
Synthesis of Novel Acyl Derivatives of 3-(4,5,6,7-Tetrabromo-1H-benzimidazol-1-yl)propan-1-ols—Intracellular TBBi-Based CK2 Inhibitors with Proapoptotic Properties

 , , ,
, , , 
Abstract
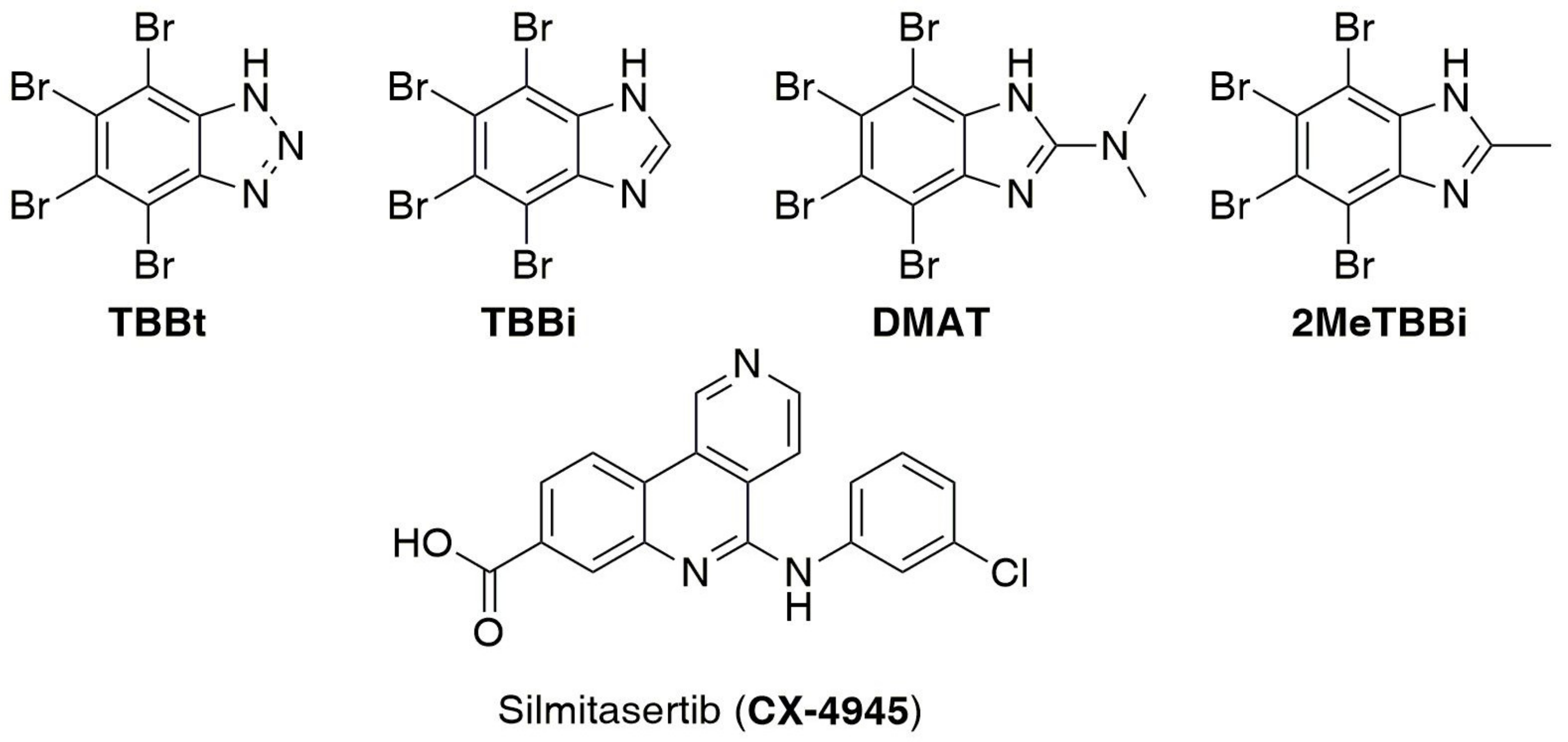
1. Introduction
2. Results
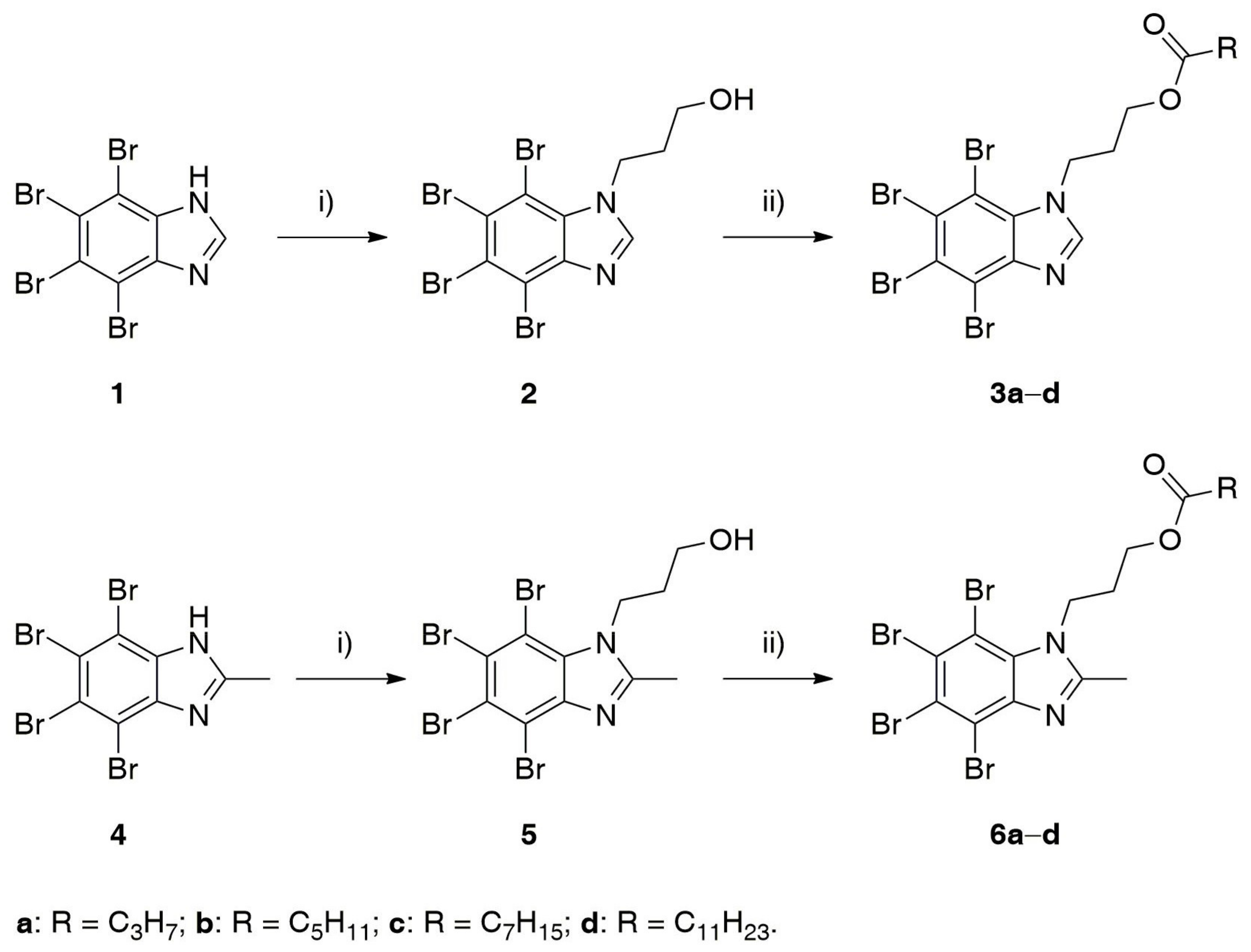
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Inhibition of Recombinant CK2 and PIM1
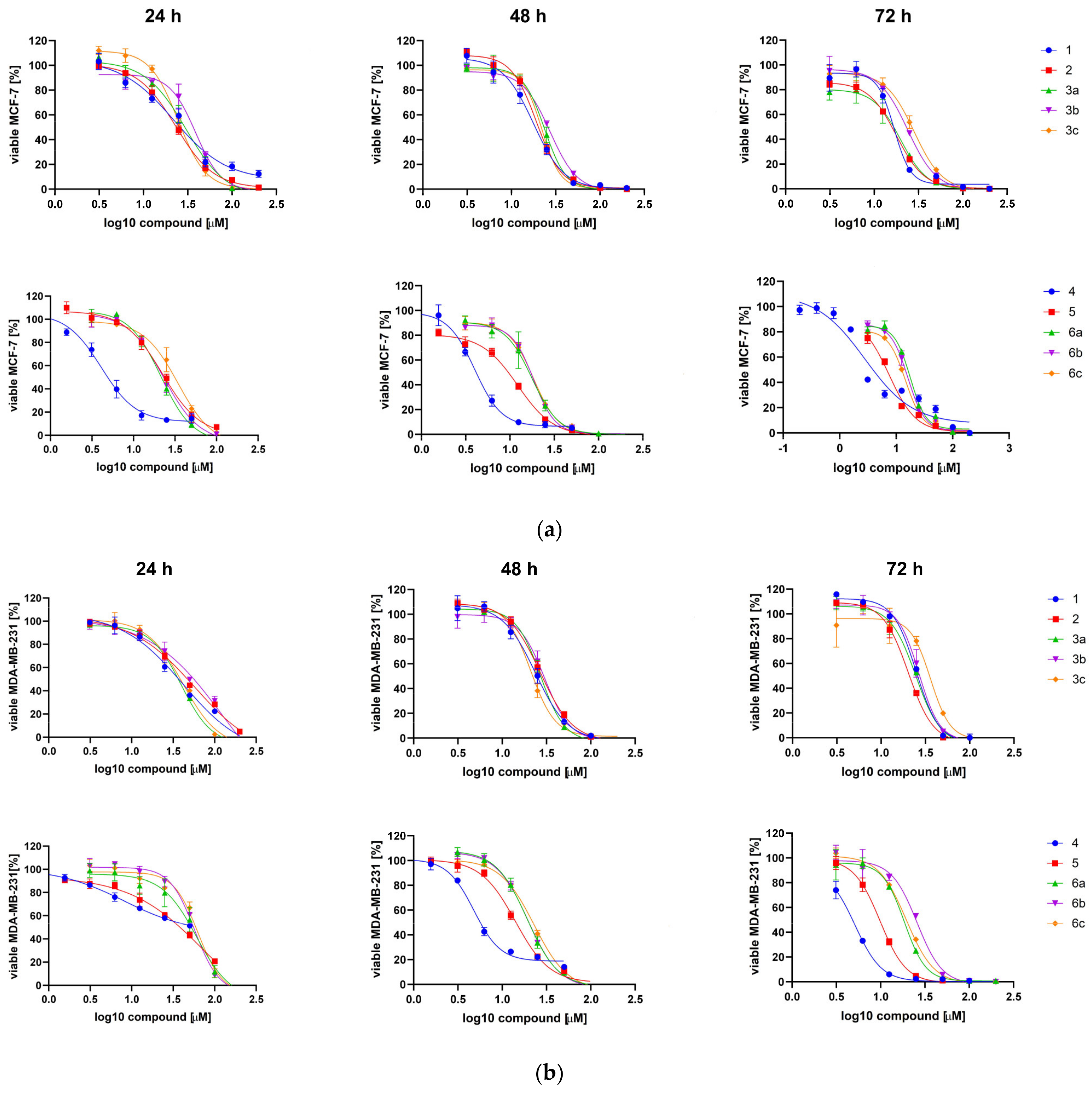
2.2.2. Cytotoxic Effect of TBBi Derivatives toward Breast Cancer Cells
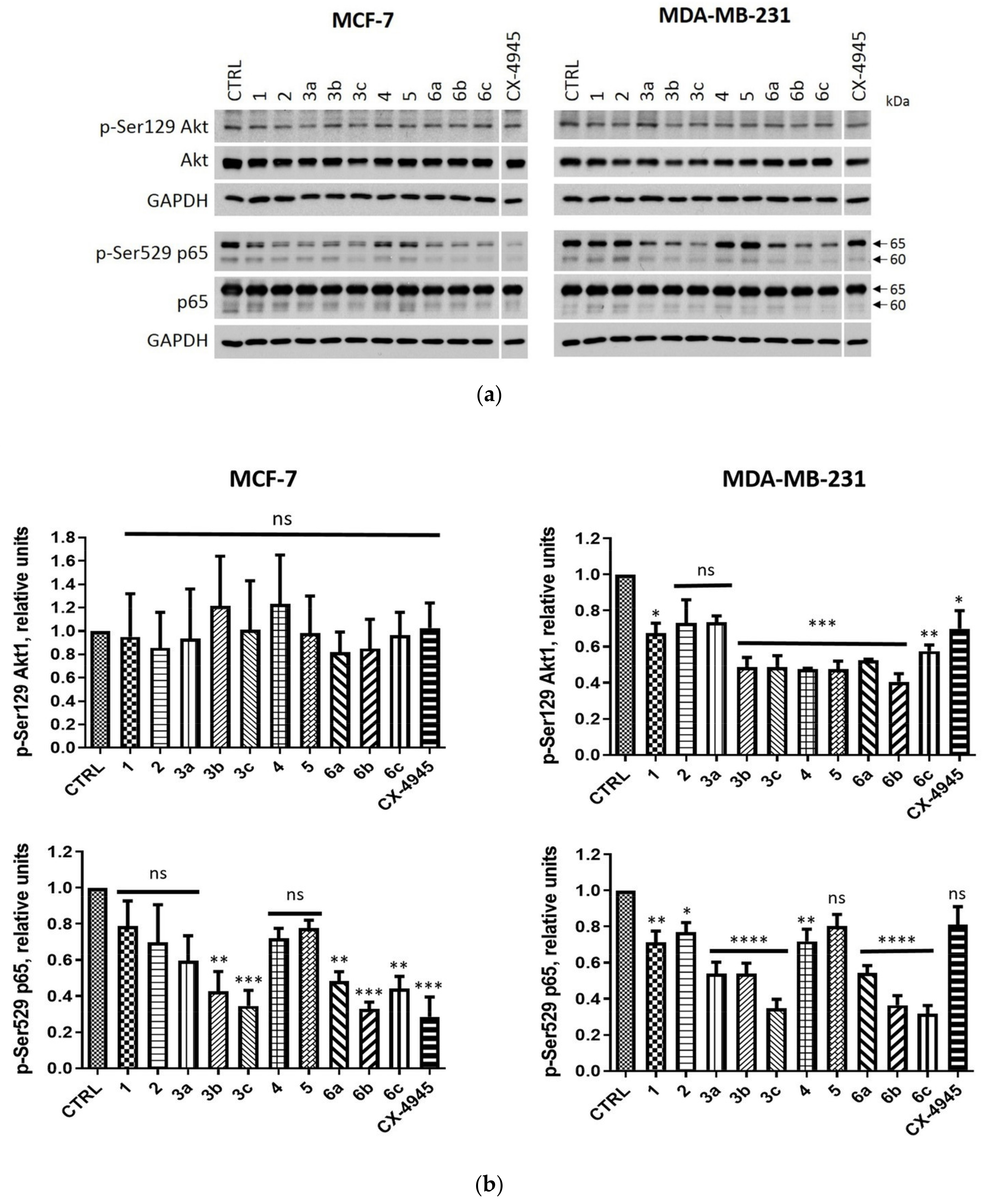
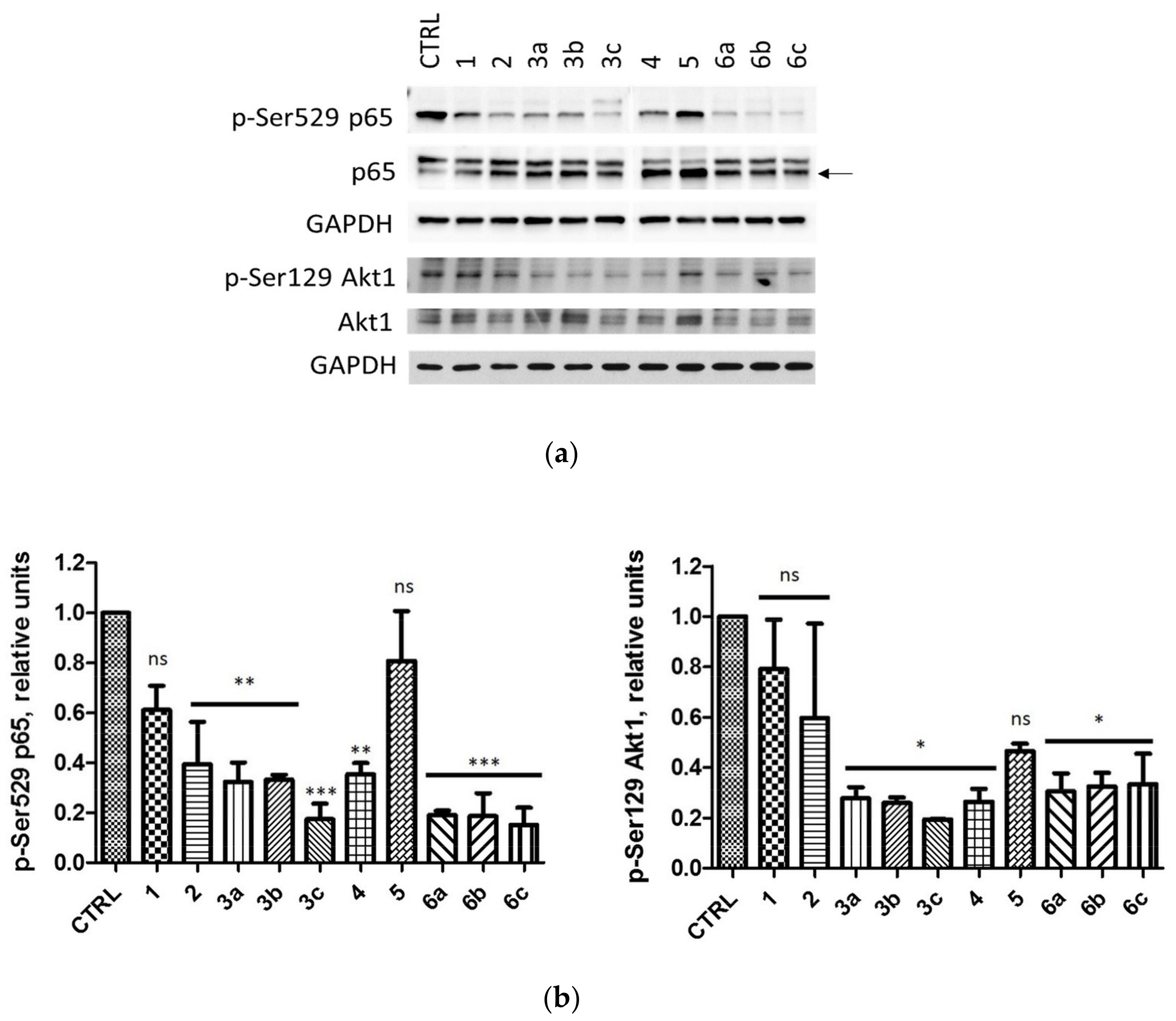
2.2.3. Intracellular Inhibition of Protein Kinase CK2 in MCF-7 and MDA-MB-231 Cells
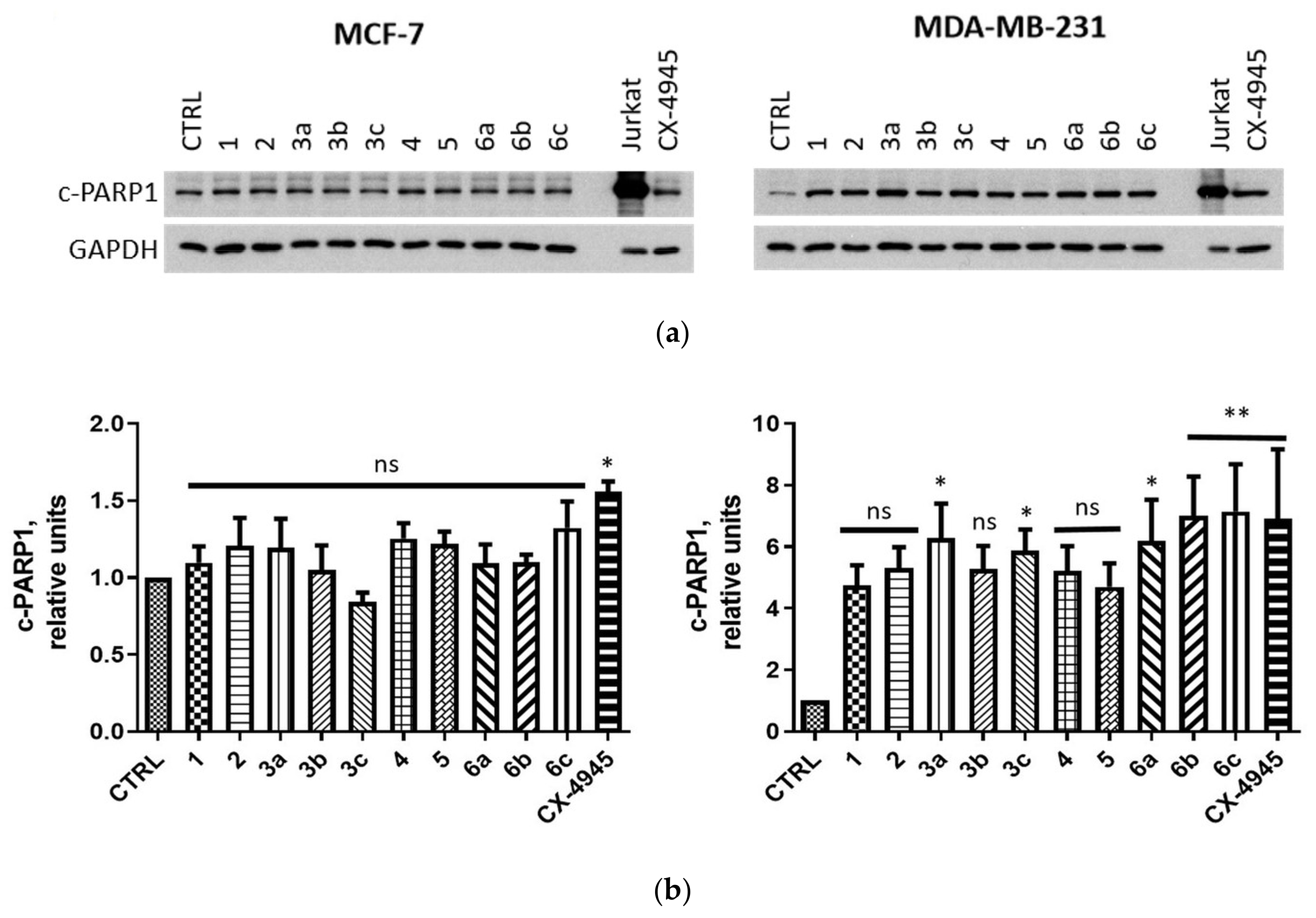
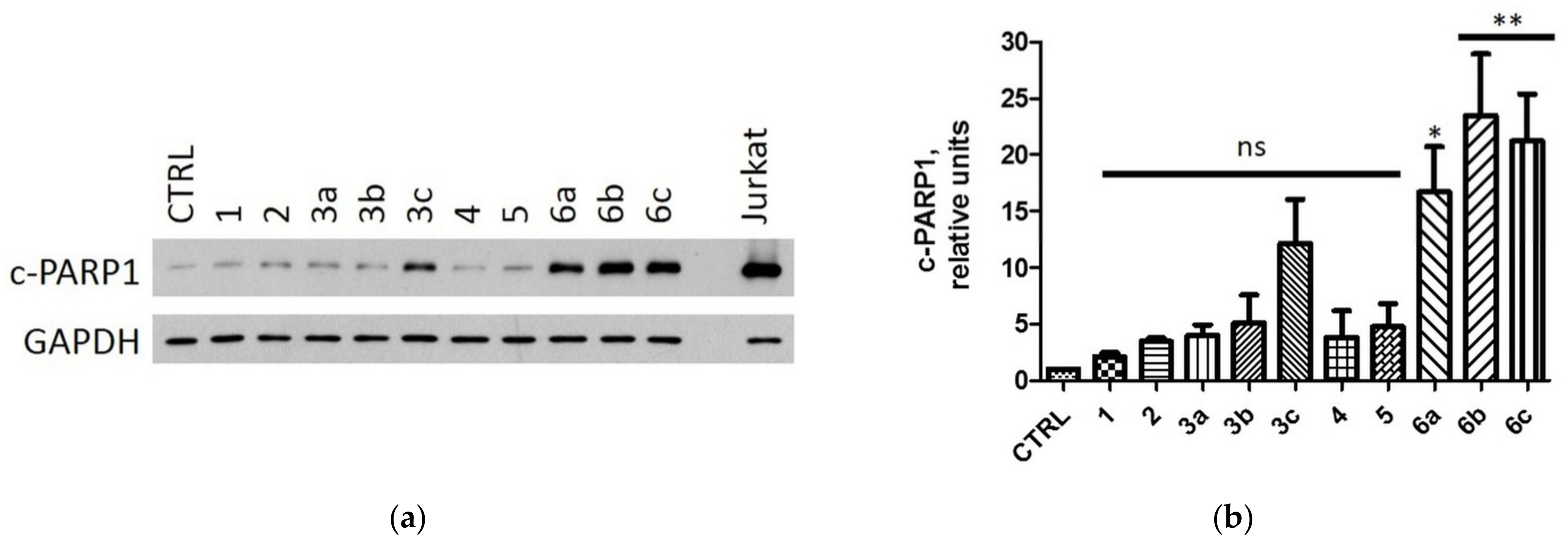
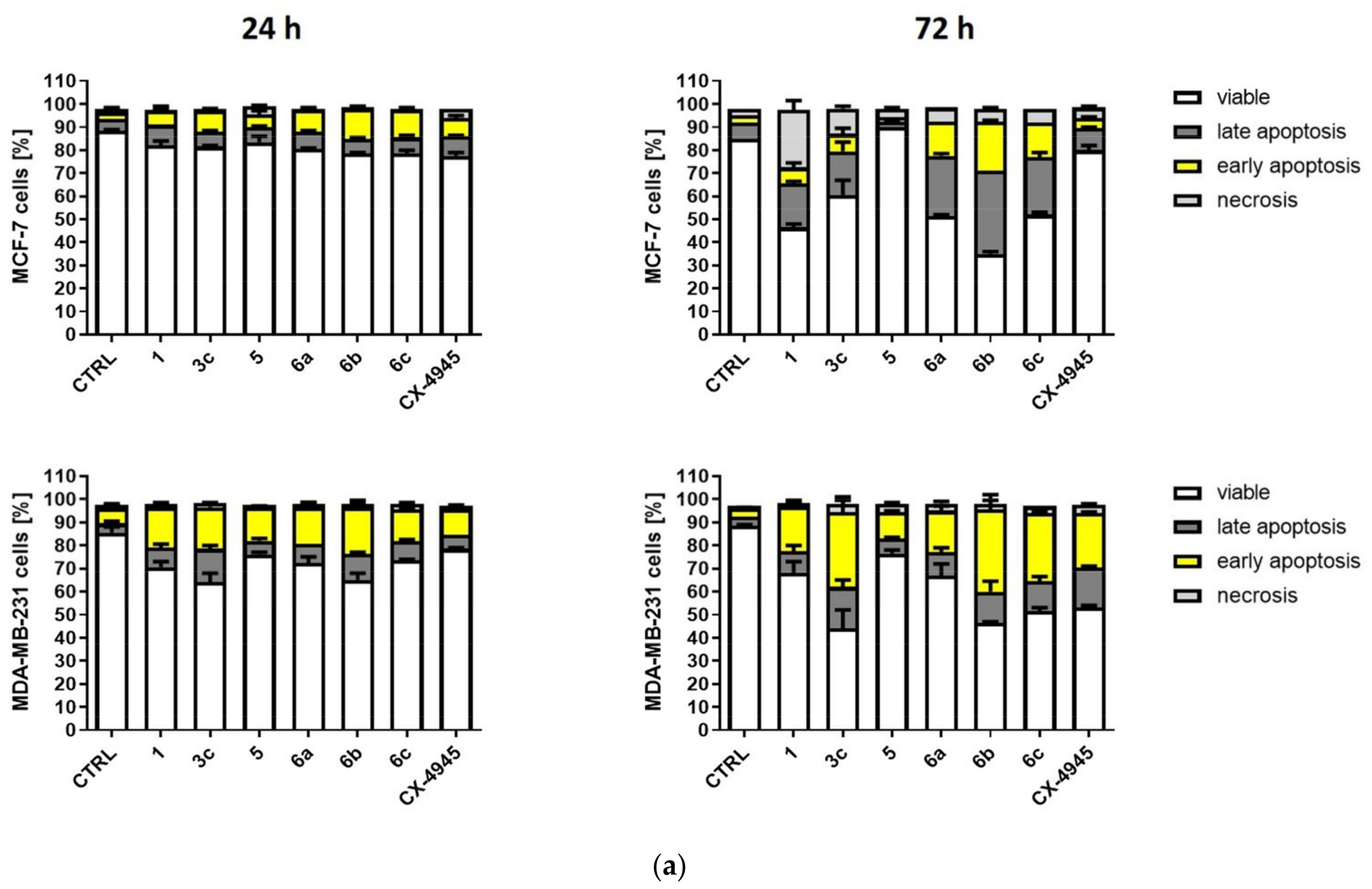
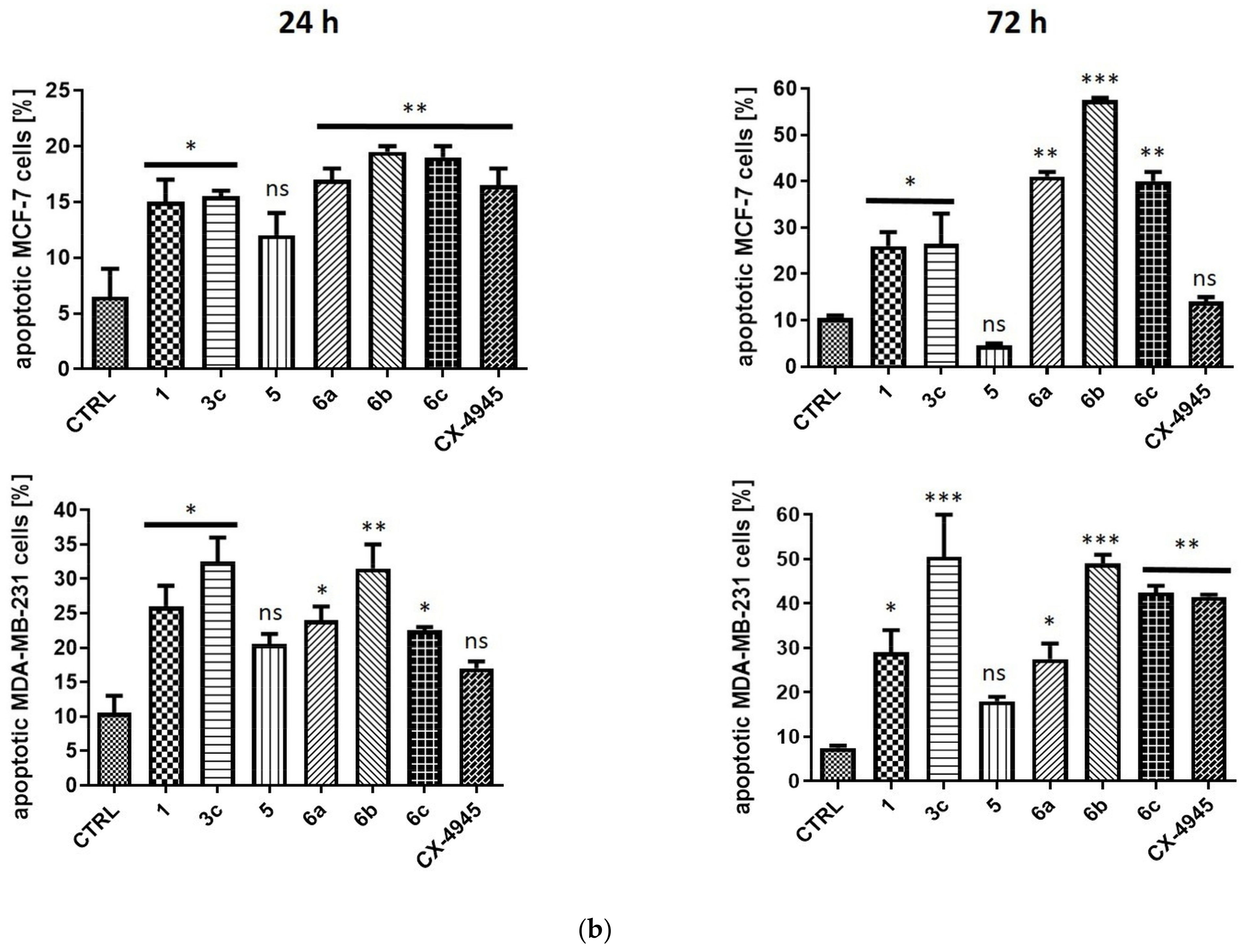
2.2.4. Induction of Apoptosis in MCF-7 and MDA-MB-231 Cells
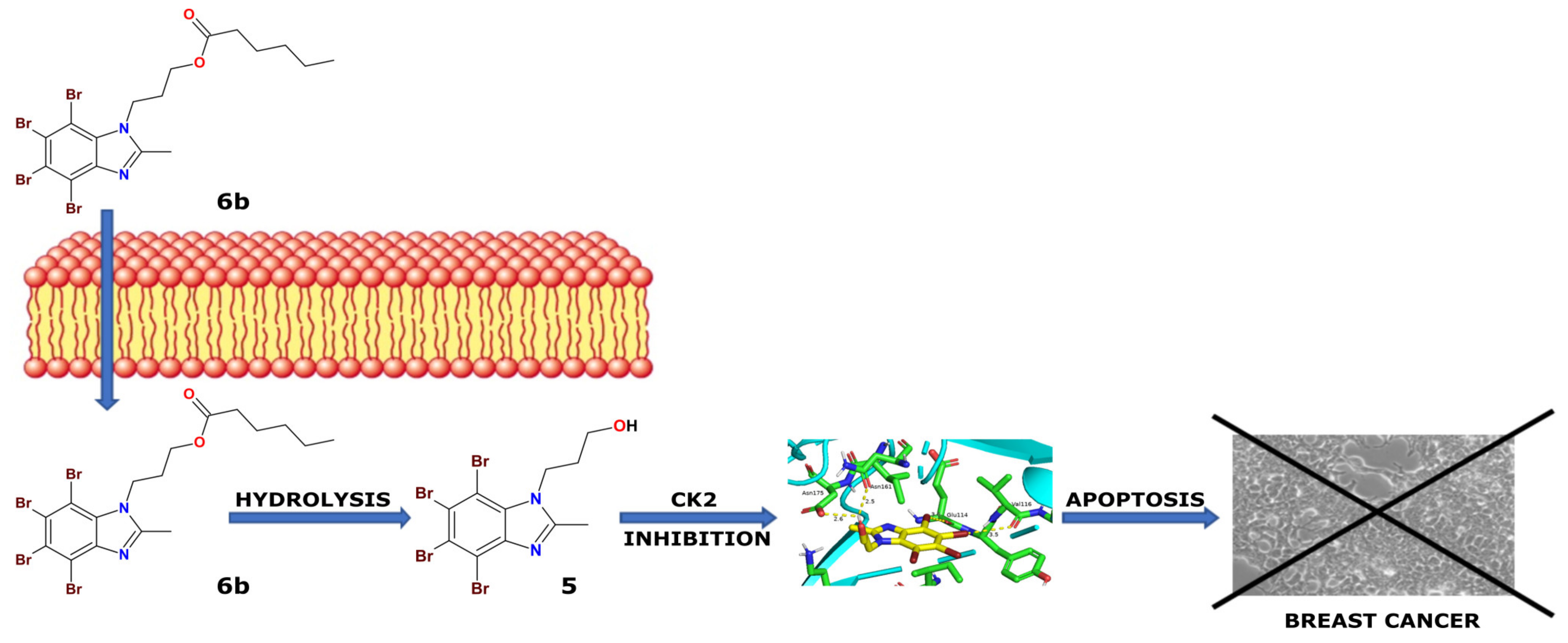
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
4.1.2. General Procedure for N-alkylation of 1 and 4
4.1.3. General Procedure for Esterification of 2 and 5
4.2. Biological Evaluation
4.2.1. Reagents and Antibodies
4.2.2. Inhibition of Recombinant CK2 and PIM1
4.2.3. Cell Culture and Agents Treatment
4.2.4. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT)-Based Viability Assay
4.2.5. Western Blotting
4.2.6. Densitometry
4.2.7. Detection of Apoptosis by Flow Cytometry
4.2.8. Cell Cycle Progression by Flow Cytometry
4.2.9. Statistical Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer—The road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef]
- Johnson, R.; Sabnis, N.; McConathy, W.J.; Lacko, A.G. The potential role of nanotechnology in therapeutic approaches for triple negative breast cancer. Pharmaceutics 2013, 5, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Emami, F.; Pathak, S.; Nguyen, T.T.; Shrestha, P.; Maharjan, S.; Kim, J.O.; Jeong, J.H.; Yook, S. Photoimmunotherapy with cetuximab-conjugated gold nanorods reduces drug resistance in triple negative breast cancer spheroids with enhanced infiltration of tumor-associated macrophages. J. Control. Release Off. J. Control. Release Soc. 2021, 329, 645–664. [Google Scholar] [CrossRef]
- Muley, H.; Fado, R.; Rodriguez-Rodriguez, R.; Casals, N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem. Pharmacol. 2020, 177, 113959. [Google Scholar] [CrossRef]
- Keihan Shokooh, M.; Emami, F.; Jeong, J.H.; Yook, S. Bio-inspired and smart nanoparticles for triple negative breast cancer microenvironment. Pharmaceutics 2021, 13, 287. [Google Scholar] [CrossRef]
- Cabrejos, M.E.; Allende, C.C.; Maldonado, E. Effects of phosphorylation by protein kinase CK2 on the human basal components of the RNA polymerase ii transcription machinery. J. Cell. Biochem. 2004, 93, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B.; Christenson, E.; Litchfield, D.W.; Krebs, E.G.; Eisenman, R.N. Myb DNA binding inhibited by phosphorylation at a site deleted during oncogenic activation. Nature 1990, 344, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, A.; Hingorani, K.; Negi, S.; Olson, M.O. Role of protein kinase CK2 phosphorylation in the molecular chaperone activity of nucleolar protein b23. J. Biol. Chem. 2003, 278, 9107–9115. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; Franchin, C.; Salizzato, V.; Cesaro, L.; Arrigoni, G.; Matricardi, L.; Pinna, L.A.; Donella-Deana, A. Protein kinase CK2 potentiates translation efficiency by phosphorylating eif3j at ser127. Biochim. Biophys. Acta 2015, 1853, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Roher, N.; Miró, F.; Gil, C.; Trujillo, R.; Aguilera, J.; Plana, M.; Itarte, E. Association of Protein Kinase CK2 with Eukaryotic Translation Initiation Factor Eif-2 and with Grp94/Endoplasmin. In A Molecular and Cellular View of Protein Kinase CK2; Springer: Boston, MA, USA, 1999; pp. 97–104. [Google Scholar]
- Gandin, V.; Masvidal, L.; Cargnello, M.; Gyenis, L.; McLaughlan, S.; Cai, Y.; Tenkerian, C.; Morita, M.; Balanathan, P.; Jean-Jean, O.; et al. mTORC1 and CK2 coordinate ternary and eIF4F complex assembly. Nat. Commun. 2016, 7, 11127. [Google Scholar] [CrossRef]
- Niechi, I.; Silva, E.; Cabello, P.; Huerta, H.; Carrasco, V.; Villar, P.; Cataldo, L.R.; Marcelain, K.; Armisen, R.; Varas-Godoy, M.; et al. Colon cancer cell invasion is promoted by protein kinase CK2 through increase of endothelin-converting enzyme-1c protein stability. Oncotarget 2015, 6, 42749–42760. [Google Scholar] [CrossRef]
- Patsoukis, N.; Li, L.; Sari, D.; Petkova, V.; Boussiotis, V.A. Pd-1 increases pten phosphatase activity while decreasing pten protein stability by inhibiting casein kinase 2. Mol. Cell. Biol. 2013, 33, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Vilk, G.; Canton, D.A.; Litchfield, D.W. Phosphorylation regulates the stability of the regulatory CK2beta subunit. Oncogene 2002, 21, 3754–3764. [Google Scholar] [CrossRef]
- Shen, J.; Channavajhala, P.; Seldin, D.C.; Sonenshein, G.E. Phosphorylation by the protein kinase CK2 promotes calpain-mediated degradation of ikappabalpha. J. Immunol. 2001, 167, 4919–4925. [Google Scholar] [CrossRef]
- Scaglioni, P.P.; Yung, T.M.; Choi, S.; Baldini, C.; Konstantinidou, G.; Pandolfi, P.P. CK2 mediates phosphorylation and ubiquitin-mediated degradation of the pml tumor suppressor. Mol. Cell. Biochem. 2008, 316, 149–154. [Google Scholar] [CrossRef]
- Homma, M.K.; Homma, Y. Cell cycle and activation of CK2. Mol. Cell. Biochem. 2008, 316, 49–55. [Google Scholar] [CrossRef]
- Ahmed, K. Joining the cell survival squad: An emerging role for protein kinase CK2. Trends Cell Biol. 2002, 12, 226–230. [Google Scholar] [CrossRef]
- Piazza, F.A.; Ruzzene, M.; Gurrieri, C.; Montini, B.; Bonanni, L.; Chioetto, G.; Di Maira, G.; Barbon, F.; Cabrelle, A.; Zambello, R.; et al. Multiple myeloma cell survival relies on high activity of protein kinase CK2. Blood 2006, 108, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Turowec, J.P.; Duncan, K.E.; Vilk, G.; Wu, C.; Luscher, B.; Li, S.S.; Gloor, G.B.; Litchfield, D.W. A peptide-based target screen implicates the protein kinase CK2 in the global regulation of caspase signaling. Sci. Signal. 2011, 4, ra30. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Akashi, M.; Matsuda, M.; Goto, K.; Miyata, Y.; Node, K.; Nishida, E. Involvement of the protein kinase CK2 in the regulation of mammalian circadian rhythms. Sci. Signal. 2009, 2, ra26. [Google Scholar] [CrossRef]
- Nunez de Villavicencio-Diaz, T.; Rabalski, A.J.; Litchfield, D.W. Protein kinase CK2: Intricate relationships within regulatory cellular networks. Pharmaceuticals 2017, 10, 27. [Google Scholar] [CrossRef]
- Salvi, M.; Sarno, S.; Cesaro, L.; Nakamura, H.; Pinna, L.A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim. Biophys. Acta 2009, 1793, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: Mechanistic rationale for drug combination therapy. Mol. Cancer Therap. 2012, 11, 994–1005. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef]
- Landesman-Bollag, E.; Romieu-Mourez, R.; Song, D.H.; Sonenshein, G.E.; Cardiff, R.D.; Seldin, D.C. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 2001, 20, 3247–3257. [Google Scholar] [CrossRef]
- Way, T.D.; Kao, M.C.; Lin, J.K. Apigenin induces apoptosis through proteasomal degradation of her2/neu in her2/neu-overexpressing breast cancer cells via the phosphatidylinositol 3-kinase/akt-dependent pathway. J. Biol. Chem. 2004, 279, 4479–4489. [Google Scholar] [CrossRef] [PubMed]
- Romieu-Mourez, R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. Sonenshein, Protein kinase CK2 promotes aberrant activation of nuclear factor-κb, transformed phenotype, and survival of breast cancer cells. Cancer Res. 2002, 62, 6770–6778. [Google Scholar]
- Gray, G.K.; McFarland, B.C.; Rowse, A.L.; Gibson, S.A.; Benveniste, E.N. Therapeutic CK2 inhibition attenuates diverse prosurvival signaling cascades and decreases cell viability in human breast cancer cells. Oncotarget 2014, 5, 6484–6496. [Google Scholar] [CrossRef] [PubMed]
- Lukowska-Chojnacka, E.; Winska, P.; Wielechowska, M.; Poprzeczko, M.; Bretner, M. Synthesis of novel polybrominated benzimidazole derivatives-potential CK2 inhibitors with anticancer and proapoptotic activity. Bioorg. Med. Chem. 2016, 24, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, K.; Winska, P.; Skierka, K.; Wielechowska, M.; Bretner, M. Synthesis, in vitro antiproliferative activity and kinase profile of new benzimidazole and benzotriazole derivatives. Bioorg. Chem. 2017, 72, 1–10. [Google Scholar] [CrossRef]
- Gallagher, T.F.; Fukushima, D.K.; Barry, M.C.; Dobriner, K. Recent Progress in Hormone Research; Academic Press: New York, NY, USA, 1951; Volume 6. [Google Scholar]
- Borowiecki, P.; Kraszewski, M. Highly efficient, solvent-free esterification of testosterone promoted by a recyclable polymer-supported tosylic acid catalyst under microwave irradiation. Arkivoc 2019, 2019, 288–305. [Google Scholar] [CrossRef]
- Samuels, L.T.; West, C.D. Vitamins and Hormones; Academic Press: New York, NY, USA, 1952; Volume 10. [Google Scholar]
- Glazko, A.J.E.; Edgerton, W.H.; Dill, W.A.; Lenz, W.R. Chloromycetln palmitate-a synthetic ester of chloromycetin. Antibiot. Chemother. 1952, 2, 234–242. [Google Scholar]
- Luber, A.D.; Flaherty, J.F., Jr. Famciclovir for treatment of herpesvirus infections. Ann. Pharmacother. 1996, 30, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Jarvest, R.L.; Sutton, D.; Vere Hodge, R.A. Famciclovir. Discovery and development of a novel antiherpesvirus agent. Pharm. Biotechnol. 1998, 11, 313–343. [Google Scholar]
- Takahashi, M.; Uehara, T.; Nonaka, M.; Minagawa, Y.; Yamazaki, R.; Haba, M.; Hosokawa, M. Synthesis and evaluation of haloperidol ester prodrugs metabolically activated by human carboxylesterase. Eur. J. Pharm. Sci. 2019, 132, 125–131. [Google Scholar] [CrossRef]
- Song, X.; Lorenzi, P.L.; Landowski, C.P.; Vig, B.S.; Hilfinger, J.M.; Amidon, G.L. Amino acid ester prodrugs of the anticancer agent gemcitabine: Synthesis, bioconversion, metabolic bioevasion, and hpept1-mediated transport. Mol. Pharm. 2005, 2, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.R.; Chamberlain, S.D.; Jones, L.A.; de Miranda, P.; Reynolds, D.J.; Peoples, M.E.; Krenitsky, T.A. 5′prime;-ester prodrugs of the varicella-zoster antiviral agent, 6-methoxypurine arabinoside. Antivir. Chem. Chemother. 2016, 3, 141–146. [Google Scholar] [CrossRef]
- Mahmoudian, M.; Eaddy, J.; Dawson, M. Enzymic acylation of 506u78 (2-amino-9-β-d-arabinofuranosyl-6-methoxy-9h-purine), a powerful new anti-leukaemic agent. Biotechnol. Appl. Microbiol. 1999, 29, 229–233. [Google Scholar]
- Zhang, X.; Li, X.; You, Q.; Zhang, X. Prodrug strategy for cancer cell-specific targeting: A recent overview. Eur. J. Med. Chem. 2017, 139, 542–563. [Google Scholar] [CrossRef] [PubMed]
- Abet, V.; Filace, F.; Recio, J.; Alvarez-Builla, J.; Burgos, C. Prodrug approach: An overview of recent cases. Eur. J. Med. Chem. 2017, 127, 810–827. [Google Scholar] [CrossRef]
- Otera, J.; Nishikido, J. Esterification: Methods, Reactions, and Applications, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009. [Google Scholar]
- Walther, R.; Rautio, J.; Zelikin, A.N. Prodrugs in medicinal chemistry and enzyme prodrug therapies. Adv. Drug Deliv. Rev. 2017, 118, 65–77. [Google Scholar] [CrossRef]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nature Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.L. Prodrugs in Drug Design and Development. In Textbook of Drug Design and Discovery; Stromgaard, K., Krogsgaard-Larsen, P., Madsen, U., Eds.; CRC Press: Boca Raton, FL, USA, 2017; pp. 155–173. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Borowiecki, P.; Winska, P.; Bretner, M.; Gizinska, M.; Koronkiewicz, M.; Staniszewska, M. Synthesis of novel proxyphylline derivatives with dual anti-candida albicans and anticancer activity. Eur. J. Med. Chem. 2018, 150, 307–333. [Google Scholar] [CrossRef]
- Chojnacki, K.; Winska, P.; Wielechowska, M.; Lukowska-Chojnacka, E.; Tolzer, C.; Niefind, K.; Bretner, M. Biological properties, and structural study of new aminoalkyl derivatives of benzimidazole and benzotriazole, dual inhibitors of CK2 and PIM1 kinases. Bioorg. Chem. 2018, 80, 266–275. [Google Scholar] [CrossRef]
- Najda-Bernatowicz, A.; Lebska, M.; Orzeszko, A.; Kopanska, K.; Krzywinska, E.; Muszynska, G.; Bretner, M. Synthesis of new analogs of benzotriazole, benzimidazole and phthalimide—potential inhibitors of human protein kinase CK2. Bioorg. Med. Chem. 2009, 17, 1573–1578. [Google Scholar] [CrossRef]
- Janeczko, M.; Orzeszko, A.; Kazimierczuk, Z.; Szyszka, R.; Baier, A. CK2alpha and CK2alpha’ subunits differ in their sensitivity to 4,5,6,7-tetrabromo- and 4,5,6,7-tetraiodo-1h-benzimidazole derivatives. Eur. J. Med. Chem. 2012, 47, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Wawro, A.M.; Wielechowska, M.; Bretner, M. Synthesis of new optically pure tetrabromobenzotriazole derivatives via lipase-catalyzed transesterification. J. Mol. Catal. B Enzym. 2013, 87, 44–50. [Google Scholar] [CrossRef]
- Borowiecki, P.; Wawro, A.M.; Winska, P.; Wielechowska, M.; Bretner, M. Synthesis of novel chiral TBBt derivatives with hydroxyl moiety. Studies on inhibition of human protein kinase CK2alpha and cytotoxicity properties. Eur. J. Med. Chem. 2014, 84, 364–374. [Google Scholar] [CrossRef]
- Lukowska-Chojnacka, E.; Staniszewska, M.; Bondaryk, M.; Maurin, J.K.; Bretner, M. Lipase-catalyzed kinetic resolution of novel antifungal n-substituted benzimidazole derivatives. Chirality 2016, 28, 347–354. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Chojnacki, K.; Lindenblatt, D.; Winska, P.; Wielechowska, M.; Toelzer, C.; Niefind, K.; Bretner, M. Synthesis, biological properties and structural study of new halogenated azolo[4,5-b]pyridines as inhibitors of CK2 kinase. Bioorg. Chem. 2021, 106, 104502. [Google Scholar] [CrossRef]
- Kumar, M.; Jaiswal, R.K.; Yadava, P.K.; Singh, R.P. An assessment of poly (ADP-ribose) polymerase-1 role in normal and cancer cells. BioFactors 2020, 46, 894–905. [Google Scholar] [CrossRef]
- Bursch, W.; Karwan, A.; Mayer, M.; Dornetshuber, J.; Frohwein, U.; Schulte-Hermann, R.; Fazi, B.; Di Sano, F.; Piredda, L.; Piacentini, M.; et al. Cell death and autophagy: Cytokines, drugs, and nutritional factors. Toxicology 2008, 254, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Viht, K.; Saaver, S.; Vahter, J.; Enkvist, E.; Lavogina, D.; Sinijarv, H.; Raidaru, G.; Guerra, B.; Issinger, O.G.; Uri, A. Acetoxymethyl ester of tetrabromobenzimidazole-peptoid conjugate for inhibition of protein kinase CK2 in living cells. Bioconjug. Chem. 2015, 26, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Kuo, P.C. The tumor microenvironment. Surg. Oncol. 2012, 21, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Douglas, L.; Delaney, A.; Houghton, J.A. Casein kinase ii (CK2) enhances death-inducing signaling complex (disc) activity in trail-induced apoptosis in human colon carcinoma cell lines. Oncogene 2005, 24, 2050–2058. [Google Scholar] [CrossRef]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of protein kinase ck2 as a key target in cancer therapy. BioFactors 2010, 36, 187–195. [Google Scholar] [CrossRef]
- Wang, S.; He, M.; Li, L.; Liang, Z.; Zou, Z.; Tao, A. Cell-in-cell death is not restricted by caspase-3 deficiency in mcf-7 cells. J. Breast Cancer 2016, 19, 231–241. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | Residual Activity of the Enzyme [%] | Ki [µM] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CK2α | CK2 α2β2 | PIM1 | CK2α | CK2 α2β2 | PIM1 | ||||
| 5 µM | 10 µM | 5 µM | 10 µM | 5 µM | 10 µM | ||||
| 1 | 19.3 ± 0.2 | 12.4 ± 0.1 | 9.6 ± 2.6 | 5.0 ± 1.2 | 11.2 ± 0.2 | 5.7 ± 0.5 | 0.58 [57] | 0.15 | 0.27 |
| 2 | 26.1 ± 5.3 | 18.5 ± 0.3 | 16.0 ± 1.8 | 7.6 ± 0.2 | 17.2 ± 2.3 | 10.1 ± 1.0 | 0.27 [58] | 0.28 ± 0.06 | 0.42 ± 0.02 |
| 3a | 71.0 ± 9.5 | 59.1 ± 3.7 | 58.0 ± 4.0 | 34.8 ± 3.4 | 65.0 ± 2.8 | 24.4 ± 0.9 | – | – | 2.23 ± 0.07 |
| 3b | 89.3 ± 8.0 | 83.0 ± 13.0 | 85.5 ± 8.3 | 83.1 ± 7.5 | 85.0 ± 0.1 | 74.2 ± 11.2 | – | – | – |
| 3c | 86.1 ± 4.2 | 79.2 ± 7.6 | 95.3 ± 2.9 | 92.7 ± 3.5 | 99.1 ± 15.7 | 95.4 ± 4.0 | – | – | – |
| 4 | 18.8 ± 1.1 | 7.7 ± 3.3 | 6.0 ± 0.9 | 2.9 ± 0.2 | 8.0 ± 0.9 | 4.5 ± 0.2 | 1.11 ± 0.30 [34] | 0.11 ± 0.04 | 0.21 ± 0.02 |
| 5 | 26.1 ± 3.5 | 16.3 ± 1.0 | 14.2 ± 2.0 | 9.1 ± 0.6 | 9.3 ± 2.6 | 3.4 ± 0.3 | 0.20 ± 0.08 | 0.05 ± 0.01 | 0.15 ± 0.08 |
| 6a | 81.5 ± 8.0 | 68.4 ± 9.8 | 75.6 ± 5.9 | 49.6 ± 0.8 | 38.8 ± 3.1 | 19.0 ± 0.1 | – | – | 1.94 ± 0.16 |
| 6b | 76.8 ± 1.9 | 74.3 ± 0.1 | 85.4 ± 0.6 | 71.4 ± 0.2 | 62.4 ± 8.3 | 46.4 ± 2.7 | – | – | – |
| 6c | 89.9 ± 5.2 | 84.3 ± 10.4 | 90.2 ± 4.7 | 80.8 ± 1.9 | 95.2 ± 9.4 | 79.8 ± 8.6 | – | – | – |
| CX-4945 | 0 | 0 | 0 | 0 | 2.3 ± 0.1 | 1.2 ± 0.1 | – | – | – |
| Cpd. | EC50 ± SD [μM] | Log P * | Log S * | |||||
|---|---|---|---|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | |||||||
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | |||
| 1 | 20.83 ± 1.49 | 18.82 ± 1.60 | 16.86 ± 1.36 | 51.90 ± 14.43 | 25.11 ± 2.04 | 24.92 ± 0.25 | 4.33 | −6.23 |
| 2 | 24.09 ± 1.39 | 20.08 ± 1.36 | 19.60 ± 1.68 | 54.62 ± 4.32 | 27.19 ± 0.49 | 20.57 ± 0.86 | 3.93 | −5.37 |
| 3a | 29.34 ± 1.24 | 23.05 ± 0.77 | 21.51 ± 1.86 | 40.80 ± 1.58 | 27.84 ± 1.31 | 24.52 ± 0.48 | 5.51 | −6.55 |
| 3b | 39.56 ± 1.06 | 28.38 ± 1.71 | 24.77 ± 1.33 | 95.58 ± 20.40 | 32.21 ± 3.26 | 27.30 ± 1.26 | 6.40 | −7.49 |
| 3c | 24.34 ± 0.25 | 21.14 ± 1.23 | 29.85 ± 1.45 | 45.11 ± 5.43 | 23.68 ± 3.97 | 32.77 ± 3.25 | 7.29 | −8.43 |
| 3d | >100 | >100 | >100 | >100 | >100 | >100 | 9.07 | −10.28 |
| 4 | 4.12 ± 0.03 | 4.20 ± 0.29 | 2.66 ± 0.19 | 12.78 ± 6.60 | 5.35 ± 0.82 | 4.73 ± 0.48 | 4.46 | −5.90 |
| 5 | 17.62 ± 6.35 | 15.96 ± 5.31 | 4.90 ± 2.71 | 57.84 ± 20.79 | 21.03 ± 9.85 | 9.99 ± 0.44 | 4.05 | −5.04 |
| 6a | 20.15 ± 1.74 | 18.07 ± 0.52 | 16.47 ± 1.13 | 52.17 ± 11.68 | 20.49 ± 1.97 | 16.85 ± 4.07 | 5.64 | −6.21 |
| 6b | 22.35 ± 1.12 | 18.59 ± 0.18 | 18.31 ± 2.86 | 51.43 ± 8.22 | 21.16 ± 2.40 | 22.06 ± 3.18 | 6.53 | −7.15 |
| 6c | 25.63 ± 12.30 | 20.46 ± 2.92 | 14.85 ± 2.14 | 61.28 ± 2.23 | 29.83 ± 9.85 | 21.30 ± 2.24 | 7.42 | −8.08 |
| 6d | >100 | >100 | >100 | >100 | >100 | >100 | 9.19 | −9.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chojnacki, K.; Wińska, P.; Karatsai, O.; Koronkiewicz, M.; Milner-Krawczyk, M.; Wielechowska, M.; Rędowicz, M.J.; Bretner, M.; Borowiecki, P. Synthesis of Novel Acyl Derivatives of 3-(4,5,6,7-Tetrabromo-1H-benzimidazol-1-yl)propan-1-ols—Intracellular TBBi-Based CK2 Inhibitors with Proapoptotic Properties. Int. J. Mol. Sci. 2021, 22, 6261. https://doi.org/10.3390/ijms22126261
Chojnacki K, Wińska P, Karatsai O, Koronkiewicz M, Milner-Krawczyk M, Wielechowska M, Rędowicz MJ, Bretner M, Borowiecki P. Synthesis of Novel Acyl Derivatives of 3-(4,5,6,7-Tetrabromo-1H-benzimidazol-1-yl)propan-1-ols—Intracellular TBBi-Based CK2 Inhibitors with Proapoptotic Properties. International Journal of Molecular Sciences. 2021; 22(12):6261. https://doi.org/10.3390/ijms22126261
Chicago/Turabian StyleChojnacki, Konrad, Patrycja Wińska, Olena Karatsai, Mirosława Koronkiewicz, Małgorzata Milner-Krawczyk, Monika Wielechowska, Maria Jolanta Rędowicz, Maria Bretner, and Paweł Borowiecki. 2021. "Synthesis of Novel Acyl Derivatives of 3-(4,5,6,7-Tetrabromo-1H-benzimidazol-1-yl)propan-1-ols—Intracellular TBBi-Based CK2 Inhibitors with Proapoptotic Properties" International Journal of Molecular Sciences 22, no. 12: 6261. https://doi.org/10.3390/ijms22126261
APA StyleChojnacki, K., Wińska, P., Karatsai, O., Koronkiewicz, M., Milner-Krawczyk, M., Wielechowska, M., Rędowicz, M. J., Bretner, M., & Borowiecki, P. (2021). Synthesis of Novel Acyl Derivatives of 3-(4,5,6,7-Tetrabromo-1H-benzimidazol-1-yl)propan-1-ols—Intracellular TBBi-Based CK2 Inhibitors with Proapoptotic Properties. International Journal of Molecular Sciences, 22(12), 6261. https://doi.org/10.3390/ijms22126261