C.; Beyer, C.; Lin, N.-Y.; Distler, A.; Zerr, P.; Palumbo, K.; Susok, L.; Kreuter, A.; Distler, O.; et al. Inhibition of casein kinase II reduces TGFβ induced fibroblast activation and ameliorates experimental fibrosis. Ann. Rheum. Dis. 2014, 74, 936–943. [Google Scholar] [CrossRef]
- Pedroza, M.; Le, T.T.; Lewis, K.M.; Karmouty-Quintana, H.; To, S.; George, A.T.; Blackburn, M.R.; Tweardy, D.J.; Agarwal, S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016, 30, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escrivá, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef] [Green Version]
- Beyer, C.; Distler, J.H.W. Tyrosine kinase signaling in fibrotic disorders: Translation of basic research to human disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Tzouvelekis, A.; Ntolios, P.; Karameris, A.; Vilaras, G.; Boglou, P.; Koulelidis, A.; Archontogeorgis, K.; Kaltsas, K.; Zacharis, G.; Sarikloglou, E.; et al. Increased Expression of Epidermal Growth Factor Receptor (EGF-R) in Patients with Different Forms of Lung Fibrosis. BioMed Res. Int. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liao, S.; Bennett, S.; Tang, H.; Song, D.; Wood, D.; Zhan, X.; Xu, J. STAT3 and its targeting inhibitors in osteosarcoma. Cell Prolif. 2021, 54, e12974. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Benedict, S.H.; Clark, R.A.F.; King, T.E. Pathogenesis of Pulmonary Fibrosis in Interstitial Lung Disease: Alveolar Macrophage PDGF(B) Gene Activation and Up-regulation by Interferon Gamma. Am. Rev. Respir. Dis. 1991, 143, 167–173. [Google Scholar] [CrossRef]
- Homma, S.; Nagaoka, I.; Abe, H.; Takahashi, K.; Seyama, K.; Nukiwa, T.; Kira, S. Localization of platelet-derived growth factor and insulin-like growth factor I in the fibrotic lung. Am. J. Respir. Crit. Care Med. 1995, 152, 2084–2089. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, L.; Mandal, J.; Molino, A.; Stolz, D.; Tamm, M.; Lu, S.; Roth, M. PDGF-BB induces PRMT1 expression through ERK1/2 dependent STAT1 activation and regulates remodeling in primary human lung fibroblasts. Cell Signal 2016, 28, 307–315. [Google Scholar] [CrossRef]
- Li, P.; Huang, T.; Zou, Q.; Liu, D.; Wang, Y.; Tan, X.; Wei, Y.; Qiu, H. FGFR2 Promotes Expression of PD-L1 in Colorectal Cancer via the JAK/STAT3 Signaling Pathway. J. Immunol. 2019, 202, 3065–3075. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Xu, Y.; He, B.; Ma, R.; Wang, Y.; Chang, Y.; Xie, Y.; Wu, L.; Huang, J.; Xiao, Z. Ionizing radiation modulates vascular endothelial growth factor expression through STAT3 signaling pathway in rat neonatal primary astrocyte cultures. Brain Behav. 2020, 10, e01529. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Xia, L.; Lu, J. Interleukin-4 in rheumatoid arthritis patients with interstitial lung disease: A pilot study. Indian J. Med. Res. 2013, 138, 919–921. [Google Scholar]
- Jakubzick, C.; Kunkel, S.L.; Puri, R.K.; Hogaboam, C.M. Therapeutic Targeting of IL-4- and IL-13-Responsive Cells in Pulmonary Fibrosis. Immunol. Res. 2004, 30, 339–350. [Google Scholar] [CrossRef]
- Jakubzick, C.; Choi, E.S.; Carpenter, K.J.; Kunkel, S.L.; Evanoff, H.; Martinez, F.J.; Flaherty, K.R.; Toews, G.B.; Colby, T.V.; Travis, W.D.; et al. Human Pulmonary Fibroblasts Exhibit Altered Interleukin-4 and Interleukin-13 Receptor Subunit Expression in Idiopathic Interstitial Pneumonia. Am. J. Pathol. 2004, 164, 1989–2001. [Google Scholar] [CrossRef] [Green Version]
- Passalacqua, G.; Mincarini, M.; Colombo, D.; Troisi, G.; Ferrari, M.; Bagnasco, D.; Balbi, F.; Riccio, A.M.; Canonica, G.W. IL-13 and idiopathic pulmonary fibrosis: Possible links and new therapeutic strategies. Pulm. Pharmacol. Ther. 2017, 45, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Prêle, C.M.; Yao, E.; O’Donoghue, R.J.J.; Mutsaers, S.E.; Knight, D.A. STAT3: A central mediator of pulmonary fibrosis? Proc. Am. Thorac. Soc. 2012, 9, 177–182. [Google Scholar] [CrossRef]
- Park, C.S.; Chung, S.W.; Ki, S.Y.; Lim, G.-I.; Uh, S.-T.; Kim, Y.H.; Choi, D.I.; Park, J.S.; Lee, D.W.; Kitaichi, M. Increased Levels of Interleukin-6 Are Associated with Lymphocytosis in Bronchoalveolar Lavage Fluids of Idiopathic Nonspecific Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2000, 162, 1162–1168. [Google Scholar] [CrossRef]
- Mozaffarian, A.; Brewer, A.W.; Trueblood, E.S.; Luzina, I.G.; Todd, N.W.; Atamas, S.P.; Arnett, H.A. Mechanisms of Oncostatin M-Induced Pulmonary Inflammation and Fibrosis. J. Immunol. 2008, 181, 7243–7253. [Google Scholar] [CrossRef]
- O’Donoghue, R.J.J.; Knight, D.A.; Richards, C.D.; Prele, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- E Lindahl, G.; Stock, C.J.; Shi-Wen, X.; Leoni, P.; Sestini, P.; Howat, S.L.; Bou-Gharios, G.; Nicholson, A.G.; Denton, C.P.; Grutters, J.C.; et al. Microarray profiling reveals suppressed interferon stimulated gene program in fibroblasts from scleroderma-associated interstitial lung disease. Respir. Res. 2013, 14, 80. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Surinkaew, S.; Naud, P.; Qi, X.-Y.; Gillis, M.-A.; Shi, Y.-F.; Tardif, J.-C.; Dobrev, D.; Nattel, S. JAK-STAT signalling and the atrial fibrillation promoting fibrotic substrate. Cardiovasc. Res. 2017, 113, 310–320. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.-M.; Weigt, S.S.; Saggar, R.; Palchevskiy, V.; Volkmann, E.R.; Liang, L.L.; Ross, D.; Ardehali, A.; Lynch, J.P.; Belperio, J.A. Endotype–phenotyping may predict a treatment response in progressive fibrosing interstitial lung disease. EBioMedicine 2019, 50, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paukku, K.; Valgeirsdóttir, S.; Saharinen, P.; Bergman, M.; Heldin, C.H.; Silvennoinen, O. Platelet-derived growth factor (PDGF)-induced activation of signal transducer and activator of transcription (Stat) 5 is mediated by PDGF beta-receptor and is not dependent on c-src, fyn, jak1 or jak2 kinases. Biochem. J. 2000, 345, 759–766. [Google Scholar] [CrossRef]
- Jaskiewicz, K.; Mycroft, K.; Maskey-Warzechowska, M.; Paralusz, K.; Siemiez, N.; Nejman-Gryz, P.; Barnas, M.; Krenke, R.; Gorska, K. Exhaled Biomarkers in Idiopathic Pulmonary Fibrosis—A Six-Month Follow-Up Study in Patients Treated with Pirfenidone. J. Clin. Med. 2020, 9, 2523. [Google Scholar] [CrossRef] [PubMed]
- Simler, N.R.; E Brenchley, P.; Horrocks, A.W.; Greaves, S.M.; Hasleton, P.S.; Egan, J.J. Angiogenic cytokines in patients with idiopathic interstitial pneumonia. Thorax 2004, 59, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Chen, R.; Liu, X.; Xie, J.; Si, K.; Duan, L. Effects of Matrine on JAK-STAT Signaling Transduction Pathways in Bleomycin-Induced Pulmonary Fibrosis. Afr. J. Tradit. Complement. Altern. Med. 2013, 10, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Li, Y.; Wang, X.-L.; Li, X.-Z.; Chen, Y.-W.; Yang, L.-L.; Ming, H.-X. Effect of Total Flavonoids of Oxytropis falcata Bunge on the Expression of p-JAK1-and p-STAT1-Related Proteins in Idiopathic Pulmonary Fibrosis. Evidence-Based Complement. Altern. Med. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Milara, J.; Ballester, B.; Morell, A.; Ortiz, J.L.; Escrivá, J.; Fernández, E.; Perez-Vizcaino, F.; Cogolludo, A.; Pastor, E.; Artigues, E.; et al. JAK2 mediates lung fibrosis, pulmonary vascular remodelling and hypertension in idiopathic pulmonary fibrosis: An experimental study. Thorax 2018, 73, 519–529. [Google Scholar] [CrossRef]
- Shi, K.; Jiang, J.; Ma, T.; Xie, J.; Duan, L.; Chen, R.; Song, P.; Yu, Z.; Liu, C.; Zhu, Q.; et al. Dexamethasone attenuates bleomycin-induced lung fibrosis in mice through TGF-β, Smad3 and JAK-STAT pathway. Int. J. Clin. Exp. Med. 2014, 7, 2645–2650. [Google Scholar]
- Fan, X.-M.; Wang, Z.-L.; Li, Z.-H. STAT1 activation and STAT1-dependent immune-response gene ICAM-1 expression in alveolar macrophages of rats suffered from interstitial pulmonary fibrosis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin. J. Cell. Mol. Immunol. 2003, 19, 3–6. [Google Scholar]
- Shieh, J.-M.; Tseng, H.-Y.; Jung, F.; Yang, S.-H.; Lin, J.-C. Elevation of IL-6 and IL-33 Levels in Serum Associated with Lung Fibrosis and Skeletal Muscle Wasting in a Bleomycin-Induced Lung Injury Mouse Model. Mediat. Inflamm. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Li, H.; Zhao, X.; Wang, J.; Zong, M.; Yang, H. Bioinformatics analysis of gene expression profile data to screen key genes involved in pulmonary sarcoidosis. Gene 2017, 596, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Casanova, N.; Pouladi, N.; Wang, T.; Lussier, Y.; Knox, K.S.; Garcia, J.G.N. Identification of Jak-STAT signaling involvement in sarcoidosis severity via a novel microRNA-regulated peripheral blood mononuclear cell gene signature. Sci. Rep. 2017, 7, 4237. [Google Scholar] [CrossRef]
- Schischmanoff, P.O.; Naccache, J.-M.; Carrere, A.; Richardson, S.; Kambouchner, M.; Raphael, M.; Valeyre, D.; Fagard, R. Progressive pulmonary sarcoidosis is associated with over-expression of TYK2 and p21Waf1/Cip1. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2006, 23, 101–107. [Google Scholar]
- Suzuki, T.; McCarthy, C.; Carey, B.C.; Borchers, M.; Beck, D.; Wikenheiser-Brokamp, K.A.; Black, D.; Chalk, C.; Trapnell, B.C. Increased Pulmonary GM-CSF Causes Alveolar Macrophage Accumulation. Mechanistic Implications for Desquamative Interstitial Pneumonitis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Le, T.-T.T.; Karmouty-Quintana, H.; Melicoff, E.; Weng, T.; Chen, N.-Y.; Pedroza, M.; Zhou, Y.; Davies, J.; Philip, K.; Molina, J.G.; et al. Blockade of IL-6 Trans Signaling Attenuates Pulmonary Fibrosis. J. Immunol. 2014, 193, 3755–3768. [Google Scholar] [CrossRef] [Green Version]
- Fabre, A.; Marchal, S.; Forbes, L.R.; Vogel, T.P.; Barlogis, V.; Triolo, V.; Rohrlich, P.-S.; Bérard, E.; Frankel, D.; Ambrosetti, D.; et al. STAT3 Gain of Function: A New Kid on the Block in Interstitial Lung Diseases. Am. J. Respir. Crit. Care Med. 2018, 197, e22–e23. [Google Scholar] [CrossRef] [PubMed]
- Forbes, L.R.; Vogel, T.P.; Cooper, M.A.; Castro-Wagner, J.; Schussler, E.; Weinacht, K.G.; Plant, A.S.; Su, H.C.; Allenspach, E.J.; Slatter, M.; et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J. Allergy Clin. Immunol. 2018, 142, 1665–1669. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, M.; Scaglia, P.; Keselman, A.; Martucci, L.; Karabatas, L.; Domené, S.; Martin, A.; Pennisi, P.; Blanco, M.; Sanguineti, N.; et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol. Cell. Endocrinol. 2018, 473, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.D.; Vogel, T.P.; Forbes, L.; Mauro, S.-K.; Stray-Pedersen, A.; Niemela, J.E.; Lyons, J.; Engelhardt, K.R.; Zhang, Y.; Topcagic, N.; et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015, 125, 591–599. [Google Scholar] [CrossRef]
- Giovannini-Chami, L.; Vogel, T.P.; Forbes, L.R.; Fabre, A.; Trojani, M.-C.; Leroy, S.; Antunes, O.; Vincent-Mefitiot, N.; Hiéronimus, S.; Baque-Juston, M.; et al. STAT3 gain of function: A new aetiology of severe rheumatic disease. Rheumatology 2018, 58, 365–367. [Google Scholar] [CrossRef]
- Silva-Carmona, M.; Vogel, T.P.; Marchal, S.; Guesmi, M.; Dubus, J.-C.; Leroy, S.; Fabre, A.; Barlogis, V.; Forbes, L.R.; Giovannini-Chami, L. Successful Treatment of Interstitial Lung Disease in STAT3 Gain-of-Function Using JAK Inhibitors. Am. J. Respir. Crit. Care Med. 2020, 202, 893–897. [Google Scholar] [CrossRef]
- Fabre, A.; Marchal, S.; Barlogis, V.; Mari, B.; Barbry, P.; Rohrlich, P.-S.; Forbes, L.R.; Vogel, T.P.; Giovannini-Chami, L. Clinical Aspects of STAT3 Gain-of-Function Germline Mutations: A Systematic Review. J. Allergy Clin. Immunol. Pr. 2019, 7, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Jägle, S.; Heeg, M.; Grün, S.; Rensing-Ehl, A.; Maccari, M.E.; Klemann, C.; Jones, N.; Lehmberg, K.; Bettoni, C.; Warnatz, K.; et al. Distinct molecular response patterns of activating STAT3 mutations associate with penetrance of lymphoproliferation and autoimmunity. Clin. Immunol. 2020, 210, 108316. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.L.; Al Remeithi, S.S.; Towe, C.T.; Dauber, A.; Backeljauw, P.F.; Tyzinski, L.; Kumar, A.R.; Hwa, V. Developmental Adaptive Immune Defects Associated with STAT5B Deficiency in Three Young Siblings. J. Clin. Immunol. 2021, 41, 136–146. [Google Scholar] [CrossRef]
- Hwa, V. STAT5B deficiency: Impacts on human growth and immunity. Growth Horm. IGF Res. 2016, 28, 16–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeau, K.; Hwa, V.; Rosenfeld, R.G. STAT5b Deficiency: An Unsuspected Cause of Growth Failure, Immunodeficiency, and Severe Pulmonary Disease. J. Pediatr. 2011, 158, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, T.; Dotta, L.; Giacomelli, M.; Vairo, D.; Badolato, R. STAT mutations as program switchers: Turning primary immunodeficiencies into autoimmune diseases. J. Leukoc. Biol. 2016, 101, 29–38. [Google Scholar] [CrossRef]
- Stock, C.J.W.; De Lauretis, A.; Visca, D.; Daccord, C.; Kokosi, M.; Kouranos, V.; Margaritopoulos, G.; George, P.M.; Molyneaux, P.L.; Nihtyanova, S.; et al. Defining genetic risk factors for scleroderma-associated interstitial lung disease. Clin. Rheumatol. 2020, 39, 1173–1179. [Google Scholar] [CrossRef] [Green Version]
- Kagan, P.; Sultan, M.; Tachlytski, I.; Safran, M.; Ben-Ari, Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS ONE 2017, 12, e0176173. [Google Scholar] [CrossRef]
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-L.; Zhang, X.; Bai, J.; Gai, L.; Ye, X.-L.; Zhang, L.; Xu, Q.; Zhang, Y.-X.; Xu, L.; Li, H.-P.; et al. Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: Potential roles in the inhibition of epithelial–mesenchymal transition and fibroblast activation. Cell Death Dis. 2013, 4, e665. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.-Y.; Zeng, Y.; Lei, Z.; Wang, L.; Yang, H.; Liu, Z.; Zhao, J.; Zhang, H.-T. JAK/STAT3 signaling is required for TGF-β-induced epithelial-mesenchymal transition in lung cancer cells. Int. J. Oncol. 2014, 44, 1643–1651. [Google Scholar] [CrossRef] [Green Version]
- Colomiere, M.; Ward, A.C.; Riley, C.; Trenerry, M.K.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial–mesenchymal transition in ovarian carcinomas. Br. J. Cancer 2008, 100, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, M.; Liu, Y.; Hu, H.; Pan, Y.; Zou, W.; Fan, X.; Hu, X. Transforming growth factor β1 promotes migration and invasion in HepG2 cells: Epithelial-to-mesenchymal transition via JAK/STAT3 signaling. Int. J. Mol. Med. 2017, 41, 129–136. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.; Gong, Y.; Chen, Y.; Yu, D.; Wang, X.; Zhang, X.; Dou, Y.; Liu, D.; Cheng, G.; Lu, S.; et al. IL-6 promotes epithelial-to-mesenchymal transition of human peritoneal mesothelial cells possibly through the JAK2/STAT3 signaling pathway. Am. J. Physiol. Physiol. 2017, 313, F310–F318. [Google Scholar] [CrossRef]
- Li, M.; Luan, F.; Zhao, Y.; Hao, H.; Zhou, Y.; Han, W.; Fu, X. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp. Biol. Med. 2016, 241, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.; Jones, M.; Davies, D.; Wang, Y. Epithelial-Mesenchymal Transition Contributes to Pulmonary Fibrosis via Aberrant Epithelial/Fibroblastic Cross-Talk. J. Lung Heal. Dis. 2019, 3, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Fintha, A.; Gasparics, Á.; Rosivall, L.; Sebe, A. Therapeutic Targeting of Fibrotic Epithelial-Mesenchymal Transition–An Outstanding Challenge. Front. Pharmacol. 2019, 10, 388. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Lomas, N.J.; Watts, K.L.; Akram, K.M.; Forsyth, N.R.; A Spiteri, M. Idiopathic pulmonary fibrosis: Immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int. J. Clin. Exp. Pathol. 2012, 5, 58–71. [Google Scholar] [PubMed]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.W.; Blokland, K.; Pathinayake, P.S.; Burgess, J.K.; Mutsaers, S.E.; Prele, C.M.; Schuliga, M.; Grainge, C.L.; Knight, D.A. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L162–L172. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Kasembeli, M.M.; Robinson, P.; Tweardy, D.J. Targeting Janus Kinases and Signal Transducer and Activator of Transcription 3 to Treat Inflammation, Fibrosis, and Cancer: Rationale, Progress, and Caution. Pharmacol. Rev. 2020, 72, 486–526. [Google Scholar] [CrossRef] [Green Version]
- Waters, D.W.; Blokland, K.; Pathinayake, P.S.; Wei, L.; Schuliga, M.; Jaffar, J.; Westall, G.P.; Hansbro, P.M.; Prele, C.M.; Mutsaers, S.E.; et al. STAT3 Regulates the Onset of Oxidant-induced Senescence in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Barbas-Filho, J.V.; Ferreira, M.A.; Sesso, A.; Kairalla, R.A.; Carvalho, C.R.; Capelozzi, V.L. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J. Clin. Pathol. 2001, 54, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Uhal, B.D. The role of apoptosis in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 138–144. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Thannickal, V.J.; Horowitz, J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Devgan, G.; Darnell, J.E.; Bromberg, J.F. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc. Natl. Acad. Sci. USA 2001, 98, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.-M.; Seeger, W.; et al. Epithelial Endoplasmic Reticulum Stress and Apoptosis in Sporadic Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Lawson, W.E.; Cheng, D.-S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [Green Version]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.-S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [Green Version]
- Corvol, H.; Flamein, F.; Epaud, R.; Clement, A.; Guillot, L. Lung alveolar epithelium and interstitial lung disease. Int. J. Biochem. Cell Biol. 2009, 41, 1643–1651. [Google Scholar] [CrossRef]
- Waris, G.; Tardif, K.D.; Siddiqui, A. Endoplasmic reticulum (ER) stress: Hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem. Pharmacol. 2002, 64, 1425–1430. [Google Scholar] [CrossRef]
- Ahyi, A.-N.N.; Quinton, L.J.; Jones, M.R.; Ferrari, J.D.; Pepper-Cunningham, Z.A.; Mella, J.R.; Remick, D.G.; Mizgerd, J.P. Roles of STAT3 in Protein Secretion Pathways during the Acute-Phase Response. Infect. Immun. 2013, 81, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-Dependent Activation of JAK1 and STAT3 Contributes to Endoplasmic Reticulum Stress-Induced Inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, D.N.; Ashley, S.L.; Moore, B.B. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L590–L601. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Nho, R.S.; Hergert, P. IPF Fibroblasts Are Desensitized to Type I Collagen Matrix-Induced Cell Death by Suppressing Low Autophagy via Aberrant Akt/mTOR Kinases. PLoS ONE 2014, 9, e94616. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasarmidi, E.; Sarantoulaki, S.; Trachalaki, A.; Margaritopoulos, G.; Bibaki, E.; Spandidos, D.; Tzanakis, N.; Antoniou, K. Investigation of key autophagy-and mitophagy-related proteins and gene expression in BALF cells from patients with IPF and RA-ILD. Mol. Med. Rep. 2018, 18, 3891–3897. [Google Scholar] [CrossRef] [Green Version]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, Z.; Liangkun, Y.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Zhanggui, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebba, A. Tocilizumab: The first interleukin-6-receptor inhibitor. Am. J. Heal. Pharm. 2008, 65, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.W.; Ryerson, C.J.; Guler, S.A. Progression of fibrosing interstitial lung disease. Respir. Res. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- You, H.; Xu, D.; Zhao, J.; Li, J.; Wang, Q.; Tian, X.; Li, M.; Zeng, X. JAK Inhibitors: Prospects in Connective Tissue Diseases. Clin. Rev. Allergy Immunol. 2020, 59, 334–351. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.; Villarino, A.; Gadina, M.; McInnes, I.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Strand, V.; Van Vollenhoven, R.F.; Lee, E.B.; Fleischmann, R.; Zwillich, S.H.; Gruben, D.; Koncz, T.; Wilkinson, B.; Wallenstein, G. Tofacitinib or adalimumab versus placebo: Patient-reported outcomes from a phase 3 study of active rheumatoid arthritis. Rheumatology 2016, 55, 1031–1041. [Google Scholar] [CrossRef] [Green Version]
- Kremer, J.M.; Bloom, B.J.; Breedveld, F.C.; Coombs, J.H.; Fletcher, M.P.; Gruben, D.; Krishnaswami, S.; Burgos-Vargas, R.; Wilkinson, B.; Zerbini, C.A.F.; et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009, 60, 1895–1905. [Google Scholar] [CrossRef]
- Wang, W.; Bhattacharyya, S.; Marangoni, R.G.; Carns, M.; Dennis-Aren, K.; Yeldandi, A.; Wei, J.; Varga, J. The JAK/STAT pathway is activated in systemic sclerosis and is effectively targeted by tofacitinib. J. Scleroderma Relat. Disord. 2020, 5, 40–50. [Google Scholar] [CrossRef]
- Sendo, S.; Saegusa, J.; Yamada, H.; Nishimura, K.; Morinobu, A. Tofacitinib facilitates the expansion of myeloid-derived suppressor cells and ameliorates interstitial lung disease in SKG mice. Arthritis Res. 2019, 21, 1–9. [Google Scholar] [CrossRef]
- Lescoat, A.; Lelong, M.; Jeljeli, M.; Piquet-Pellorce, C.; Morzadec, C.; Ballerie, A.; Jouneau, S.; Jego, P.; Vernhet, L.; Batteux, F.; et al. Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: Perspectives for scleroderma-associated interstitial lung disease. Biochem. Pharmacol. 2020, 178, 114103. [Google Scholar] [CrossRef]
- Kurasawa, K.; Arai, S.; Namiki, Y.; Tanaka, A.; Takamura, Y.; Owada, T.; Arima, M.; Maezawa, R. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology 2018, 57, 2114–2119. [Google Scholar] [CrossRef] [PubMed]
- Conca, W.; Weheba, I.; Abouzied, M.-E.; Abdelsayed, A.; Aleyouni, Y.; Al-Mutairy, E.; Bakshi, N.; Khalid, M. Iacta Alea Est: The Inexorable Advance of Tofacitinib in the Treatment of Dermatomyositis-Associated Rapidly Progressive Interstitial Lung Disease. A Case Report. Front. Pharmacol. 2020, 11, 585761. [Google Scholar] [CrossRef]
- E Fragoulis, G.; McInnes, I.B.; Siebert, S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology 2019, 58, i43–i54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, J.W.; Al-Fayoumi, S.; Taylor, J.; Velichko, S.; O’Mahony, A. Comparative phenotypic profiling of the JAK2 inhibitors ruxolitinib, fedratinib, momelotinib, and pacritinib reveals distinct mechanistic signatures. PLoS ONE 2019, 14, e0222944. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6764664/ (accessed on 14 April 2021). [CrossRef] [PubMed]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.D.; Flanagan, M.E.; Telliez, J.-B. Discovery and Development of Janus Kinase (JAK) Inhibitors for Inflammatory Diseases. J. Med. Chem. 2014, 57, 5023–5038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, R.; Chen, C.-W.; Mallano, T.; Dees, C.; Distler, A.; Reich, A.; Bergmann, C.; Ramming, A.; Gelse, K.; et al. JAK1-dependent transphosphorylation of JAK2 limits the antifibrotic effects of selective JAK2 inhibitors on long-term treatment. Ann. Rheum. Dis. 2017, 76, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Bader-Meunier, B.; Hadchouel, A.; Berteloot, L.; Polivka, L.; Béziat, V.; Casanova, J.-L.; Lévy, R. Effectiveness and safety of ruxolitinib for the treatment of refractory systemic idiopathic juvenile arthritis like associated with interstitial lung disease: A case report. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Baricitinib: First Global Approval. Drugs 2017, 77, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Balci, S.; Ekinci, R.M.K.; de Jesus, A.A.; Goldbach-Mansky, R.; Yilmaz, M. Baricitinib experience on STING-associated vasculopathy with onset in infancy: A representative case from Turkey. Clin. Immunol. 2020, 212, 108273. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Perillo, F.; Metella Refini, R.; Bergantini, L.; Bellisai, F.; Selvi, E.; Cameli, P.; Manganelli, S.; Conticini, E.; Cantarini, L.; et al. Efficacy of baricitinib in treating rheumatoid arthritis: Modulatory effects on fibrotic and inflammatory biomarkers in a real-life setting. Int. Immunopharmacol. 2020, 86, 106748. [Google Scholar] [CrossRef]
- Wu, C.; Zheng, S.; Chen, Y.; Zheng, M. Single-cell RNA Expression Profiling of ACE2, the Putative Receptor of Wuhan 2019-nCoV, in the Nasal Tissue. MedRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Satarker, S.; Tom, A.A.; Shaji, R.A.; Alosious, A.; Luvis, M.; Nampoothiri, M. JAK-STAT Pathway Inhibition and their Implications in COVID-19 Therapy. Postgrad. Med. 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.; Davidson, S.M.; Yellon, D.M. The cytokine storm of COVID-19: A spotlight on prevention and protection. Expert Opin. Ther. Targets 2020, 24, 723–730. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Li, X.; Ma, X. Acute Respiratory Failure in COVID-19: Is it “Typical” ARDS? Crit Care [Internet]. 2020. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7202792/ (accessed on 27 May 2021).
- Atabati, E.; Dehghani-Samani, A.; Mortazavimoghaddam, S.G. Association of COVID-19 and other viral infections with interstitial lung diseases, pulmonary fibrosis, and pulmonary hypertension: A narrative review. Can. J. Respir. Ther. 2020, 56, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Myall, K.J.; Mukherjee, B.; Castanheira, A.M.; Lam, J.L.; Benedetti, G.; Mak, S.M.; Preston, R.; Thillai, M.; Dewar, A.; Molyneaux, P.L.; et al. Persistent Post-COVID-19 Interstitial Lung Disease. An Observational Study of Corticosteroid Treatment. Ann. Am. Thorac. Soc. 2021, 18, 799–806. [Google Scholar] [CrossRef]
- Gagiannis, D.; Steinestel, J.; Hackenbroch, C.; Schreiner, B.; Hannemann, M.; Bloch, W.; Umathum, V.G.; Gebauer, N.; Rother, C.; Stahl, M.; et al. Clinical, Serological, and Histopathological Similarities Between Severe COVID-19 and Acute Exacerbation of Connective Tissue Disease-Associated Interstitial Lung Disease (CTD-ILD). Front. Immunol. 2020, 11, 587517. [Google Scholar] [CrossRef] [PubMed]
- Polak, S.B.; Van Gool, I.C.; Cohen, D.; Von Der Thüsen, J.H.; Van Paassen, J. A systematic review of pathological findings in COVID-19: A pathophysiological timeline and possible mechanisms of disease progression. Mod. Pathol. 2020, 33, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, J.; Tan, K.; Yang, W.; Zhao, H.; Wang, G. Discharge may not be the end of treatment: Pay attention to pulmonary fibrosis caused by severe COVID-19. J. Med. Virol. 2021, 93, 1378–1386. [Google Scholar] [CrossRef]
- Yanhong, R.; Shiyao, W.; Min, L.; Youmin, G.; Huaping, D. When COVID-19 encounters interstitial lung disease: Challenges and management. Chin. J. Tuberc. Respir. Dis. Chin. Med. J. 2020, 43, E039. [Google Scholar]
- Hui, D.S.; Joynt, G.; Wong, K.T.; Gomersall, C.; Li, T.S.; Antonio, G.; Ko, F.W.S.; Chan, M.C.; Chan, D.P.; Tong, M.W.; et al. Impact of severe acute respiratory syndrome (SARS) on pulmonary function, functional capacity and quality of life in a cohort of survivors. Thorax 2005, 60, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Antonio, G.E.; Wong, K.T.; Hui, D.; Wu, A.; Lee, N.; Yuen, E.H.Y.; Leung, C.B.; Rainer, T.; Cameron, P.; Chung, S.S.C.; et al. Thin-Section CT in Patients with Severe Acute Respiratory Syndrome Following Hospital Discharge: Preliminary Experience. Radiology 2003, 228, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.-C.; Ng, A.W.-K.; Lee, L.S.-U.; Kaw, G.; Kwek, S.-K.; Leow, M.K.-S.; Earnest, A. 1-Year Pulmonary Function and Health Status in Survivors of Severe Acute Respiratory Syndrome. Chest 2005, 128, 1393–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.M.; Lee, E.Y.; Singh, R.; Enani, M.A.; Al Dossari, K.; Van Gorkom, K.; Larsson, S.G.; Langer, R.D. Follow-up chest radiographic findings in patients with MERS-CoV after recovery. Indian J. Radiol. Imaging 2017, 27, 342–349. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Qiao, W.; Zhang, J.; Qi, Z. Baricitinib, a drug with potential effect to prevent SARS-COV-2 from entering target cells and control cytokine storm induced by COVID-19. Int. Immunopharmacol. 2020, 86, 106749. [Google Scholar] [CrossRef]
- Seif, F.; Aazami, H.; Khoshmirsafa, M.; Kamali, M.; Mohsenzadegan, M.; Pornour, M.; Mansouri, D. JAK Inhibition as a New Treatment Strategy for Patients with COVID-19. Int. Arch. Allergy Immunol. 2020, 181, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, M.N.; Aydemir, H.B.; Korkmaz, E.M.; Budak, M.; Cekin, N.; Pinarbasi, E. Computationally predicted SARS-COV-2 encoded microRNAs target NFKB, JAK/STAT and TGFB signaling pathways. Gene Rep. 2021, 22, 101012. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Lu, S.; Yu, M.; Zhu, L.; Zhu, C.; Li, C.; Fang, J.; Zhu, X.; Wang, X. The potential involvement of JAK-STAT signaling pathway in the COVID-19 infection assisted by ACE2. Gene 2021, 768, 145325. [Google Scholar] [CrossRef]
- Meletiadis, J.; Tsiodras, S.; Tsirigotis, P. Interleukin-6 Blocking vs. JAK-STAT Inhibition for Prevention of Lung Injury in Patients with COVID-19. Infect. Dis. Ther. 2020, 9, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Huang, L.; Wang, N.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020, 146, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Novartis Provides Update on RUXCOVID Study of Ruxolitinib for Hospitalized Patients with COVID-19 [Internet]. Novartis. Available online: https://www.novartis.com/news/media-releases/novartis-provides-update-ruxcovid-study-ruxolitinib-hospitalized-patients-covid-19 (accessed on 19 March 2021).
- Cantini, F.; Niccoli, L.; Matarrese, D.; Nicastri, E.; Stobbione, P.; Goletti, D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 2020, 81, 318–356. [Google Scholar] [CrossRef]
- Bronte, V.; Ugel, S.; Tinazzi, E.; Vella, A.; De Sanctis, F.; Canè, S.; Batani, V.; Trovato, R.; Fiore, A.; Petrova, V.; et al. Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J. Clin. Investig. 2020, 130, 6409–6416. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, Y.-X.; Jiang, L.-J.; Chen, Q.; Wang, T.; Ye, D.-W. Targeting JAK-STAT Signaling to Control Cytokine Release Syndrome in COVID-19. Trends Pharmacol. Sci. 2020, 41, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Lechowicz, K.; Drożdżal, S.; Machaj, F.; Rosik, J.; Szostak, B.; Zegan-Barańska, M.; Biernawska, J.; Dabrowski, W.; Rotter, I.; Kotfis, K. COVID-19: The Potential Treatment of Pulmonary Fibrosis Associated with SARS-CoV-2 Infection. J. Clin. Med. 2020, 9, 1917. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, B.J.; Yang, J.C.; Wang, M.Y.; Chen, C.; Luo, G.X.; He, W.F. Research advances in the mechanism of pulmonary fibrosis induced by coronavirus disease 2019 and the corresponding therapeutic measures. Zhonghua Shao Shang Za Zhi 2020, 36, 691–697. [Google Scholar] [PubMed]


| Major Idiopathic Interstitial Pneumonias | Histologic Findings | Radiologic Pattern |
|---|---|---|
| Idiopathic pulmonary fibrosis (IPF) | Heterogeneous areas of patchy lung fibrosis and UIP. [3] | Basal and peripheral reticular opacities with honeycombing and traction bronchiectasis. [6] |
| Idiopathic nonspecific interstitial pneumonia (NSIP) | Symmetric and homogeneous UIP. | Patchy ground-glass opacities and scattered micronodules. [7] |
| Respiratory bronchiolitis–interstitial lung disease (RB–ILD) | Alveolar macrophages within the bronchioles. | Centrilobular nodules. Central and peripheral bronchial wall thickening. [8] |
| Desquamative interstitial pneumonia (DIP) | Alveolar spaces with macrophages and desquamated alveolar cells. | Extensive and diffuse ground-glass opacities with peripheral and lower lobe predominance. [3] |
| Cryptogenic organizing pneumonia (COP) | Tissue polyps within the alveolar ducts and alveoli, with preservation of the lung architecture. | Patchy peripheral or peribronchial consolidations predominant in the lower lung lobes and multiple nodules. [9] |
| Acute interstitial pneumonia (AIP) | Diffuse alveolar damage. | Extensive ground-glass opacities and areas of consolidation. [10] |
| JAK/STAT-Induced Pathway | ILD, ILD Animal Model and/or Cell Type |
|---|---|
| IL-4 → JAK1/JAK3 → STAT6 | AECs from BLM-induced fibrosis mouse models [20,21] |
| IL-13 → JAK1/TYK2 → STAT6 | AECs from BLM-induced fibrosis mouse model [20,21] |
| IL-6 → STAT3 | IPF fibroblasts, normal lung fibroblasts and BLM-induced mouse model [22,23,24] |
| IL-11 → STAT3 | BLM-induced fibrosis mouse model, human lung fibroblasts [25,26,27] |
| IL-31 → STAT1 | Mouse pulmonary fibrosis [28] |
| TGF-β1 → STAT3 | Human fibroblasts, AECs, SS mouse model [29,30] |
| TGF-β1 → JAK2 → STAT3 | IPF AECs and fibroblasts, SS [31,32] |
| EGF → STAT3 | IPF, COP, NSIP [33,34] |
| PDGF → STAT1 | ILDs alveolar macrophages, IPF BALF [35,36,37] |
| FGF → STAT1/STAT3/STAT5 | Progressive fibrosing ILD [38,39] |
| VEGF → STAT3 | IPF, Idiopathic interstitial pneumonia [40,41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montero, P.; Milara, J.; Roger, I.; Cortijo, J. Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6211. https://doi.org/10.3390/ijms22126211
Montero P, Milara J, Roger I, Cortijo J. Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms. International Journal of Molecular Sciences. 2021; 22(12):6211. https://doi.org/10.3390/ijms22126211
Chicago/Turabian StyleMontero, Paula, Javier Milara, Inés Roger, and Julio Cortijo. 2021. "Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms" International Journal of Molecular Sciences 22, no. 12: 6211. https://doi.org/10.3390/ijms22126211
APA StyleMontero, P., Milara, J., Roger, I., & Cortijo, J. (2021). Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms. International Journal of Molecular Sciences, 22(12), 6211. https://doi.org/10.3390/ijms22126211