Abstract
Hyperhomocysteinemia (HHcy) is remarkably common among the aging population. The relation between HHcy and the development of neurodegenerative diseases, such as Alzheimer’s disease (AD) and eye diseases, and age-related macular degeneration (AMD) and diabetic retinopathy (DR) in elderly people, has been established. Disruption of the blood barrier function of the brain and retina is one of the most important underlying mechanisms associated with HHcy-induced neurodegenerative and retinal disorders. Impairment of the barrier function triggers inflammatory events that worsen disease pathology. Studies have shown that AD patients also suffer from visual impairments. As an extension of the central nervous system, the retina has been suggested as a prominent site of AD pathology. This review highlights inflammation as a possible underlying mechanism of HHcy-induced barrier dysfunction and neurovascular injury in aging diseases accompanied by HHcy, focusing on AD.
1. Introduction
Elevated levels of plasma homocysteine (Hcy), known as hyperhomocysteinemia (HHcy), is a major risk factor for neurodegenerative and cardiovascular disorders [1]. Over the last decade, elevated levels of amino acid Hcy have been frequently reported in patient with aging diseases. This relatively large incidence of HHcy in the elderly population is attributed to lowered nutritional absorption and decreased metabolic function with advanced age [2]. The percentage of the aging population has been increasing in the last 10 years and is expected to continue to grow for another 20 years, and this is attributed to improved life expectancies [3]. Among the most common 10 diseases affecting the aging population over the age of 65 are vision loss disabilities, such as diabetic retinopathy (DR) and age-related macular degeneration (AMD), neurodegenerative diseases, such as Alzheimer’s disease (AD), and osteoarthritis or osteoporosis [3], as shown in Figure 1.
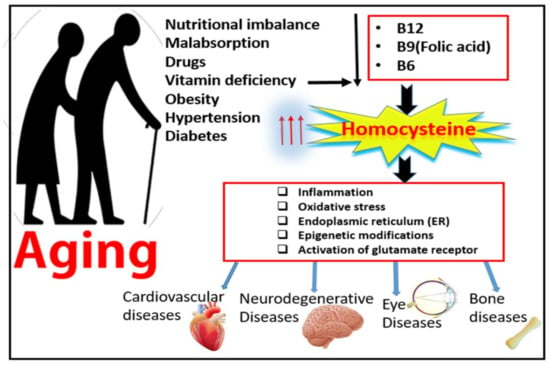
Figure 1.
Effect of aging on homocysteine metabolism and possible mechanisms of hyperhomocysteinemia-associated aging diseases.
Our and others’ work reported that HHcy causes disruption of the blood barrier function in both the brain [4,5,6,7] and retina [8,9,10,11,12,13,14]. The blood–brain barrier (BBB) separates the brain from the circulatory system and is made of tightly packed endothelial cells that line the cerebral vessels, separating blood stream components from the neuronal brain tissue [15]. This barrier is tightly maintained via specialized tight junctions, gap junctions, and adherent junctions and is vital for various features, such as: preventing entry of harmful substances to the neuronal tissue of the brain, performing selective transportation and trafficking of molecules into and out of the brain, and allowing specific ion transporter channels to regulate ionic transporters [16]. Similarly to the BBB, the blood–retinal barrier (BRB) regulates fluids and molecular movement between the ocular vascular and retinal tissues and prevents leakage of macromolecules and other potentially harmful agents into the retina. An intact BRB is vital for retinal structural and functional integrity as it plays an essential role in the maintenance of the retinal neuron microenvironment. The BRB consists of two components, an inner and outer BRB. Vision is negatively affected in clinical situations associated with BRB breakdown, such as DR, in which the inner BRB is altered [17,18,19], and AMD, in which the outer BRB is altered [18,19,20].
The current review emphasizes the involvement of HHcy-induced barrier dysfunction (BBB and BRB) in the development and progression of the most common vision loss diseases (DR and AMD) and the most common neurodegenerative (AD) disease in the elderly population and the possible underlying mechanisms that impair the barrier function. Our previously published work over the last few years has proposed many mechanisms for HHcy-induced barrier dysfunction, such as endoplasmic reticulum (ER) stress [21], activation of oxidative stress [11], induction of epigenetic modifications [9], induction of inflammation [22], and activation of a glutamate receptor, the N-methyl-D-aspartate receptor (NMDAR) [13].
2. Homocysteine and Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of neurodegenerative disease and is the major cause of dementia, accounting for 60–70% of cases [23]. Worldwide, more than 26 million individuals have been diagnosed with AD. As the population ages, the prevalence of AD is expected to increase to effect over 100 million by 2050 [24]. Unfortunately, there is still no available effective treatment for AD, however, controlling the risk factors can still reduce the number of cases and associated cost, especially as AD is a devastating disease for the patients and their families and puts a huge financial burden on the whole of society [25]. The symptoms of this disease may start as simple symptoms, such as early forgetfulness, then deteriorate over time to gradual worsening in language, orientation, and behavior and late severe loss of memory and some bodily functions until eventual death [26]. The etiology of AD is complex and multi-factorial and still poorly understood. The main pathological features of AD are the intracellular accumulation of neurofibrillary tangles composed of hyperphosphorylated tau protein and increased production and deposition of amyloid-β (Aβ) with concomitant loss of synapses and neurons [27]. In addition, it is associated with BBB dysfunction [28,29]. AD progression has also been related to a gradual damage in function and structure in the hippocampus and neocortex areas of the brain involved in memory and cognition. These changes are concomitant with NMDAR activation and oxidative stress, ultimately resulting in AD pathology [30].
Hcy was elevated in patients with AD compared with normal controls and has been suggested as an independent risk factor for AD [31,32,33], to reduce the size and volume of the hippocampus and cortex of healthy elderly people [34], to sensitize hippocampal neurons to excitotoxins in animal models [35], to enhance neuronal death in mouse models of cerebral stroke [36], and to induce a dose-dependent increase in apoptotic cell death in cultured hippocampal neurons [35]. Indeed, HHcy was reported to double the risk of developing AD [37]. The underlying cellular mechanisms by which elevated Hcy induce neuronal death or exacerbate the consequences of other insults are still unclear. However, some potential mechanisms have been suggested for HHcy-induced brain damage and explain the connections between HHcy and AD, such as increasing cellular oxidative stress and hypo-methylation of DNA and proteins [38,39,40,41] ER stress [42], cerebrovascular damage [43], neuroinflammation [44], Aβ elevation [45,46], and tau protein phosphorylation [47].
One main connection between HHcy and AD pathology is the impairment of the BBB [48]. Elevated levels of Hcy were reported to compromise BBB integrity when the BBB was evaluated in a mouse model of HHcy [4,6], and was also reported to increase permeability of the BBB by NMDA receptor-dependent regulation of tight junctions [4]. Furthermore, microvascular disorders associated with HHcy were suggested as direct causal mechanisms linking vitamin B deficiency (B6, B12, and folic acid), resulting in HHcy and neurological dysfunction in AD [42]. In the early stages of AD, the impairment of BBB homeostasis induces the production of pro-inflammatory cytokines, which worsens synapse destruction and the accumulation and activation of microglia, while in the late stage of AD, amyloid deposits are frequently observed in larger blood vessels as well as smaller cerebral capillaries [49,50].
3. Homocysteine and Neurovascular Eye Diseases
Hcy was linked to many visual disorders. Over the last decade, Hcy was notably reported to be elevated in patients with retinal neurovascular diseases such as DR [51,52,53,54] and AMD [55,56,57,58]. Accumulating evidence from pervious publications linked HHcy to many vasculopathies, including endothelial dysfunction, vessel wall malformations, loss of extracellular matrix collagen, and disruption of the BRB in rodents and humans [59]. There is an association between HHcy and diabetes-induced microangiopathies (diabetic nephropathy, retinopathy, and macular edema) [60,61,62,63]. Furthermore, impaired endothelial cell function has been reported in HHcy both in vitro and in vivo [64]; however, the underlying cellular and molecular mechanisms have not yet been clearly defined.
Our previous studies on a mouse model of HHcy caused by deficiency of the cystathionine-β-synthase enzyme (CBS) (cbs−/− and cbs+/− mice) reported an association between HHcy and retinal vascular dysfunction such as pathological neovascularization, central retinal vein occlusion, pericyte loss, and gliosis. Gliosis is a reactive change in glial cells in response to damage to the central nervous system (CNS) and involves the hypertrophy or proliferation of several different types of glial cells, including astrocytes [12] and microglia [22]. Besides the cbs (genetic) model of HHcy, we used another model of HHcy by injecting L-Homocysteine thiolactone hydrochloride locally in wild type (B57-BL6) mouse eyes (intravitreal injection) and we were able to confirm the changes we previously observed in the CBS mice. We reported retinal pigment epithelial (RPE) function disruption (barrier and phagocytic functions) by HHcy. We also reported the development of AMD-like features in mouse models of HHcy, and these features included vacuolization, hypopigmentation, atrophy, increased thickness of the basal laminar membrane, hyporeflective lucency, disturbed RPE–photoreceptor relationship, and development of choroidal neovascularization (CNV) [10].
4. Mechanisms of Homocysteine-Induced Neurovascular/Neurodegenerative Changes in the Central Nervous System (CNS)
The relation between elevated Hcy and neurovascular/neurodegenerative diseases has been established. Many mechanisms were proposed and studied for HHcy-induced vascular dysfunction, such as impaired endothelial function [65,66,67,68] oxidative stress [4,69,70], ER stress [71,72,73,74], inflammation [75,76,77,78,79], epigenetic modification [80,81,82,83], and activation of matrix metalloproteinase [84,85,86,87,88]. However, the exact mechanism of HHcy-induced vascular and barrier dysfunction is still undetermined.
Our work over the last decade has studied the effect of HHcy on retinal vasculature and reported that HHcy induced retina ischemia, tissue hypoxia, neovascularization, BRB dysfunction, and subsequent retinal hyperpermeability [8,10,12]. Moreover, this was followed by further studies to determine the underlying mechanisms of HHcy-induced vascular and BRB dysfunction. Given the anatomical fact that the retina is a part of the central nervous system, our studies on the BRB confirmed the effect of HHcy on the BBB. Remarkably, a growing body of evidence has demonstrated that AD affects both the brain and retina, with a significant correlation between pathological changes observed in both organs [89], highlighting the fact that retinal, neuronal, and microvascular alterations in AD could provide a unique window to the brain [90]. The retina is considered as an accessible “extension of the brain”, thereby it can be used as a non-invasive surrogate for detection and monitoring of AD-related pathological changes [91]. Based on these reports, we aimed to investigate if accumulation of β-amyloid and P-Tau, which are a characteristic pathological events of AD brains, occurs in the retina under HHcy conditions. Interestingly, our immunofluorescence data showed increased β-amyloid and P-Tau expression in RPE cells treated with Hcy (20 µM, 50 µM, 100 µM) in vitro and in primary RPE cells isolated from the genetic mouse model of HHcy (cbs) in vivo, as shown in Figure 2.
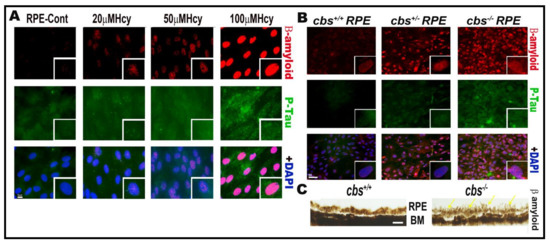
Figure 2.
Hcy induces accumulation of β-amyloid and P-Tau proteins in retinal RPE. IF staining showed increased expression of β-amyloid (red) and P-Tau (green) in (A) RPE treated with and without Hcy (20 μM, 50 μM, and 100 μM). (B) Primary RPE cells isolated from cbs+/− and cbs−/− mouse retina. (C) Histological staining of retinal sections with thioflavin S stain showing more accumulation of β-amyloid protein in RPE area of the cbs−/− mouse retina. The cbs+/− mice (heterozygous) represent mild/moderate HHcy, have about a 4- to 7-fold increase in plasma Hcy level, and show a mild retinal phenotype and normal life span, while the cbs−/− mice (homozygous with no copies of cbs) represent severe HHcy, have about a 30-fold increase in plasma Hcy with severe retinal phenotype and a short life span of ~3 to 5 weeks. Scale bars: 50 μm.
The current review will highlight the role of inflammation as a possible mechanism of HHcy-induced neurovascular changes and barrier dysfunction in aging diseases linked to HHcy, such as DR, AMD, and AD.
5. Homocysteine and Activation of Inflammation in Aging CNS Diseases
Accumulating evidence underscores the role of inflammation in the pathogenesis of aging diseases [92]. A strong association between HHcy and inflammation has been reported in various studies using human and experimental models [92,93,94]. Furthermore, HHcy was reported to show pro-inflammatory properties that led to visual dysfunction and optic nerve damage [51]. The association between HHcy and the induction of inflammation was attributed to two facts, first, the research that linked HHcy and the induction of inflammatory elements including leukocyte adhesion, expression of adhesion molecules, oxidative stress, reduced nitric oxide bioavailability, and endothelial dysfunction [95]. Additionally, another fact is that HHcy has been reported in association with various inflammatory diseases [96,97,98,99,100].
Given the acknowledged involvement of both HHcy and inflammation in age-related diseases, it is important to understand the role of HHcy in the induction of CNS inflammation. In this context, a recent study implies activation of Hcy/cytotoxic ceramide signaling as an underlying mechanism that propagates neuroinflmmation, degeneration, and apoptosis in AD [101]. Sun et al. reported an association between HHcy and AD development in elderly adults via potential mechanisms, including promoting inflammatory reactions, suppression of memory-related proteins, tau hyperphosphorylation, and Aβ accumulation [102]. Braun et al. found that HHcy altered inflammatory milieu, enhanced parenchymal plaque deposition, and triggered microglia function via upregulation of the expression of multiple “homeostatic” microglial genes in an experimental mouse model of AD [103]. In vitro, Hcy triggered a dose-dependent release of pro-inflammatory cytokines from adult astrocytes. Additionally, similar data was demonstrated in vivo, where an experimental rat model of mild HHcy showed inflammatory conditions in the cerebral cortex. Another suggested role for pro-inflammatory cytokines in Hcy-induced cognitive impairment is downstream activation of a pro-inflammatory-mediated increase in matrix metalloproteinase 9 (MMP9) which subsequently results in degradation of tight junctions, microhemorrhages, and, ultimately, cognitive impairment [104]. Further, auto-oxidation of Hcy, leading to cellular oxidative stress, could mediate HHcy-induced neurotoxicity through the formation of reactive oxygen species, causing neuroinflammation and apoptosis [105]. Additionally, accumulating evidence from other studies has reported that HHcy mediated vascular inflammation via activation of NF-κB in vascular smooth muscle cells [106] and contributed to the induction of cerebral ischemia via induction of cerebral microglia activation [107] and upregulation of pro-inflammatory cytokines [108].
Hcy and other pro-inflammatory factors such as interleukin-6 (IL-6), C-reactive protein (CRP), and alpha-1-antichymotrypsin (ACT) have been linked to neuroinflammation and cognitive decline [109]. Kommer et al. reported HHcy as an independent factor of lower level of information processing, general cognitive functioning, and fluid intelligence. Interestingly, the strongest negative correlation between HHcy and immediate recall was observed in persons with a high level of serum IL-6. HHcy was also negatively associated with retention in persons in the highest CRP tertile and with a faster rate of decline in persons in the lower and middle tertiles of CRP. Additionally, in the middle tertile of ACT, HHcy was associated with lower information processing speed and faster decline [110]. These results suggested that a combination of elevated Hcy and inflammation may be useful as a predictor of cognitive impairment.
Various signaling pathways have been linked to Hcy-induced production of pro-inflammatory cytokines. Zou et al. reported that HHcy promoted DNA synthesis and activation of microglia via activation of the p38MAPK/NADPH oxidase/ROS pathway. Activated microglia in turn produce a diverse range of neurotoxic and pro-inflammatory factors [107]. Another possible link has been reported between HHcy and lowered cystathionine γ-lyase expression and H2S production in macrophages via triggering DNA hypermethylation [111]. Moroever, increased ROS and suppressed NO production by HHcy could explain the downstream inflammatory cascade [112]. This was evidenced by the ability of antioxidants such as N-acetyl cysteine and vitamin C and E to reduce Hcy-induced inflammation in animal models [113].
The disruption of the redox system in the aging population may be related to melatonin suppression. Increasing age is often associated with diminished endogenous melatonin production. Melatonin is a free radical scavenger that has shown the ability to protect the brain against various neurological injuries by direct free radical scavenging. Therefore, age-associated low melatonin production contributes to aggrevated Hcy-induced cerebral injury [114]. In this context, melatonin attenuated Hcy-induced cerebral lipid and protein oxidation in rat brains [115]. Another mechanism proposed for melatonin’s protective effect is the inhibition of apoptosis. Melatonin was found to inhibit Hcy-induced neural apoptosis by inhibiting mitochondrial cytochrome c release and restoring the anti-apoptotic/apoptotic protein balance. Additionally, melatonin inhibited Hcy-triggered DNA fragmentation by cleavage of poly(ADP-ribose) polymerase in hippocampal neurons of hyperhomocysteinemic rats [116]. Moreover, melatonin was able to modulate adhesion molecule expression in neural cells and improve learning and memory performances in hyperhomocysteinemic rats [117].
Consistent with these reports, we also reported the role of inflammation in aging retinal and brain diseases associated with HHcy such as DR, AMD, and AD [22]. Our recently published research showed that HHcy is accompanied by inflammation in both the retina and brain [22]. Indeed, mice genetically overexpressing Hcy demonstrated microglia activation and upregulated inflammatory markers in both retina and brain tissue. Moreover, similar results were obtained in vitro in Hcy-treated retinal pigment epithelial cells, human retinal endothelial cells, and monocyte cell lines. Analysis of supernatants from the aforementioned cells after treatment with Hcy indicated increased levels of pro-inflammatory cytokines along with downregulation of anti-inflammatory cytokines. Furthermore, nuclear translocation of transcription factor NF-κB that controls the expression of numerous inflammatory cytokines was also evaluated in vivo and in vitro. Interestingly, our data showed Hcy-induced NF-κB activation and nuclear translocation of NF-κB from the cytoplasmic to the nuclear compartments of cells under elevated Hcy conditions. These results suggested that HHcy induced inflammatory responses in the mouse brain, and retina and cultured retinal and microglial cells. Consequently, elimination of extra Hcy or prevention/attenuation of inflammation could be a promising intervention for alleviating damage associated with HHcy in age-related diseases such as DR, AMD, and AD.
Hcy acts as an agonist for metabotropic glutamate receptors as well as for NMDA receptors [118,119,120,121]. Considerable evidence supports the involvement of the NMDA receptor in the pathogenesis of AD [24,120,122,123,124]. Interestingly, its role in HHcy-induced neuronal degeneration [125,126] and BBB dysfunction was reported [4,127,128]. Our recent work highlighted its role in retinal vascular pathology and BRB dysfunction as well [13]. Interestingly, NMDA receptor activation has been linked to neuroinflammation and neurodegeneration involved in AD pathology [23]. On the other hand, inflammation, in addition to other factors, including oxidative stress, tau hyperphosphorylation, and Aβ deposition, has been associated with increased activity and/or sensitivity of the brain glutamatergic system, leading ultimately to neuronal dysfunction and cell death in AD [129]. It is believed that pro-inflammatory cytokines are released by Aβ-activated microglia. This in turn leads to disruption of redox and glutamate homeostasis and activation/sensitization of NMDA receptors, leading to neuronal cell death. Interestingly, NMDA receptor activation triggers Aβ production and deposition, leading to a vicious cycle. This viscous cascade could be blocked by memantine [130]. Gérard and Hansson reported that NMDA receptor activation is essential for triggering Ca2+ signaling and pro-inflammatory cytokine secretion in cultured astrocytes, contributing to reactive astrogliosis [131]. In brain microglia, activation of NMDA receptors triggered an inflammatory response and neuronal cell death, contributing significantly to cortical damage. This damage was markedly attenuated via pharmacological inhibition or genetically induced loss of NMDAR function in microglia [132]. All these data emphasize the crucial role of NMDA–inflammation crosstalk in the pathogenesis of AD.
Currently, there is no cure for AD. The Alzheimer’s Disease Medications Fact Sheet published by the National Institute on Aging shows few treatments options for AD. Mainly, two categories include the FDA-approved prescription drugs for the treatment of patients with AD, such as cholinesterase inhibitors, which are used for mild to moderate AD, and an inhibitor for NMDA receptors, memantine, for the treatment of moderate to severe AD. Memantine is an FDA-approved NMDAR antagonist, which possibly functions through suppressing extra-synaptic NMDAR signaling. Further studies are needed to help elucidate the molecular mechanisms of how glutamate and NMDARs function in the etiology of AD [23].
NMDA receptor activation has been related to synaptic dysfunction in AD. Synaptic NMDARs are neuroprotective and are required for the survival of neurons. However, over-activation of NMDARs located outside of the synapse play a key role in antagonizing the synaptic pro-survival signaling pathway by causing compression on the presynaptic and postsynaptic neurons and glial cells, loss of mitochondrial membrane potential, and cell death, which shift the balance toward excitotoxicity and neurodegeneration [24,31,58,133].
HHcy was reported to induce NDMDA receptor-dependent vascular inflammation, BBB disruption, and hippocampus synaptic dysfunction in mice. However, pharmacological inhibition of NMDA receptors mitigated theses effects [134,135]. Indeed, NMDA receptor stimulation mediates HHcy-induced oxidative injury in nerve terminals [136], neuronal cell death [35], and tau protein phosphorylation [137]. Therefore, Hcy–NMDA receptor stimulation may be the one key mechanism that may be fundamental for Hcy-induced neuronal damage in AD. Elevated Hcy levels in the brain overstimulate NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, resulting in higher production of free radicals, increased levels of cytoplasmic calcium, and activation of caspases, leading to cellular death. These events have also been reported to play a major role in BBB disruption through the increased activity of matrix metalloproteinases (MMPs) [138]. Interestingly, HHcy has been reported to induce CRP gene and protein expression, which in turn augmented NMDA receptor expression in rat vascular smooth muscles [139]. Therefore, Hcy can elicit a pro-inflammatory response in vascular smooth muscle cells of brain small arteries via stimulating CRP production accompanied by NMDA-ROS-MAPK-NF-κB signal pathway activation [113].
We believe that HHcy activates NMDARs and induces subsequent activation of many mechanisms, such as oxidative stress, ER stress, and inflammation. We also believe that there is a link between the changes occurring in aging eye diseases and AD disease with HHcy. Consequently, that led us to evaluate the eyes of mice with HHcy (CBS mice) for well-known markers of AD, such as beta amyloid (Aβ) protein and tau protein (T-tau), and tau phosphorylated at threonine 181 (P-tau181). Our data showed marked elevation of the AD marker in RPE cells by Hcy treatment in a dose-dependent manner, as shown in Figure 2. These two biomarkers have been incorporated into research diagnostic criteria for AD and have added importance in the differential diagnosis of AD and related conditions [140]. AD is characterized by altered levels of those makers in the cerebrospinal fluid (CSF) [141] and also in the plasma [142]. Various clnical and preclincal studies have linked HHcy to accumualtion of those markers in the brain of AD patients. Indeed, Hcy level was independently correlated with plasma Aβ 40 and 42 levels in AD patients [143]. Additionally, homocysteic acid (HA), an oxidized Hcy metabolite, induced accumulation of neurotoxic Aβ 42 in rat cortical neurons [144]. Further, Hcy increased vulnerability of vascular smooth muscle cells to Aβ, implying a role in increasing the risk of cerebral amyloid angiopathy, a morphological hallmark of AD [145]. This Hcy effect was explained by potentiation of c-secretase enzyme activity by an endoplasmic protein—Hcy-related protein (HERP)—that is formed in the presence of Hcy, promoting accumulation of Aβ proteins in the brain [43]. Another proposed mechanism is Hcy-induced upregulation of the presenilin 1 (PS1) gene—a key factor for Aβ formation in AD—by DNA hypomethylation. PS1 promotes amyloid precursor protein (APP) synthesis, triggering Aβ protein accumulation [146].
The crucial role of HHcy in AD pathology has been further evidenced by clinical studies that demonstrated the efficacy of B vitamin supplementation on AD development, as deficiency of these vitamins causes Hcy accumulation in the body. A randomized controlled trial showed that folic acid supplementation improved cognition and mitigated inflammation in AD patients [147]. A high intake of folic acid has also been linked to a decrease in the risk of AD development in elderly people [148]. Moreover, a recent cohort study concluded that inadequate vitamin B12 intake intensified cognitive decline in patients with dementia [149]. Another randomized control trial demonstrated the clinical benefits of B vitamin supplementation for cognitive decline in AD patients (folic acid, B6, B12) [150].
In conclusion, it is obvious that both brain and retinal tissue are subjected to a linked cascade of events during pathogenesis of AD. HHcy-induced inflammation plays a crucial role during these events, resulting ultimately in neurodegeneration, barrier dysfunction, and cognitive and retinal impairment. Studying common pathogenic features between the brain and retina during AD development could lead to better understanding of HHcy’s role in different cellular mechanisms, including inflammation, and hence the accelerated development of therapeutic interventions that target Hcy itself or its downstream key signaling pathways.
Author Contributions
Conceptualization, A.T.; methodology, A.T., Y.Z., P.R.; investigation, A.T., Y.Z., P.R.; resources, A.T.; writing—original draft preparation, A.T., N.M.E.; writing—review and editing A.T., N.M.E.; supervision A.T.; project administration, A.T.; funding acquisition, A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the American Heart Association (AHA) Scientist Development Grant award #16SDG3070001, and NEI grant awards 1R01EY029751-02 and P30EY031631.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Smith, A.D.; Refsum, H. Homocysteine—From disease biomarker to disease prevention. J. Intern. Med. 2021. [Google Scholar] [CrossRef]
- Azzini, E.; Ruggeri, S.; Polito, A. Homocysteine: Its Possible Emerging Role in At-Risk Population Groups. Int. J. Mol. Sci. 2020, 21, 1421. [Google Scholar] [CrossRef] [PubMed]
- Jaul, E.; Barron, J. Age-Related Diseases and Clinical and Public Health Implications for the 85 Years Old and Over Population. Front. Public Health 2017, 5, 335. [Google Scholar] [CrossRef]
- Beard, R.S., Jr.; Bearden, S.E. Vascular complications of cystathionine beta-synthase deficiency: Future directions for homocysteine-to-hydrogen sulfide research. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H13–H26. [Google Scholar] [CrossRef] [PubMed]
- Lominadze, D.; Roberts, A.M.; Tyagi, N.; Moshal, K.S.; Tyagi, S.C. Homocysteine causes cerebrovascular leakage in mice. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1206–H1213. [Google Scholar] [CrossRef]
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J.; Dole, V.S.; Loscalzo, J.; Handy, D.E.; Wagner, D.D. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Kamat, P.K.; Familtseva, A.; Chaturvedi, P.; Muradashvili, N.; Narayanan, N.; Tyagi, S.C.; Tyagi, N. Role of microRNA29b in blood-brain barrier dysfunction during hyperhomocysteinemia: An epigenetic mechanism. J. Cereb. Blood Flow Metab. 2014, 34, 1212–1222. [Google Scholar] [CrossRef]
- Tawfik, A.; Al-Shabrawey, M.; Roon, P.; Sonne, S.; Covar, J.A.; Matragoon, S.; Ganapathy, P.S.; Atherton, S.S.; El-Remessy, A.; Ganapathy, V.; et al. Alterations of retinal vasculature in cystathionine-beta-synthase mutant mice, a model of hyperhomocysteinemia. Investig. Ophthamol. Vis. Sci. 2013, 54, 939–949. [Google Scholar] [CrossRef]
- Elmasry, K.; Mohamed, R.; Sharma, I.; Elsherbiny, N.M.; Liu, Y.; Al-Shabrawey, M.; Tawfik, A. Epigenetic modifications in hyperhomocysteinemia: Potential role in diabetic retinopathy and age-related macular degeneration. Oncotarget 2018, 9, 12562–12590. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Mander, S.; Hussein, K.A.; Elsherbiny, N.M.; Smith, S.B.; Al-Shabrawey, M.; Tawfik, A. Hyperhomocysteinemia disrupts retinal pigment epithelial structure and function with features of age-related macular degeneration. Oncotarget 2016, 7, 8532–8545. [Google Scholar] [CrossRef]
- Mohamed, R.; Sharma, I.; Ibrahim, A.S.; Saleh, H.; Elsherbiny, N.M.; Fulzele, S.; Elmasry, K.; Smith, S.B.; Al-Shabrawey, M.; Tawfik, A. Hyperhomocysteinemia Alters Retinal Endothelial Cells Barrier Function and Angiogenic Potential via Activation of Oxidative Stress. Sci. Rep. 2017, 7, 11952. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Markand, S.; Al-Shabrawey, M.; Mayo, J.N.; Reynolds, J.; Bearden, S.E.; Ganapathy, V.; Smith, S.B. Alterations of retinal vasculature in cystathionine-beta-synthase heterozygous mice: A model of mild to moderate hyperhomocysteinemia. Am. J. Pathol. 2014, 184, 2573–2585. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Mohamed, R.; Kira, D.; Alhusban, S.; Al-Shabrawey, M. N-Methyl-D-aspartate receptor activation, novel mechanism of homocysteine-induced blood-retinal barrier dysfunction. J. Mol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Samra, Y.A.; Elsherbiny, N.M.; Al-Shabrawey, M. Implication of Hyperhomocysteinemia in Blood Retinal Barrier (BRB) Dysfunction. Biomolecules 2020, 10, 1119. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Shakib, M.; Cunha-Vaz, J.G. Studies on the permeability of the blood-retinal barrier. IV. Junctional complexes of the retinal vessels and their role in the permeability of the blood-retinal barrier. Exp. Eye Res. 1966, 5, 229–234. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.G. The blood-retinal barriers. Doc. Ophthalmol. 1976, 41, 287–327. [Google Scholar] [CrossRef]
- Cunha-Vaz, J. The Blood-Retinal Barrier in the Management of Retinal Disease: EURETINA Award Lecture. Ophthalmologica 2017, 237, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schultz, H.; Song, Y.; Baumann, B.H.; Kapphahn, R.J.; Montezuma, S.R.; Ferrington, D.A.; Dunaief, J.L. Increased serum proteins in non-exudative AMD retinas. Exp. Eye Res. 2019, 186, 107686. [Google Scholar] [CrossRef]
- Tawfik, A.; Smith, S.B. Increased ER stress as a mechanism of retinal neurovasculopathy in mice with severe hyperhomocysteinemia. Austin J. Clin. Ophthalmol. 2014, 1, 1023. [Google Scholar]
- Elsherbiny, N.M.; Sharma, I.; Kira, D.; Alhusban, S.; Samra, Y.A.; Jadeja, R.; Martin, P.; Al-Shabrawey, M.; Tawfik, A. Homocysteine Induces Inflammation in Retina and Brain. Biomolecules 2020, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, S.; Coley, N.; Aisen, P.; Carrillo, M.C.; DeKosky, S.; Durga, J.; Fillit, H.; Frisoni, G.B.; Froelich, L.; Gauthier, S.; et al. Methodological issues in primary prevention trials for neurodegenerative dementia. J. Alzheimers Dis. 2009, 16, 235–270. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Gonzalez, A.; Andrade, V.; Cortes, N.; Tapia, J.P.; Guzman-Martinez, L. Alzheimer’s Disease in the Perspective of Neuroimmunology. Open Neurol. J. 2018, 12, 50–56. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Morishita, R. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: Relevance to pathogenesis and therapy. Front. Aging Neurosci. 2014, 6, 171. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Lyros, E.; Bakogiannis, C.; Liu, Y.; Fassbender, K. Molecular links between endothelial dysfunction and neurodegeneration in Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 18–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S. Homocysteine and Alzheimer’s disease. Lancet Neurol. 2003, 2, 425–428. [Google Scholar] [CrossRef]
- Zhuo, J.M.; Wang, H.; Pratico, D. Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol. Sci. 2011, 32, 562–571. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and Dementia: An International Consensus Statement. J. Alzheimers Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef]
- Den Heijer, T.; Vermeer, S.E.; Clarke, R.; Oudkerk, M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Homocysteine and brain atrophy on MRI of non-demented elderly. Brain 2003, 126, 170–175. [Google Scholar] [CrossRef]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.; Penix, L.; Mattson, M.P. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Ahmadi, M.; Kruman, I.; Biniszkiewicz, D.; Meisel, A.; Gertz, K. Folate deficiency increases postischemic brain injury. Stroke 2005, 36, 321–325. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Beydoun, H.A.; Gamaldo, A.A.; Teel, A.; Zonderman, A.B.; Wang, Y. Epidemiologic studies of modifiable factors associated with cognition and dementia: Systematic review and meta-analysis. BMC Public Health 2014, 14, 643. [Google Scholar] [CrossRef]
- Ho, P.I.; Collins, S.C.; Dhitavat, S.; Ortiz, D.; Ashline, D.; Rogers, E.; Shea, T.B. Homocysteine potentiates beta-amyloid neurotoxicity: Role of oxidative stress. J. Neurochem. 2001, 78, 249–253. [Google Scholar] [CrossRef]
- Robinson, N.; Grabowski, P.; Rehman, I. Alzheimer’s disease pathogenesis: Is there a role for folate? Mech. Ageing Dev. 2018, 174, 86–94. [Google Scholar] [CrossRef]
- Streck, E.L.; Vieira, P.S.; Wannmacher, C.M.; Dutra-Filho, C.S.; Wajner, M.; Wyse, A.T. In vitro effect of homocysteine on some parameters of oxidative stress in rat hippocampus. Metab. Brain Dis. 2003, 18, 147–154. [Google Scholar] [CrossRef]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef]
- Troen, A.M.; Shea-Budgell, M.; Shukitt-Hale, B.; Smith, D.E.; Selhub, J.; Rosenberg, I.H. B-vitamin deficiency causes hyperhomocysteinemia and vascular cognitive impairment in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12474–12479. [Google Scholar] [CrossRef]
- Sai, X.; Kawamura, Y.; Kokame, K.; Yamaguchi, H.; Shiraishi, H.; Suzuki, R.; Suzuki, T.; Kawaichi, M.; Miyata, T.; Kitamura, T.; et al. Endoplasmic reticulum stress-inducible protein, Herp, enhances presenilin-mediated generation of amyloid beta-protein. J. Biol. Chem. 2002, 277, 12915–12920. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Givvimani, S.; Sathnur, P.B.; Tyagi, S.C.; Tyagi, N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 2013, 252, 302–319. [Google Scholar] [CrossRef]
- Zhuo, J.M.; Portugal, G.S.; Kruger, W.D.; Wang, H.; Gould, T.J.; Pratico, D. Diet-induced hyperhomocysteinemia increases amyloid-beta formation and deposition in a mouse model of Alzheimer’s disease. Curr. Alzheimer. Res. 2010, 7, 140–149. [Google Scholar] [CrossRef]
- Pacheco-Quinto, J.; Rodriguez de Turco, E.B.; DeRosa, S.; Howard, A.; Cruz-Sanchez, F.; Sambamurti, K.; Refolo, L.; Petanceska, S.; Pappolla, M.A. Hyperhomocysteinemic Alzheimer’s mouse model of amyloidosis shows increased brain amyloid beta peptide levels. Neurobiol. Dis. 2006, 22, 651–656. [Google Scholar] [CrossRef]
- Sontag, E.; Nunbhakdi-Craig, V.; Sontag, J.M.; Diaz-Arrastia, R.; Ogris, E.; Dayal, S.; Lentz, S.R.; Arning, E.; Bottiglieri, T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci. 2007, 27, 2751–2759. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Clearing amyloid through the blood-brain barrier. J. Neurochem. 2004, 89, 807–811. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Ajith, T.A.; Ranimenon. Homocysteine in ocular diseases. Clin. Chim. Acta 2015, 450, 316–321. [Google Scholar] [CrossRef]
- Dong, N.; Shi, H.; Tang, X. Plasma homocysteine levels are associated with macular thickness in type 2 diabetes without diabetic macular edema. Int. Ophthalmol. 2018, 38, 737–746. [Google Scholar] [CrossRef]
- Lei, X.; Zeng, G.; Zhang, Y.; Li, Q.; Zhang, J.; Bai, Z.; Yang, K. Association between homocysteine level and the risk of diabetic retinopathy: A systematic review and meta-analysis. Diabetol. Metab. Syndr. 2018, 10, 61. [Google Scholar] [CrossRef]
- Lei, X.W.; Li, Q.; Zhang, J.Z.; Zhang, Y.M.; Liu, Y.; Yang, K.H. The Protective Roles of Folic Acid in Preventing Diabetic Retinopathy Are Potentially Associated with Suppressions on Angiogenesis, Inflammation, and Oxidative Stress. Ophthalm. Res. 2019, 1–13. [Google Scholar] [CrossRef]
- Gopinath, B.; Flood, V.M.; Rochtchina, E.; Wang, J.J.; Mitchell, P. Homocysteine, folate, vitamin B-12, and 10-y incidence of age-related macular degeneration. Am. J. Clin. Nutr. 2013, 98, 129–135. [Google Scholar] [CrossRef]
- Christen, W.G.; Cook, N.R.; Chiuve, S.E.; Ridker, P.M.; Gaziano, J.M. Prospective study of plasma homocysteine, its dietary determinants, and risk of age-related macular degeneration in men. Ophthalm. Epidemiol. 2018, 25, 79–88. [Google Scholar] [CrossRef]
- Christen, W.G.; Chasman, D.I.; Cook, N.R.; Chiuve, S.E.; Ridker, P.M.; Buring, J.E. Homocysteine, B Vitamins, MTHFR Genotype, and Incident Age-related Macular Degeneration. Ophthalmol. Retina 2018, 2, 508–510. [Google Scholar] [CrossRef]
- Rochtchina, E.; Wang, J.J.; Flood, V.M.; Mitchell, P. Elevated serum homocysteine, low serum vitamin B12, folate, and age-related macular degeneration: The Blue Mountains Eye Study. Am. J. Ophthalmol. 2007, 143, 344–346. [Google Scholar] [CrossRef]
- Beard, R.S., Jr.; Reynolds, J.J.; Bearden, S.E. Hyperhomocysteinemia increases permeability of the blood-brain barrier by NMDA receptor-dependent regulation of adherens and tight junctions. Blood 2011, 118, 2007–2014. [Google Scholar] [CrossRef]
- Aydin, E.; Demir, H.D.; Ozyurt, H.; Etikan, I. Association of plasma homocysteine and macular edema in type 2 diabetes mellitus. Eur. J. Ophthalmol. 2008, 18, 226–232. [Google Scholar] [CrossRef]
- Ukinc, K.; Ersoz, H.O.; Karahan, C.; Erem, C.; Eminagaoglu, S.; Hacihasanoglu, A.B.; Yilmaz, M.; Kocak, M. Methyltetrahydrofolate reductase C677T gene mutation and hyperhomocysteinemia as a novel risk factor for diabetic nephropathy. Endocrine 2009, 36, 255–261. [Google Scholar] [CrossRef]
- Yang, G.; Lu, J.; Pan, C. The impact of plasma homocysteine level on development of retinopathy in type 2 diabetes mellitus. Zhonghua Nei Ke Za Zhi 2002, 41, 34–38. [Google Scholar] [PubMed]
- Vaccaro, O.; Perna, A.F.; Mancini, F.P.; Iovine, C.; Cuomo, V.; Sacco, M.; Tufano, A.; Rivellese, A.A.; Ingrosso, D.; Riccardi, G. Plasma homocysteine and microvascular complications in type 1 diabetes. Nutr. Metab. Cardiovasc. Dis. 2000, 10, 297–304. [Google Scholar] [PubMed]
- Cheng, Z.; Yang, X.; Wang, H. Hyperhomocysteinemia and Endothelial Dysfunction. Curr. Hypertens. Rev. 2009, 5, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Keller, C.; Hoffmann, U.; Loscalzo, J. Endothelial dysfunction and atherothrombosis in mild hyperhomocysteinemia. Vasc. Med. 2002, 7, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Heydrick, S.; Zhang, Y.Y.; Bierl, C.; Cap, A.; Loscalzo, J. Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthase-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 34–41. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, F.; Tan, H.; Liao, D.; Bryan, R.M., Jr.; Randhawa, J.K.; Rumbaut, R.E.; Durante, W.; Schafer, A.I.; Yang, X.; et al. Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2515–2521. [Google Scholar] [CrossRef]
- Zhang, H.S.; Cao, E.H.; Qin, J.F. Homocysteine induces cell cycle G1 arrest in endothelial cells through the PI3K/Akt/FOXO signaling pathway. Pharmacology 2005, 74, 57–64. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J. 2004, 3, 4. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc. Diabetol. 2002, 1, 3. [Google Scholar] [CrossRef]
- Wang, X.C.; Sun, W.T.; Yu, C.M.; Pun, S.H.; Underwood, M.J.; He, G.W.; Yang, Q. ER stress mediates homocysteine-induced endothelial dysfunction: Modulation of IKCa and SKCa channels. Atherosclerosis 2015, 242, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shang, F.; Li, X.; Lu, S.; Niu, X.; Zhang, Z.; Liu, J.; Li, X.; Zhao, L. Blocking PERK resuces vascular smooth muscle cells from homocysteine-induced ER stress and apoptosis. Front. Biosci. Landmark Ed. 2020, 25, 536–548. [Google Scholar] [PubMed]
- Hu, H.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, K.; Sun, H. Alpha-lipoic acid defends homocysteine-induced endoplasmic reticulum and oxidative stress in HAECs. Biomed. Pharmacother. 2016, 80, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.O.; Son, Y.; Lee, J.H.; Choi, S.W.; Kim, S.H.; Cheong, Y.K.; Chung, H.T.; Pae, H.O. Both nitric oxide and nitrite prevent homocysteine-induced endoplasmic reticulum stress and subsequent apoptosis via cGMP-dependent pathway in neuronal cells. Biochem. Biophys. Res. Commun. 2017, 493, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, Z.; Liu, K.; Liu, X.; Sun, H. Catalpol Inhibits Homocysteine-induced Oxidation and Inflammation via Inhibiting Nox4/NF-kappaB and GRP78/PERK Pathways in Human Aorta Endothelial Cells. Inflammation 2019, 42, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Koller, A.; Szenasi, A.; Dornyei, G.; Kovacs, N.; Lelbach, A.; Kovacs, I. Coronary Microvascular and Cardiac Dysfunction Due to Homocysteine Pathometabolism; A Complex Therapeutic Design. Curr. Pharm. Des. 2018, 24, 2911–2920. [Google Scholar] [CrossRef]
- Dobrynina, L.A.; Kalashnikova, L.A.; Shabalina, A.A.; Kostyreva, M.V.; Aleksandrova, E.V.; Gafarova, M.E.; Shamtieva, K.V. Indicators of homeostasis, inflammation and homocysteine in ischemic stroke in the young age. Zhurnal Nevrologii i Psikhiatrii Imeni SS Korsakova 2017, 117, 25–33. [Google Scholar] [CrossRef]
- AnandBabu, K.; Sen, P.; Angayarkanni, N. Oxidized LDL, homocysteine, homocysteine thiolactone and advanced glycation end products act as pro-oxidant metabolites inducing cytokine release, macrophage infiltration and pro-angiogenic effect in ARPE-19 cells. PLoS ONE 2019, 14, e0216899. [Google Scholar] [CrossRef]
- Kumar, M.; Sandhir, R. Hydrogen sulfide suppresses homocysteine-induced glial activation and inflammatory response. Nitric Oxide 2019, 90, 15–28. [Google Scholar] [CrossRef]
- Kamat, P.K.; Mallonee, C.J.; George, A.K.; Tyagi, S.C.; Tyagi, N. Homocysteine, Alcoholism, and Its Potential Epigenetic Mechanism. Alcohol Clin. Exp. Res. 2016, 40, 2474–2481. [Google Scholar] [CrossRef]
- Perla-Kajan, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mohammad, G.; Sahajpal, N. Faulty homocysteine recycling in diabetic retinopathy. Eye Vis. 2020, 7, 4. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. Homocysteine Disrupts Balance between MMP-9 and Its Tissue Inhibitor in Diabetic Retinopathy: The Role of DNA Methylation. Int. J. Mol. Sci. 2020, 21, 1771. [Google Scholar] [CrossRef]
- Wang, Z.S.; Jin, H.; Wang, D.M. Influence of hydrogen sulfide on zymogen activation of homocysteine-induced matrix metalloproteinase-2 in H9C2 cardiocytes. Asian Pac. J. Trop. Med. 2016, 9, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Bescond, A.; Augier, T.; Chareyre, C.; Garcon, D.; Hornebeck, W.; Charpiot, P. Influence of homocysteine on matrix metalloproteinase-2: Activation and activity. Biochem. Biophys. Res. Commun. 1999, 263, 498–503. [Google Scholar] [CrossRef]
- Shi, Y.F.; Chi, J.F.; Tang, W.L.; Xu, F.K.; Liu, L.B.; Ji, Z.; Lv, H.T.; Guo, H.Y. Effects of rosuvastatin on the production and activation of matrix metalloproteinase-2 and migration of cultured rat vascular smooth muscle cells induced by homocysteine. J. Zhejiang Univ. Sci. B 2013, 14, 696–704. [Google Scholar] [CrossRef]
- Kundu, S.; Tyagi, N.; Sen, U.; Tyagi, S.C. Matrix imbalance by inducing expression of metalloproteinase and oxidative stress in cochlea of hyperhomocysteinemic mice. Mol. Cell. Biochem. 2009, 332, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, A.A.; Weintraub, S.; Fawzi, W. Retinal Imaging in Alzheimer’s Disease: In Search of the Holy Grail. Ophthalmology 2020, 127, 119–121. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Shi, C.; Shen, M.; Lu, F. Advances in retina imaging as potential biomarkers for early diagnosis of Alzheimer’s disease. Transl Neurodegener. 2021, 10, 6. [Google Scholar] [CrossRef]
- Bevan, R.J.; Hughes, T.R.; Williams, P.A.; Good, M.A.; Morgan, B.P.; Morgan, J.E. Retinal ganglion cell degeneration correlates with hippocampal spine loss in experimental Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Sanchez, N.; Alvarez-Rios, A.I.; Guerrero, J.M.; Garcia-Garcia, F.J.; Rodriguez-Manas, L.; Cruz-Chamorro, I.; Lardone, P.J.; Carrillo-Vico, A. Homocysteine and C-Reactive Protein Levels Are Associated With Frailty in Older Spaniards: The Toledo Study for Healthy Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Luo, H.; Zhang, L.; Huang, Y.; Liu, B.; Ma, K.; Feng, J.; Xie, J.; Zheng, J.; Hu, J.; et al. Hyperhomocysteinemia exaggerates adventitial inflammation and angiotensin II-induced abdominal aortic aneurysm in mice. Circ. Res. 2012, 111, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Li, X.; Shan, H.; Saredy, J.J.; Cueto, R.; Xia, J.; Jiang, X.; Yang, X.F.; Wang, H. Ly6C(+) Inflammatory Monocyte Differentiation Partially Mediates Hyperhomocysteinemia-Induced Vascular Dysfunction in Type 2 Diabetic db/db Mice. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2097–2119. [Google Scholar] [CrossRef] [PubMed]
- Holven, K.B.; Holm, T.; Aukrust, P.; Christensen, B.; Kjekshus, J.; Andreassen, A.K.; Gullestad, L.; Hagve, T.A.; Svilaas, A.; Ose, L.; et al. Effect of folic acid treatment on endothelium-dependent vasodilation and nitric oxide-derived end products in hyperhomocysteinemic subjects. Am. J. Med. 2001, 110, 536–542. [Google Scholar] [CrossRef]
- Phelip, J.M.; Ducros, V.; Faucheron, J.L.; Flourie, B.; Roblin, X. Association of hyperhomocysteinemia and folate deficiency with colon tumors in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2008, 14, 242–248. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Capecchi, P.L.; Selvi, E.; Lorenzini, S.; Bisogno, S.; Galeazzi, M.; Laghi Pasini, F. Hyperhomocysteinemia, inflammation and autoimmunity. Autoimmun. Rev. 2007, 6, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Wakkee, M.; Thio, H.B.; Prens, E.P.; Sijbrands, E.J.; Neumann, H.A. Unfavorable cardiovascular risk profiles in untreated and treated psoriasis patients. Atherosclerosis 2007, 190, 1–9. [Google Scholar] [CrossRef]
- Akalin, A.; Alatas, O.; Colak, O. Relation of plasma homocysteine levels to atherosclerotic vascular disease and inflammation markers in type 2 diabetic patients. Eur. J. Endocrinol. 2008, 158, 47–52. [Google Scholar] [CrossRef]
- Alvares Delfino, V.D.; de Andrade Vianna, A.C.; Mocelin, A.J.; Barbosa, D.S.; Mise, R.A.; Matsuo, T. Folic acid therapy reduces plasma homocysteine levels and improves plasma antioxidant capacity in hemodialysis patients. Nutrition 2007, 23, 242–247. [Google Scholar] [CrossRef]
- Le Stunff, H.; Veret, J.; Kassis, N.; Denom, J.; Meneyrol, K.; Paul, J.L.; Cruciani-Guglielmacci, C.; Magnan, C.; Janel, N. Deciphering the Link Between Hyperhomocysteinemia and Ceramide Metabolism in Alzheimer-Type Neurodegeneration. Front. Neurol. 2019, 10, 807. [Google Scholar] [CrossRef]
- Sun, J.; Wen, S.; Zhou, J.; Ding, S. Association between malnutrition and hyperhomocysteine in Alzheimer’s disease patients and diet intervention of betaine. J. Clin. Lab. Anal. 2017, 31. [Google Scholar] [CrossRef]
- Braun, D.J.; Dimayuga, E.; Morganti, J.M.; Van Eldik, L.J. Microglial-associated responses to comorbid amyloid pathology and hyperhomocysteinemia in an aged knock-in mouse model of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 274. [Google Scholar] [CrossRef] [PubMed]
- Price, B.R.; Wilcock, D.M.; Weekman, E.M. Hyperhomocysteinemia as a Risk Factor for Vascular Contributions to Cognitive Impairment and Dementia. Front. Aging Neurosci. 2018, 10, 350. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Vacek, J.C.; Kalani, A.; Tyagi, N. Homocysteine Induced Cerebrovascular Dysfunction: A Link to Alzheimer’s Disease Etiology. Open Neurol. J. 2015, 9, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gokkusu, C.; Tulubas, F.; Unlucerci, Y.; Ozkok, E.; Umman, B.; Aydin, M. Homocysteine and pro-inflammatory cytokine concentrations in acute heart disease. Cytokine 2010, 50, 15–18. [Google Scholar] [CrossRef]
- Zou, C.G.; Zhao, Y.S.; Gao, S.Y.; Li, S.D.; Cao, X.Z.; Zhang, M.; Zhang, K.Q. Homocysteine promotes proliferation and activation of microglia. Neurobiol. Aging 2010, 31, 2069–2079. [Google Scholar] [CrossRef]
- Chen, S.; Dong, Z.; Cheng, M.; Zhao, Y.; Wang, M.; Sai, N.; Wang, X.; Liu, H.; Huang, G.; Zhang, X. Homocysteine exaggerates microglia activation and neuroinflammation through microglia localized STAT3 overactivation following ischemic stroke. J. Neuroinflamm. 2017, 14, 187. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Van den Kommer, T.N.; Dik, M.G.; Comijs, H.C.; Jonker, C.; Deeg, D.J. Homocysteine and inflammation: Predictors of cognitive decline in older persons? Neurobiol. Aging 2010, 31, 1700–1709. [Google Scholar] [CrossRef]
- Li, J.J.; Li, Q.; Du, H.P.; Wang, Y.L.; You, S.J.; Wang, F.; Xu, X.S.; Cheng, J.; Cao, Y.J.; Liu, C.F.; et al. Homocysteine Triggers Inflammatory Responses in Macrophages through Inhibiting CSE-H2S Signaling via DNA Hypermethylation of CSE Promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef]
- Petras, M.; Tatarkova, Z.; Kovalska, M.; Mokra, D.; Dobrota, D.; Lehotsky, J.; Drgova, A. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J. Physiol. Pharmacol. 2014, 65, 15–23. [Google Scholar]
- Moretti, R.; Caruso, P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 231. [Google Scholar] [CrossRef]
- Osuna, C.; Reiter, R.J.; Garcia, J.J.; Karbownik, M.; Tan, D.X.; Calvo, J.R.; Manchester, L.C. Inhibitory effect of melatonin on homocysteine-induced lipid peroxidation in rat brain homogenates. Pharmacol. Toxicol. 2002, 90, 32–37. [Google Scholar] [CrossRef]
- Ortega-Gutierrez, S.; Fuentes-Broto, L.; Garcia, J.J.; Lopez-Vicente, M.; Martinez-Ballarin, E.; Miana-Mena, F.J.; Millan-Plano, S.; Reiter, R.J. Melatonin reduces protein and lipid oxidative damage induced by homocysteine in rat brain homogenates. J. Cell Biochem. 2007, 102, 729–735. [Google Scholar] [CrossRef]
- Baydas, G.; Reiter, R.J.; Akbulut, M.; Tuzcu, M.; Tamer, S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and anti-apoptotic protein levels. Neuroscience 2005, 135, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Ozer, M.; Yasar, A.; Tuzcu, M.; Koz, S.T. Melatonin improves learning and memory performances impaired by hyperhomocysteinemia in rats. Brain Res. 2005, 1046, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Poddar, R.; Paul, S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J. Neurochem. 2009, 110, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Zieminska, E.; Stafiej, A.; Lazarewicz, J.W. Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurones. Neurochem. Int. 2003, 43, 481–492. [Google Scholar] [CrossRef]
- Gao, L.B.; Yu, X.F.; Chen, Q.; Zhou, D. Alzheimer’s Disease therapeutics: Current and future therapies. Minerva Med. 2016, 107, 108–113. [Google Scholar] [PubMed]
- Luchowska, E.; Luchowski, P.; Paczek, R.; Ziembowicz, A.; Kocki, T.; Turski, W.A.; Wielosz, M.; Lazarewicz, J.; Urbanska, E.M. Dual effect of DL-homocysteine and S-adenosylhomocysteine on brain synthesis of the glutamate receptor antagonist, kynurenic acid. J. Neurosci. Res. 2005, 79, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lin, C.H.; Lane, H.Y. d-glutamate and Gut Microbiota in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2676. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Olivares, D.; Deshpande, V.K.; Shi, Y.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T.; Huang, X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr. Alzheimer Res. 2012, 9, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.A.; Johnson, P. Homocysteine and its derivatives as possible modulators of neuronal and non-neuronal cell glutamate receptors in Alzheimer’s disease. J. Alzheimers Dis. 2007, 11, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Ataie, A.; Ataee, R.; Shadifar, M.; Shahabi, S.; Aghajanpour, S.M.; Hosseinpour, Y. Interaction of Memantine with Homocysteine on the Apoptosis in the Rat Hippocampus cells. Int. J. Mol. Cell Med. 2012, 1, 145–152. [Google Scholar]
- Gu, S.X.; Sonkar, V.K.; Katare, P.B.; Kumar, R.; Kruger, W.D.; Arning, E.; Bottiglieri, T.; Lentz, S.R.; Dayal, S. Memantine Protects From Exacerbation of Ischemic Stroke and Blood Brain Barrier Disruption in Mild But Not Severe Hyperhomocysteinemia. J. Am. Heart Assoc. 2020, 9, e013368. [Google Scholar] [CrossRef] [PubMed]
- Fridman, O. Hyperhomocysteinemia: Atherothrombosis and neurotoxicity. Acta Physiol. Pharmacol. Ther. Latinoam. 1999, 49, 21–30. [Google Scholar]
- Kumar, K.; Kumar, A.; Keegan, R.M.; Deshmukh, R. Recent advances in the neurobiology and neuropharmacology of Alzheimer’s disease. Biomed. Pharmacother. 2018, 98, 297–307. [Google Scholar] [CrossRef]
- Parsons, C.G.; Stoffler, A.; Danysz, W. Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—Too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723. [Google Scholar] [CrossRef]
- Gerard, F.; Hansson, E. Inflammatory activation enhances NMDA-triggered Ca2+ signalling and IL-1beta secretion in primary cultures of rat astrocytes. Brain Res. 2012, 1473, 1–8. [Google Scholar] [CrossRef]
- Kaindl, A.M.; Degos, V.; Peineau, S.; Gouadon, E.; Chhor, V.; Loron, G.; Le Charpentier, T.; Josserand, J.; Ali, C.; Vivien, D.; et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann. Neurol. 2012, 72, 536–549. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine—Searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kyles, P.; Kalani, A.; Tyagi, N. Hydrogen Sulfide Ameliorates Homocysteine-Induced Alzheimer’s Disease-Like Pathology, Blood-Brain Barrier Disruption, and Synaptic Disorder. Mol. Neurobiol. 2016, 53, 2451–2467. [Google Scholar] [CrossRef] [PubMed]
- Anggono, V.; Tsai, L.H.; Gotz, J. Glutamate Receptors in Alzheimer’s Disease: Mechanisms and Therapies. Neural. Plast. 2016, 2016, 8256196. [Google Scholar] [CrossRef]
- Jara-Prado, A.; Ortega-Vazquez, A.; Martinez-Ruano, L.; Rios, C.; Santamaria, A. Homocysteine-induced brain lipid peroxidation: Effects of NMDA receptor blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotox. Res. 2003, 5, 237–243. [Google Scholar] [CrossRef]
- Ho, P.I.; Ortiz, D.; Rogers, E.; Shea, T.B. Multiple aspects of homocysteine neurotoxicity: Glutamate excitotoxicity, kinase hyperactivation and DNA damage. J. Neurosci. Res. 2002, 70, 694–702. [Google Scholar] [CrossRef]
- Lehotsky, J.; Tothova, B.; Kovalska, M.; Dobrota, D.; Benova, A.; Kalenska, D.; Kaplan, P. Role of Homocysteine in the Ischemic Stroke and Development of Ischemic Tolerance. Front. Neurosci. 2016, 10, 538. [Google Scholar] [CrossRef]
- Pang, X.; Liu, J.; Zhao, J.; Mao, J.; Zhang, X.; Feng, L.; Han, C.; Li, M.; Wang, S.; Wu, D. Homocysteine induces the expression of C-reactive protein via NMDAr-ROS-MAPK-NF-kappaB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 2014, 236, 73–81. [Google Scholar] [CrossRef]
- Lee, J.C.; Kim, S.J.; Hong, S.; Kim, Y. Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid beta (Abeta) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimers Res. Ther. 2019, 11, 34. [Google Scholar] [CrossRef]
- Shen, X.N.; Li, J.Q.; Wang, H.F.; Li, H.Q.; Huang, Y.Y.; Yang, Y.X.; Tan, L.; Dong, Q.; Yu, J.T.; Alzheimer’s Disease Neuroimaging Initiative. Plasma amyloid, tau, and neurodegeneration biomarker profiles predict Alzheimer’s disease pathology and clinical progression in older adults without dementia. Alzheimers Dement. 2020, 12, e12104. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; Gurol, M.E.; Raju, S.; Diaz-Arrastia, R.; Locascio, J.J.; Tennis, M.; Hyman, B.T.; Growdon, J.H.; Greenberg, S.M.; Bottiglieri, T. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology 2005, 65, 1402–1408. [Google Scholar] [CrossRef]
- Hasegawa, T.; Ukai, W.; Jo, D.G.; Xu, X.; Mattson, M.P.; Nakagawa, M.; Araki, W.; Saito, T.; Yamada, T. Homocysteic acid induces intraneuronal accumulation of neurotoxic Abeta42: Implications for the pathogenesis of Alzheimer’s disease. J. Neurosci. Res. 2005, 80, 869–876. [Google Scholar] [CrossRef]
- Mok, S.S.; Turner, B.J.; Beyreuther, K.; Masters, C.L.; Barrow, C.J.; Small, D.H. Toxicity of substrate-bound amyloid peptides on vascular smooth muscle cells is enhanced by homocysteine. Eur J. Biochem. 2002, 269, 3014–3022. [Google Scholar] [CrossRef]
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 gene silencing by S-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett. 2003, 541, 145–148. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Ji, L.; Wu, T.; Ji, Y.; Zhou, Y.; Zheng, M.; Zhang, M.; Xu, W.; Huang, G. Folic Acid Supplementation Mitigates Alzheimer’s Disease by Reducing Inflammation: A Randomized Controlled Trial. Mediat. Inflamm. 2016, 2016, 5912146. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.X.; Miller, J.; Green, R.; Mayeux, R. Relation of higher folate intake to lower risk of Alzheimer disease in the elderly. Arch. Neurol. 2007, 64, 86–92. [Google Scholar] [CrossRef]
- An, Y.; Feng, L.; Zhang, X.; Wang, Y.; Wang, Y.; Tao, L.; Qin, Z.; Xiao, R. Dietary intakes and biomarker patterns of folate, vitamin B6, and vitamin B12 can be associated with cognitive impairment by hypermethylation of redox-related genes NUDT15 and TXNRD1. Clin. Epigenet. 2019, 11, 139. [Google Scholar] [CrossRef]
- De Jager, C.A.; Oulhaj, A.; Jacoby, R.; Refsum, H.; Smith, A.D. Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2012, 27, 592–600. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).