
Nucleotide Excision Repair: From Molecular Defects to Neurological Abnormalities



Abstract
:1. Introduction
2. Nucleotide Excision Repair
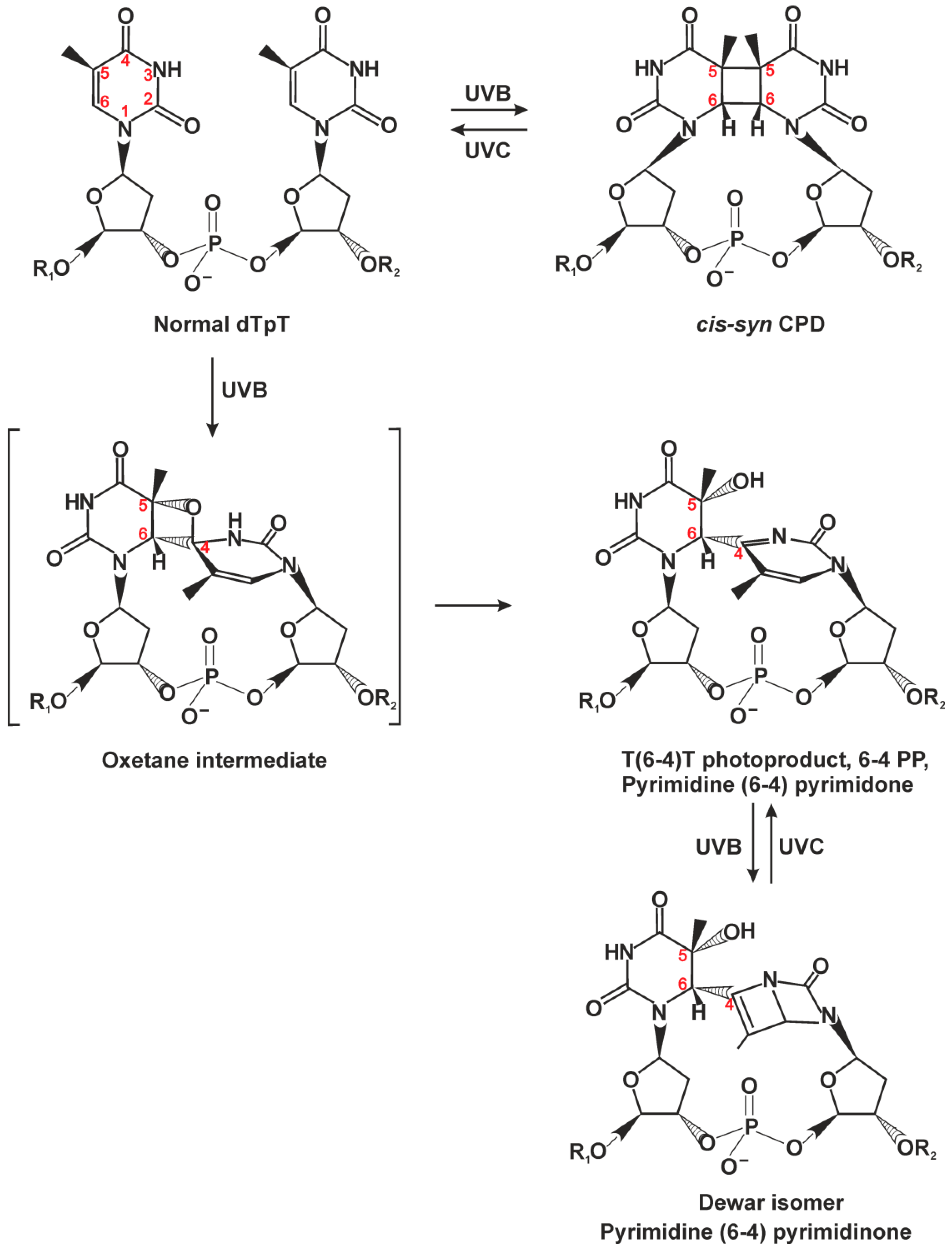
2.1. Classic NER Substrates
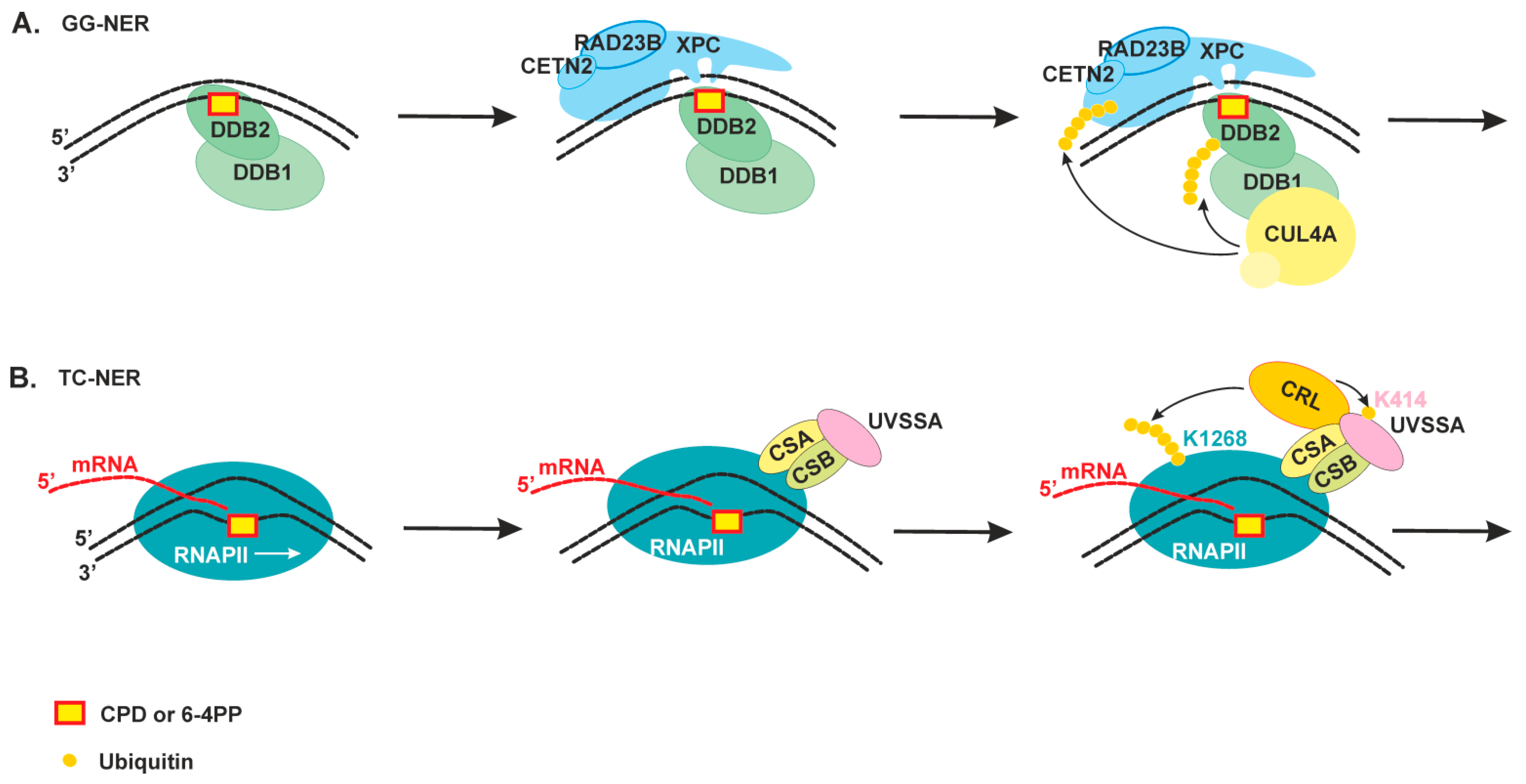
2.2. The Damage Recognition Step
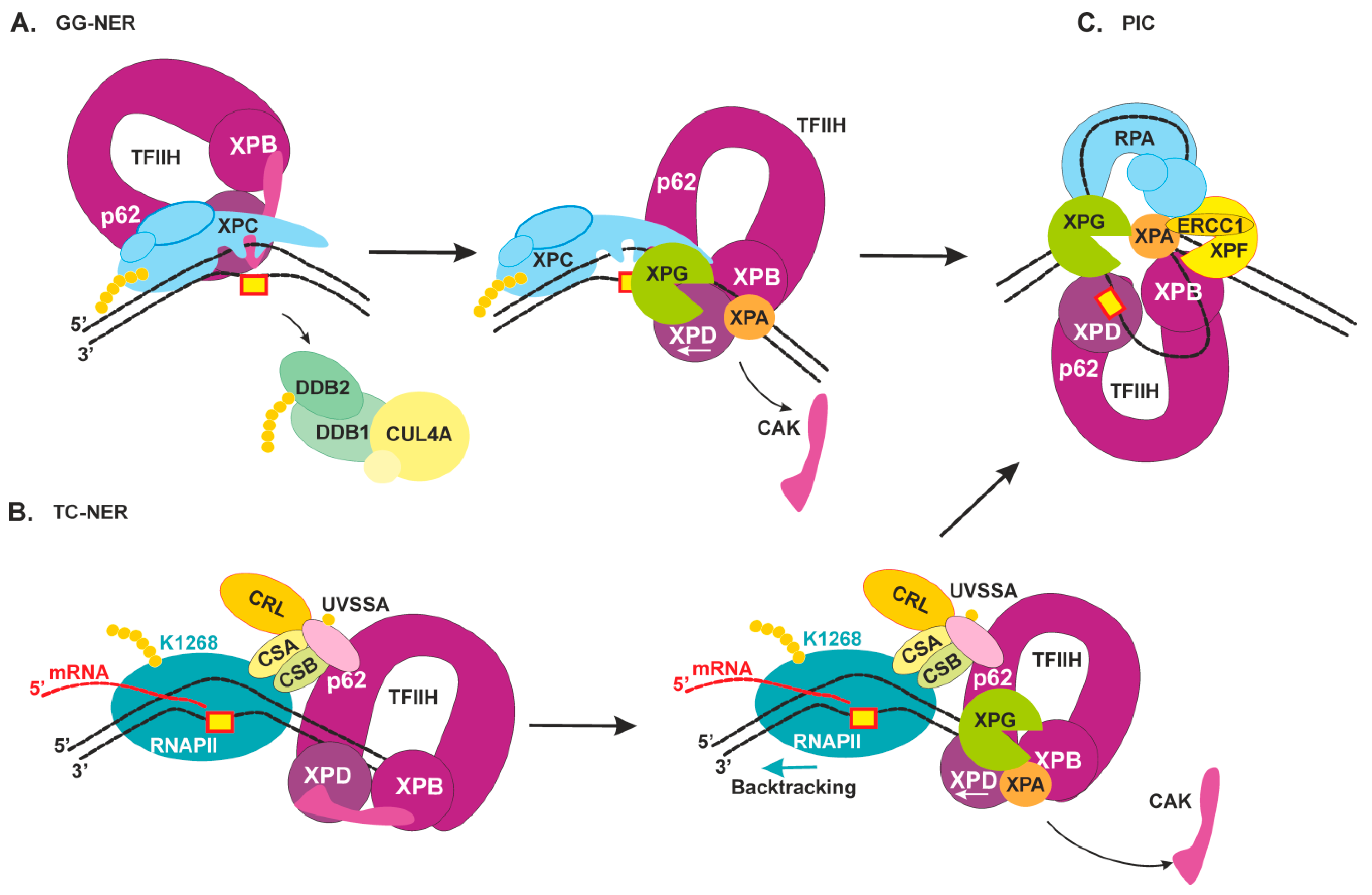
2.3. Damage Verification and Pre-Incision Complex Formation
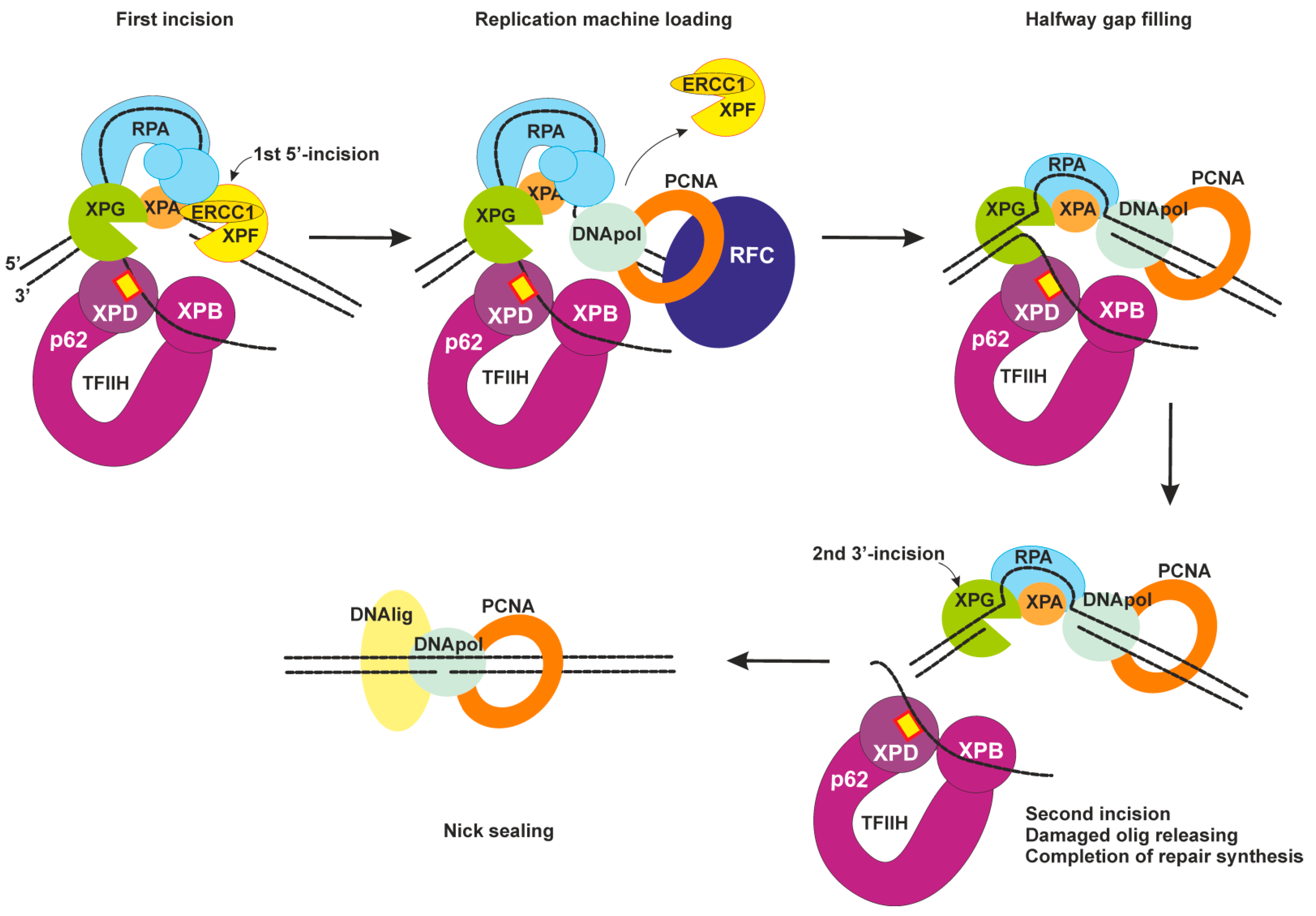
2.4. Dual Incision, Resynthesis, and Ligation
3. The Molecular Basis of Xeroderma Pigmentosum
4. XP Neurological Disease
5. Neurological Abnormalities Due to High Oxidative DNA Damage in Neurons
5.1. NER Impact in Oxidative Lesions Repair
5.2. Cyclopurines Are Bulky Lesions and Exclusive NER Substrates
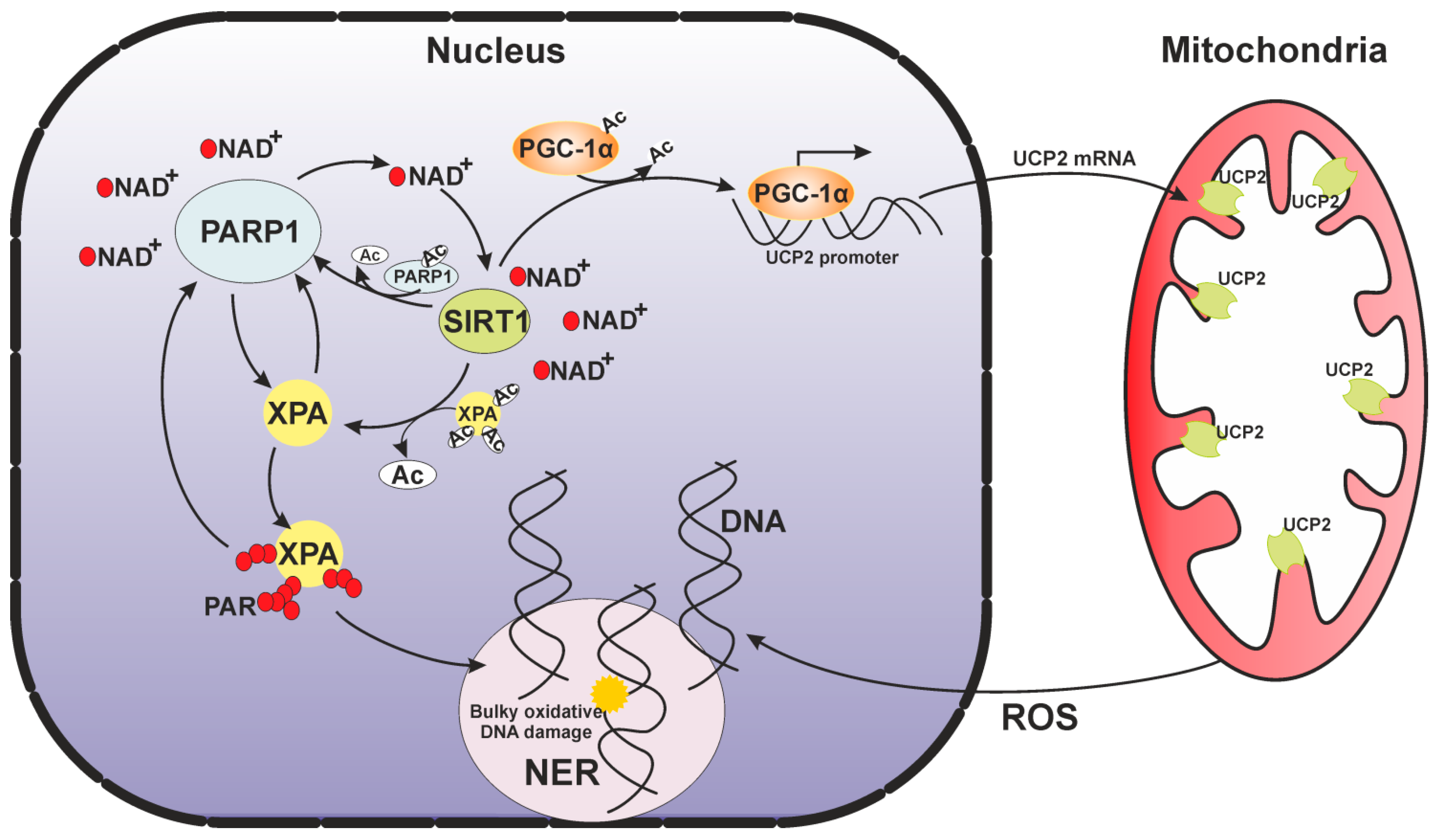
6. A Mitochondrial Echo of the Nuclear DNA Repair Deficiency
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, N.; Moreno, N.C.; Feltes, B.C.; Menck, C.F.; Houten, B.V. Cooperation and interplay between base and nucleotide excision repair pathways: From DNA lesions to proteins. Genet Mol. Biol. 2020, 43 (Suppl. 1), e20190104. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res. 2020, 48, 11227–11243. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair (Amst.) 2017, 56, 51–64. [Google Scholar] [CrossRef]
- Kusakabe, M.; Onishi, Y.; Tada, H.; Kurihara, F.; Kusao, K.; Furukawa, M.; Iwai, S.; Yokoi, M.; Sakai, W.; Sugasawa, K. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ. 2019, 41, 2. [Google Scholar] [CrossRef] [Green Version]
- Mu, H.; Geacintov, N.E.; Broyde, S.; Yeo, J.E.; Schärer, O.D. Molecular basis for damage recognition and verification by XPC-RAD23B and TFIIH in nucleotide excision repair. DNA Repair (Amst.) 2018, 71, 33–42. [Google Scholar] [CrossRef]
- Gillet, L.C.; Scharer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef]
- Son, K.; Schärer, O.D. Repair, Removal, and Shutdown: It All Hinges on RNA Polymerase II Ubiquitylation. Cell 2020, 180, 1039–1041. [Google Scholar] [CrossRef]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef]
- Rapin, I.; Lindenbaum, Y.; Dickson, D.W.; Kraemer, K.H.; Robbins, J.H. Cockayne syndrome and xeroderma pigmentosum. Neurology 2000, 55, 1442–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a light on xeroderma pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Fassihi, H.; Sethi, M.; Fawcett, H.; Wing, J.; Chandler, N.; Mohammed, S.; Craythorne, E.; Morley, A.M.; Lim, R.; Turner, S.; et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc. Natl. Acad. Sci. USA 2016, 113, E1236–E1245. [Google Scholar] [CrossRef] [Green Version]
- Bradford, P.T.; Goldstein, A.M.; Tamura, D.; Khan, S.G.; Ueda, T.; Boyle, J.; Oh, K.S.; Imoto, K.; Inui, H.; Moriwaki, S.; et al. Cancer and neurologic degeneration in xeroderma pigmentosum: Long term follow-up characterises the role of DNA repair. J. Med. Genet. 2011, 48, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. The Case for 8, 5′-Cyclopurine-2′-Deoxynucleosides as Endogenous DNA Lesions That Cause Neurodegeneration in Xeroderma Pigmentosum. Neuroscience 2007, 145, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- Okur, M.N.; Fang, E.F.; Fivenson, E.M.; Tiwari, V.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD+ signaling. Aging Cell 2020, 19, e13268. [Google Scholar] [CrossRef]
- Tiwari, V.; Baptiste, B.A.; Okur, M.N.; Bohr, V.A. Current and emerging roles of Cockayne syndrome group B (CSB) protein. Nucleic Acids Res. 2021, 49, 2418–2434. [Google Scholar] [CrossRef]
- Saki, M.; Prakash, A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Biol. Med. 2017, 107, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell Mol. Life Sci. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Bohr, V.A. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy 2014, 10, 1468–1469. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.; Baptiste, B.A.; Kim, E.; Hussain, M.; Croteau, D.L.; Bohr, V.A. DNA damage and mitochondria in cancer and aging. Carcinogenesis 2020, 41, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Mizutani, R. Structural biology of DNA (6-4) photoproducts formed by ultraviolet radiation and interactions with their binding proteins. Int. J. Mol. Sci. 2014, 15, 20321–20338. [Google Scholar] [CrossRef] [Green Version]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Kim, J.K.; Patel, D.; Choi, B.S. Contrasting structural impacts induced by cis-syn cyclobutane dimer and (6-4) adduct in DNA duplex decamers: Implication in mutagenesis and repair activity. Photochem. Photobiol. 1995, 62, 44–50. [Google Scholar] [CrossRef]
- Reardon, J.T.; Sancar, A. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 2003, 17, 2539–2551. [Google Scholar] [CrossRef] [Green Version]
- Krasikova, Y.S.; Rechkunova, N.I.; Maltseva, E.A.; Craescu, C.T.; Petruseva, I.O.; Lavrik, O.I. Influence of centrin 2 on the interaction of nucleotide excision repair factors with damaged DNA. Biochemistry 2012, 77, 346–353. [Google Scholar] [CrossRef]
- Min, J.H.; Pavletich, N.P. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 2007, 449, 570–575. [Google Scholar] [CrossRef]
- Paul, D.; Mu, H.; Zhao, H.; Ouerfelli, O.; Jeffrey, P.D.; Broyde, S.; Min, J.H. Structure and mechanism of pyrimidine-pyrimidone (6-4) photoproduct recognition by the Rad4/XPC nucleotide excision repair complex. Nucleic Acids Res. 2019, 47, 6015–6028. [Google Scholar] [CrossRef] [Green Version]
- Cheon, N.Y.; Kim, H.S.; Yeo, J.E.; Scharer, O.D.; Lee, J.Y. Single-molecule visualization reveals the damage search mechanism for the human NER protein XPC-RAD23B. Nucleic Acids Res. 2019, 47, 8337–8347. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Mu, H.; Tavakoli, A.; Dai, Q.; Chen, X.; Chakraborty, S.; He, C.; Ansari, A.; Broyde, S.; Min, J.H. Tethering-facilitated DNA’ opening’ and complementary roles of β-hairpin motifs in the Rad4/XPC DNA damage sensor protein. Nucleic Acids Res. 2020, 48, 12348–12364. [Google Scholar] [CrossRef] [PubMed]
- Buterin, T.; Meyer, C.; Giese, B.; Naegeli, H. DNA quality control by conformational readout on the undamaged strand of the double helix. Chem. Biol. 2005, 12, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Lukyanchikova, N.V.; Petruseva, I.O.; Evdokimov, A.N.; Koroleva, L.S.; Lavrik, O.I. DNA Bearing Bulky Fluorescent and Photoreactive Damage in Both Strands as Substrates of the Nucleotide Excision Repair System. Mol. Biol. (Mosk.) 2018, 52, 277–288. [Google Scholar] [CrossRef]
- Ribeiro-Silva, C.; Sabatella, M.; Helfricht, A.; Marteijn, J.A.; Theil, A.F.; Vermeulen, W.; Lans, H. Ubiquitin and TFIIH-stimulated DDB2 dissociation drives DNA damage handover in nucleotide excision repair. Nat. Commun. 2020, 11, 4868. [Google Scholar] [CrossRef]
- Sugasawa, K.; Okuda, Y.; Saijo, M.; Nishi, R.; Matsuda, N.; Chu, G.; Mori, T.; Iwai, S.; Tanaka, K.; Tanaka, K.; et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005, 121, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, Y.; Hara, Y.; Oka, Y.; Komine, O.; van den Heuvel, D.; Guo, C.; Daigaku, Y.; Isono, M.; He, Y.; Shimada, M.; et al. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180, 1228–1244.e24. [Google Scholar] [CrossRef]
- Rechkunova, N.I.; Maltseva, E.A.; Lavrik, O.I. Post-translational Modifications of Nucleotide Excision Repair Proteins and Their Role in the DNA Repair. Biochemistry 2019, 84, 1008–1020. [Google Scholar] [CrossRef]
- van der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; González-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Tufegdžić Vidaković, A.; Mitter, R.; Kelly, G.P.; Neumann, M.; Harreman, M.; Rodríguez-Martínez, M.; Herlihy, A.; Weems, J.C.; Boeing, S.; Encheva, V.; et al. Regulation of the RNAPII Pool Is Integral to the DNA Damage Response. Cell 2020, 180, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [Green Version]
- Shivji, M.K.K.; Renaudin, X.; Williams, Ç.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutakawa, S.E.; Tsai, C.L.; Yan, C.; Bralić, A.; Chazin, W.J.; Hamdan, S.M.; Schärer, O.D.; Ivanov, I.; Tainer, J.A. Envisioning how the prototypic molecular machine TFIIH functions in transcription initiation and DNA repair. DNA Repair (Amst.) 2020, 96, 102972. [Google Scholar] [CrossRef] [PubMed]
- Schilbach, S.; Hantsche, M.; Tegunov, D.; Dienemann, C.; Wigge, C.; Urlaub, H.; Cramer, P. Structures of transcription pre-initiation complex with TFIIH and Mediator. Nature 2017, 551, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Greber, B.J.; Toso, D.B.; Fang, J.; Nogales, E. The complete structure of the human TFIIH core complex. eLife 2019, 8, e44771. [Google Scholar] [CrossRef]
- He, Y.; Yan, C.; Fang, J.; Inouye, C.; Tjian, R.; Ivanov, I.; Nogales, E. Near-atomic resolution visualization of human transcription promoter opening. Nature 2016, 533, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Kokic, G.; Chernev, A.; Tegunov, D.; Dienemann, C.; Urlaub, H.; Cramer, P. Structural basis of TFIIH activation for nucleotide excision repair. Nat. Commun. 2019, 10, 2885. [Google Scholar] [CrossRef] [Green Version]
- Li, C.L.; Golebiowski, F.M.; Onishi, Y.; Samara, N.L.; Sugasawa, K.; Yang, W. Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Mol. Cell 2015, 59, 1025–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasikova, Y.S.; Rechkunova, N.I.; Maltseva, E.A.; Petruseva, I.O.; Lavrik, O.I. Localization of xeroderma pigmentosum group A protein and replication protein A on damaged DNA in nucleotide excision repair. Nucleic Acids Res. 2010, 38, 8083–8094. [Google Scholar] [CrossRef] [Green Version]
- Sugitani, N.; Sivley, R.M.; Perry, K.E.; Capra, J.A.; Chazin, W.J. XPA: A key scaffold for human nucleotide excision repair. DNA Repair (Amst.) 2016, 44, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckwitt, E.C.; Jang, S.; Carnaval Detweiler, I.; Kuper, J.; Sauer, F.; Simon, N.; Bretzler, J.; Watkins, S.C.; Carell, T.; Kisker, C.; et al. Single molecule analysis reveals monomeric XPA bends DNA and undergoes episodic linear diffusion during damage search. Nat. Commun. 2020, 11, 1356. [Google Scholar] [CrossRef]
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef]
- Krasikova, Y.S.; Rechkunova, N.I.; Maltseva, E.A.; Lavrik, O.I. RPA and XPA interaction with DNA structures mimicking intermediates of the late stages in nucleotide excision repair. PLoS ONE 2018, 13, e0190782. [Google Scholar] [CrossRef] [Green Version]
- Topolska-Woś, A.M.; Sugitani, N.; Cordoba, J.J.; Le Meur, K.V.; Le Meur, R.A.; Kim, H.S.; Yeo, J.E.; Rosenberg, D.; Hammel, M.; Schärer, O.D.; et al. A key interaction with RPA orients XPA in NER complexes. Nucleic Acids Res. 2020, 48, 2173–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Pavletich, N.P. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012, 26, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Schärer, O.D.; Wagner, G.; Ellenberger, T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007, 26, 4768–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagbemi, A.F.; Orelli, B.; Schärer, O.D. Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair (Amst.) 2011, 10, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Schärer, O.D. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009, 28, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overmeer, R.M.; Moser, J.; Volker, M.; Kool, H.; Tomkinson, A.E.; van Zeeland, A.A.; Mullenders, L.H.; Fousteri, M. Replication protein A safeguards genome integrity by controlling NER incision events. J. Cell Biol. 2011, 192, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilljam, K.M.; Müller, R.; Liabakk, N.B.; Otterlei, M. Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PLoS ONE 2012, 7, e49199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.L.; Kang, M.S.; Kim, H.Y.; Gorospe, C.M.; Kim, T.S.; Lee, S.K. The PCNA binding domain of Rad2p plays a role in mutagenesis by modulating the cell cycle in response to DNA damage. DNA Repair (Amst.) 2014, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kemp, M.G.; Reardon, J.T.; Lindsey-Boltz, L.A.; Sancar, A. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 2012, 287, 22889–22899. [Google Scholar] [CrossRef] [Green Version]
- Sertic, S.; Mollica, A.; Campus, I.; Roma, S.; Tumini, E.; Aguilera, A.; Muzi-Falconi, M. Coordinated Activity of Y Family TLS Polymerases and EXO1 Protects Non-S Phase Cells from UV-Induced Cytotoxic Lesions. Mol. Cell 2018, 70, 34–47.e4. [Google Scholar] [CrossRef] [Green Version]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Ramkumar, H.L.; Brooks, B.P.; Cao, X.; Tamura, D.; Digiovanna, J.J.; Kraemer, K.H.; Chan, C.C. Ophthalmic manifestations and histopathology of xeroderma pigmentosum: Two clinicopathological cases and a review of the literature. Surv. Ophthalmol. 2011, 56, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, A.R.; McGibbon, D.; Stefanini, M. Xeroderma pigmentosum. Orphanet J. Rare Dis. 2011, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeti, R.; Zeitlberger, A.; Peelo, C.; Fassihi, H.; Sarkany, R.P.E.; Lehmann, A.R.; Giunti, P. Xeroderma pigmentosum: Overview of pharmacology and novel therapeutic strategies for neurological symptoms. Br. J. Pharmacol. 2019, 176, 4293–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, J.T.; Bessho, T.; Kung, H.C.; Bolton, P.H.; Sancar, A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. USA 1997, 94, 9463–9468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouspikel, T.; Hanawalt, P.C. Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol. Cell Biol. 2000, 20, 1562–1570. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Nakamura, Y.; Kobayashi, N.; Iwamoto, T.; Yoshioka, A.; Kuniyasu, H.; Kishimoto, T.; Mori, T. Neurons and astrocytes exhibit lower activities of global genome nucleotide excision repair than do fibroblasts. DNA Repair (Amst.) 2007, 6, 649–657. [Google Scholar] [CrossRef]
- Laugel, V.; Dalloz, C.; Durand, M.; Sauvanaud, F.; Kristensen, U.; Vincent, M.C.; Pasquier, L.; Odent, S.; Cormier-Daire, V.; Gener, B.; et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2010, 31, 113–126. [Google Scholar] [CrossRef]
- Weidenheim, K.M.; Dickson, D.W.; Rapin, I. Neuropathology of Cockayne syndrome: Evidence for impaired development, premature aging, and neurodegeneration. Mech. Ageing Dev. 2009, 130, 619–636. [Google Scholar] [CrossRef]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Cascella, M.; Di Napoli, R.; Carbone, D.; Cuomo, G.F.; Bimonte, S.; Muzio, M.R. Chemotherapy-related cognitive impairment: Mechanisms, clinical features and research perspectives. Recenti Prog. Med. 2018, 109, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Lomeli, N.; Lepe, J.; Gupta, K.; Bota, D.A. Cognitive complications of cancer and cancer-related treatments—Novel paradigms. Neurosci. Lett. 2021, 749, 135720. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Nakane, H.; Iwamoto, T.; Krokidis, M.G.; Chatgilialoglu, C.; Tanaka, K.; Kaidoh, T.; Hasegawa, M.; Sugiura, S. High levels of oxidatively generated DNA damage 8,5’-cyclo-2’-deoxyadenosine accumulate in the brain tissues of xeroderma pigmentosum group A gene-knockout mice. DNA Repair (Amst.) 2019, 80, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.L. Oxidatively generated damage to the guanine moiety of DNA: Mechanistic aspects and formation in cells. Acc. Chem. Res. 2008, 41, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Markkanen, E. Not breathing is not an option: How to deal with oxidative DNA damage. DNA Repair (Amst.) 2017, 59, 82–105. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian DNA base excision repair: Dancing in the moonlight. DNA Repair (Amst.) 2020, 93, 102921. [Google Scholar] [CrossRef]
- Moor, N.A.; Lavrik, O.I. Coordination of DNA base excision repair by protein-protein interactions. In DNA Repair. An Update; Mognato, M., Ed.; IntechOpen: London, UK, 2019; pp. 184–217. [Google Scholar] [CrossRef] [Green Version]
- Lavrik, O.I. PARPs’ impact on base excision DNA repair. DNA Repair (Amst.) 2020, 93, 102911. [Google Scholar] [CrossRef]
- Rechkunova, N.I.; Krasikova, Y.S.; Lavrik, O.I. Interactome of Base and Nucleotide Excision DNA repair systems. Mol. Biol. 2021, 55, 181–193. [Google Scholar] [CrossRef]
- Tuo, J.; Chen, C.; Zeng, X.; Christiansen, M.; Bohr, V.A. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair (Amst.) 2002, 1, 913–927. [Google Scholar] [CrossRef]
- Muftuoglu, M.; de Souza-Pinto, N.C.; Dogan, A.; Aamann, M.; Stevnsner, T.; Rybanska, I.; Kirkali, G.; Dizdaroglu, M.; Bohr, V.A. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J. Biol. Chem. 2009, 284, 9270–9279. [Google Scholar] [CrossRef] [Green Version]
- Aamann, M.D.; Hvitby, C.; Popuri, V.; Muftuoglu, M.; Lemminger, L.; Skeby, C.K.; Keijzers, G.; Ahn, B.; Bjoras, M.; Bohr, V.A.; et al. Cockayne syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech. Ageing Dev. 2014, 135, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.K.; Muftuoglu, M.; Beck, G.; Imam, S.Z.; Bohr, V.A.; Wilson, D.M., 3rd. Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007, 35, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Klungland, A.; Höss, M.; Gunz, D.; Constantinou, A.; Clarkson, S.G.; Doetsch, P.W.; Bolton, P.H.; Wood, R.D.; Lindahl, T. Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell 1999, 3, 33–42. [Google Scholar] [CrossRef]
- Bessho, T. Nucleotide excision repair 3’ endonuclease XPG stimulates the activity of base excision repairenzyme thymine glycol DNA glycosylase. Nucleic Acids Res. 1999, 27, 979–983. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Kumar, N.; Beckwitt, E.C.; Kong, M.; Fouquerel, E.; Rapić-Otrin, V.; Prasad, R.; Watkins, S.C.; Khuu, C.; Majumdar, C.; et al. Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 2019, 26, 695–703. [Google Scholar] [CrossRef]
- Menoni, H.; Wienholz, F.; Theil, A.F.; Janssens, R.C.; Lans, H.; Campalans, A.; Radicella, J.P.; Marteijn, J.A.; Vermeulen, W. The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucleic Acids Res. 2018, 46, 7747–7756. [Google Scholar] [CrossRef] [Green Version]
- de Melo, J.T.; de Souza Timoteo, A.R.; Lajus, T.B.; Brandão, J.A.; de Souza-Pinto, N.C.; Menck, C.F.; Campalans, A.; Radicella, J.P.; Vessoni, A.T.; Muotri, A.R.; et al. XPC deficiency is related to APE1 and OGG1 expression and function. Mutat. Res. 2016, 784–785, 25–33. [Google Scholar] [CrossRef]
- Shafirovich, V.; Kropachev, K.; Kolbanovskiy, M.; Geacintov, N.E. Excision of oxidatively generated guanine lesions by competing base and nucleotide excision repair mechanisms in human cells. Chem. Res. Tox. 2019, 32, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Shafirovich, V.; Kropachev, K.; Anderson, T.; Liu, Z.; Kolbanovskiy, M.; Martin, B.D.; Sugden, K.; Shim, Y.; Chen, X.; Min, J.H.; et al. Base and nucleotide excision repair of oxidatively generated guanine lesions in DNA. J. Biol. Chem. 2016, 291, 5309–5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolbanovskiy, M.; Aharonoff, A.; Sales, A.H.; Geacintov, N.E.; Shafirovich, V. Base and Nucleotide Excision Repair Pathways in DNA Plasmids Harboring Oxidatively Generated Guanine Lesions. Chem. Res. Toxicol. 2021, 34, 154–160. [Google Scholar] [CrossRef]
- Kolbanovskiy, M.; Shim, Y.; Min, J.H.; Geacintov, N.E.; Shafirovich, V. Inhibition of Excision of Oxidatively Generated Hydantoin DNA Lesions by NEIL1 by the Competitive Binding of the Nucleotide Excision Repair Factor XPC-RAD23B. Biochemistry 2020, 59, 1728–1736. [Google Scholar] [CrossRef]
- D’Errico, M.; Parlanti, E.; Teson, M.; de Jesus, B.M.; Degan, P.; Calcagnile, A.; Jaruga, P.; Bjørås, M.; Crescenzi, M.; Pedrini, A.M.; et al. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006, 25, 4305–4315. [Google Scholar] [CrossRef] [Green Version]
- Kassam, S.N.; Rainbow, A.J. Deficient base excision repair of oxidative DNA damage induced by methylene blue plus visible light in xeroderma pigmentosum group C fibroblasts. Biochem. Biophys. Res. Commun. 2007, 359, 1004–1009. [Google Scholar] [CrossRef]
- Shimizu, Y.; Uchimura, Y.; Dohmae, N.; Saitoh, H.; Hanaoka, F.; Sugasawa, K. Stimulation of DNA Glycosylase Activities by XPC Protein Complex: Roles of Protein-Protein Interactions. J. Nucleic Acids 2010, 2010, 805698. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Hanawalt, P.C.; Spivak, G. Comet-FISH with strand-specific probes reveals transcription-coupled repair of 8-oxoGuanine in human cells. Nucleic Acids Res. 2013, 41, 7700–7712. [Google Scholar] [CrossRef] [Green Version]
- Kuraoka, I.; Bender, C.; Romieu, A.; Cadet, J.; Wood, R.D.; Lindahl, T. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3832–3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.J.; Wise, D.S.; Berry, D.A.; Kosmoski, J.V.; Smerdon, M.J.; Somers, R.L.; Mackie, H.; Spoonde, A.Y.; Ackerman, E.J.; Coleman, K.; et al. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 2000, 275, 22355–22362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatgilialoglu, C.; Ferreri, C.; Terzidis, M.A. Purine 5’,8-cyclonucleoside lesions: Chemistry and biology. Chem. Soc. Rev. 2011, 40, 1368–1382. [Google Scholar] [CrossRef]
- Iwamoto, T.; Brooks, P.J.; Nishiwaki, T.; Nishimura, K.; Kobayashi, N.; Sugiura, S.; Mori, T. Quantitative and in situ detection of oxidatively generated DNA damage 8,5′-cyclo-2′-deoxyadenosine using an immunoassay with a novel monoclonal antibody. Photochem. Photobiol. 2014, 90, 829–836. [Google Scholar]
- Theruvathu, J.A.; Jaruga, P.; Dizdaroglu, M.; Brooks, P.J. The oxidatively-induced DNA lesions 8,5′-cyclo-2′-deoxyadenosine and 8-hydroxy-2′-deoxyadenosine are strongly resistant to acid-induced hydrolysis of the glycosidic bond Mech. Ageing Dev. 2007, 128, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Das, R.S.; Samaraweera, M.; Morton, M.; Gascón, J.A.; Basu, A.K. Stability of N-glycosidic bond of (5’S)-8,5′-cyclo-2′-deoxyguanosine. Chem. Res. Toxicol. 2012, 25, 2451–2461. [Google Scholar] [CrossRef] [Green Version]
- Jaruga, P.; Dizdaroglu, M. 8,5′-Cyclopurine-2′-deoxynucleosides in DNA: Mechanisms of formation, measurement, repair and biological effects. DNA Repair (Amst.) 2008, 7, 1413–1425. [Google Scholar] [PubMed]
- Belmadoui, N.; Boussicault, F.; Guerra, M.; Ravanat, J.L.; Chatgilialoglu, C.; Cadet, J. Radiation-induced formation of purine 5′,8-cyclonucleosides in isolated and cellular DNA: High stereospecificity and modulating effect of oxygen. Org. Biomol. Chem. 2010, 8, 3211–3219. [Google Scholar] [CrossRef]
- Terzidis, M.A.; Ferreri, C.; Chatgilialoglu, C. Radiation-induced formation of purine lesions in single and double stranded DNA: Revised quantification. Front. Chem. 2015, 3, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero, C.R.; Wang, J.; Wang, Y. Induction of 8,5′-cyclo-2′-deoxyadenosine and 8,5′-cyclo-2′-deoxyguanosine in isolated DNA by Fenton-type reagents. Chem. Res. Toxicol. 2013, 26, 1361–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokidis, M.G.; Parlanti, E.; D’Errico, M.; Pascucci, B.; Pino, A.; Alimonti, A.; Pietraforte, D.; Masi, A.; Ferreri, C.; Chatgilialoglu, C. Purine DNA Lesions at Different Oxygen Concentration in DNA Repair-Impaired Human Cells (EUE-siXPA). Cells 2019, 8, 1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokidis, M.G.; D’Errico, M.; Pascucci, B.; Parlanti, E.; Masi, A.; Ferreri, C.; Chatgilialoglu, C. Oxygen-Dependent Accumulation of Purine DNA Lesions in Cockayne Syndrome Cells. Cells 2020, 9, 1671. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Geacintov, N.E.; Krokidis, M.G.; Liu, Y.; Masi, A.; Shafirovich, V.; Terzidis, M.A.; Tsegay, P.S. 5’,8-Cyclopurine Lesions in DNA Damage: Chemical, Analytical, Biological, and Diagnostic Significance. Cells 2019, 8, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Errico, M.; Parlanti, E.; Teson, M.; Degan, P.; Lemma, T.; Calcagnile, A.; Iavarone, I.; Jaruga, P.; Ropolo, M.; Pedrini, A.M.; et al. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene 2007, 26, 4336–4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marietta, C.; Gulam, H.; Brooks, P.J. A single 8,5’-cyclo-2’-deoxyadenosine lesion in a TATA box prevents binding of the TATA binding protein and strongly reduces transcription in vivo. DNA Repair (Amst.) 2002, 1, 967–975. [Google Scholar] [CrossRef]
- Pande, P.; Das, R.S.; Sheppard, C.; Kow, Y.W.; Basu, A.K. Repair efficiency of (5’S)-8,5’-cyclo-2’-deoxyguanosine and (5’S)-8,5’-cyclo-2’-deoxyadenosine depends on the complementary base. DNA Repair (Amst.) 2012, 11, 926–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropachev, K.; Ding, S.; Terzidis, M.A.; Masi, A.; Liu, Z.; Cai, Y.; Kolbanovskiy, M.; Chatgilialoglu, C.; Broyde, S.; Geacintov, N.E.; et al. Structural basis for the recognition of diastereomeric 5’,8-cyclo-2’-deoxypurine lesions by the human nucleotide excision repair system. Nucleic Acids Res. 2014, 42, 5020–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell. 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamohan, S.B.; Pillai, V.B.; Gupta, M.; Sundaresan, N.R.; Birukov, K.G.; Samant, S.; Hottinger, M.O.; Gupta, M.P. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol. Cell Biol. 2009, 29, 4116–4129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Shieh, W.M.; Ame, J.C.; Wilson, M.V.; Wang, Z.Q.; Koh, D.W.; Jacobson, M.K.; Jacobson, E.L. Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J. Biol. Chem. 1998, 273, 30069–30072. [Google Scholar] [CrossRef] [Green Version]
- Krietsch, J.; Rouleau, M.; Pic, E.; Ethier, C.; Dawson, T.M.; Dawson, V.L.; Masson, J.Y.; Poirier, G.G.; Gagne, J.P. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol. Asp. Med. 2013, 34, 1066–1087. [Google Scholar] [CrossRef] [PubMed]
- Kolthur-Seetharam, U.; Dantzer, F.; McBurney, M.W.; de Murcia, G.; Sassone-Corsi, P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle 2006, 5, 873–877. [Google Scholar] [CrossRef] [Green Version]
- Pillai, J.B.; Isbatan, A.; Imai, S.; Gupta, M.P. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 2005, 280, 43121–43130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, S.Y.; Fleuriel, C.; Leprince, D.; Rocheleau, J.V.; Piston, D.W.; Goodman, R.H. Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc. Natl. Acad. Sci. USA 2007, 104, 829–833. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, S.G.; Carter, K.M.; Bautista, R.M.; He, D.; Wang, C.; D’Orazio, J.A. Sirtuin 1-mediated deacetylation of XPA DNA repair protein enhances its interaction with ATR protein and promotes cAMP-induced DNA repair of UV damage. J. Biol. Chem. 2018, 293, 19025–19037. [Google Scholar] [CrossRef] [Green Version]
- King, B.S.; Cooper, K.L.; Liu, K.J.; Hudson, L.G. Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J. Biol. Chem. 2012, 287, 39824–39833. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.M.; Popp, O.; Gebhard, D.; Veith, S.; Fischbach, A.; Beneke, S.; Leitenstorfer, A.; Bergemann, J.; Scheffner, M.; Ferrando-May, E.; et al. Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. FEBS J. 2014, 281, 3625–3641. [Google Scholar] [CrossRef] [PubMed]
- Pleschke, J.M.; Kleczkowska, H.E.; Strohm, M.; Althaus, F.R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000, 275, 40974–40980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.H.; Mu, D.; Reardon, J.T.; Sancar, A. The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J. Biol. Chem. 1995, 270, 4896–4902. [Google Scholar] [CrossRef] [Green Version]
- Wakasugi, M.; Kasashima, H.; Fukase, Y.; Imura, M.; Imai, R.; Yamada, S.; Cleaver, J.E.; Matsunaga, T. Physical and functional interaction between DDB and XPA in nucleotide excision repair. Nucleic Acids Res. 2009, 37, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borszéková Pulzová, L.; Ward, T.A.; Chovanec, M. XPA: DNA Repair Protein of Significant Clinical Importance. Int. J. Mol. Sci. 2020, 21, 2182. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | XP | XP Neurological Disease | XP/CS | CS |
|---|---|---|---|---|
| Molecular defects | XP-A, -B, -C, -D, -E, -F and XP-V | XP-A and XP-D followed by XP-B, XP-G, and XP-F | XP-B, XP-D, XP-G | CS-A, CS-B |
| Skin sun sensitivity | + | + | + | + |
| Increased freckling | + | + | + | - |
| Sunlight—induced skin cancer | + | + | + | - |
| Photophobia | + | + | + | + |
| Anterior eye cancer | + | + | + | - |
| Retinal degeneration | - | - | + | + |
| Sensorineural deafness | - | + | + | + |
| Developmental delay | - | + | + | + |
| Progressive neurological degeneration | - | + | + | + |
| Primary neuronal degeneration | - | + | - | - |
| Demyelination of brain | - | - | + | + |
| Cerebral atrophy | - | + | + | + |
| Cerebellar atrophy | - | + | + | + |
| Calcification (basal ganglia) | - | - | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasikova, Y.; Rechkunova, N.; Lavrik, O. Nucleotide Excision Repair: From Molecular Defects to Neurological Abnormalities. Int. J. Mol. Sci. 2021, 22, 6220. https://doi.org/10.3390/ijms22126220
Krasikova Y, Rechkunova N, Lavrik O. Nucleotide Excision Repair: From Molecular Defects to Neurological Abnormalities. International Journal of Molecular Sciences. 2021; 22(12):6220. https://doi.org/10.3390/ijms22126220
Chicago/Turabian StyleKrasikova, Yuliya, Nadejda Rechkunova, and Olga Lavrik. 2021. "Nucleotide Excision Repair: From Molecular Defects to Neurological Abnormalities" International Journal of Molecular Sciences 22, no. 12: 6220. https://doi.org/10.3390/ijms22126220