
Role of Nitric Oxide in Gene Expression Regulation during Cancer: Epigenetic Modifications and Non-Coding RNAs



Abstract
:1. Role of Nitric Oxide (NO) in Epigenetic Regulation during Cancer
1.1. NO and Nitrosative Stress
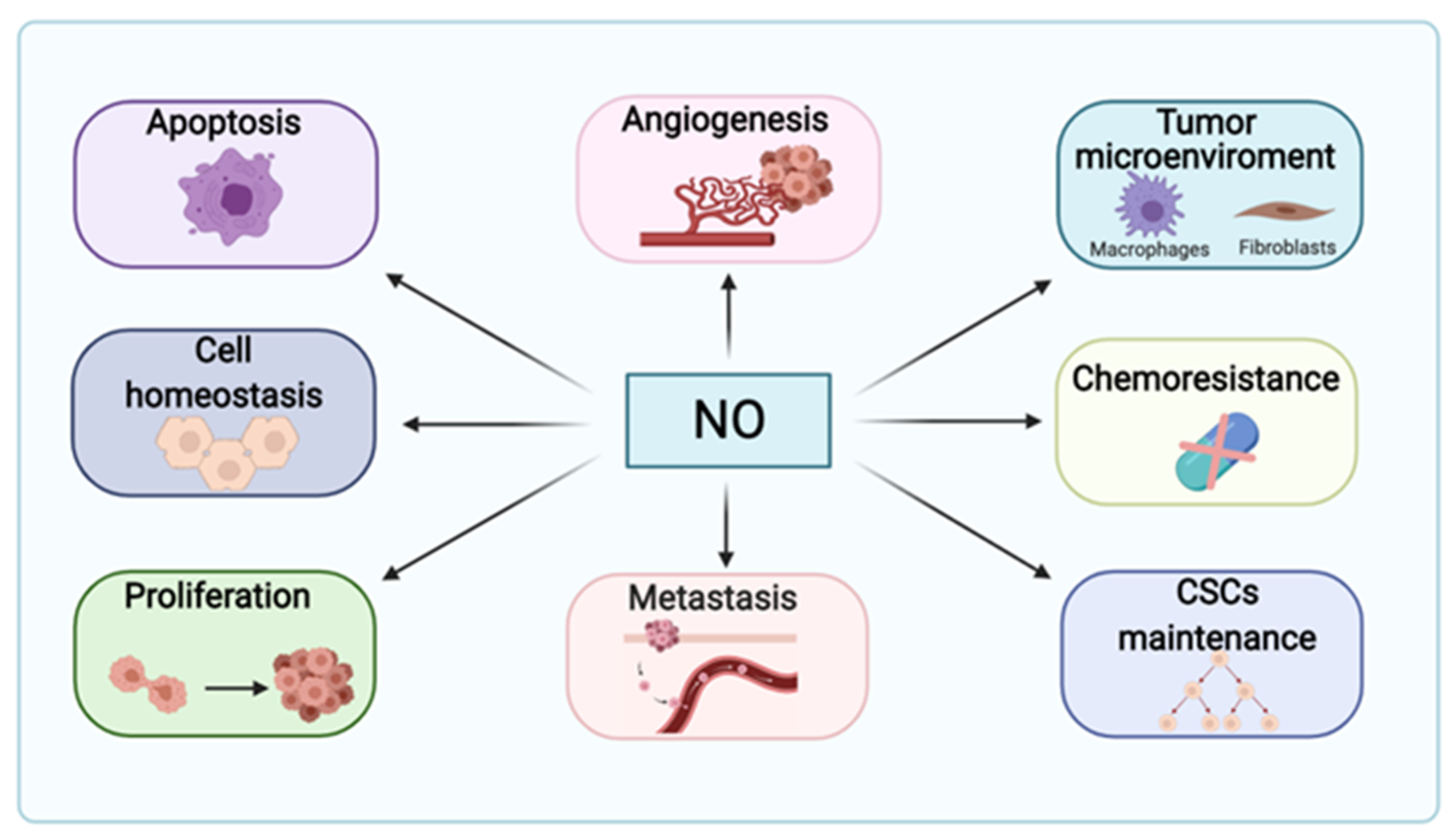
1.2. Nitric Oxide in Cancer Pathogenesis
1.3. Introducing Epigenetic Regulation Induced by NO
1.3.1. NO and DNA Methylation
1.3.2. Histone Posttranslational Modifications in Cancer
Histone Acetylation
Histone Methylation

{kind=link}
{kind=link}
{kind=link}
| Epigenetic Regulation | Enzyme | Transcriptional Role | Crosstalk between NO and Epigenetic Regulators | Impact of the Regulatory Mechanism in Carcinogenesis | References |
|---|---|---|---|---|---|
| DNA methylation | DNMT not specified | Transcriptional repression | NOS-2-derived NO reduces tumor suppression genes expression | Pro-tumoral | [45] |
| DNMT not specified | Transcriptional repression | NO induces E-cadherin methylation by IL-1B decreasing E-cadherin expression | Pro-tumoral | [44] | |
| DNMT not specified | Ectopic expression | NO causes ectopic expression of AID and enhances NOS2 expression | Pro-tumoral | [46] | |
| Histone deacetylation | HDAC6 | Transcriptional repression | NO induces HDAC6 S-nitrosation | Pro-tumoral | [60] |
| HDAC2 | Transcriptional repression | NO S-nitrosation weakens HDAC2 enzymatic function | Anti-tumoral | [58] | |
| CBP | Transcriptional repression | CBP silencing decreases NO production by downregulation NOS-3 | Anti-tumoral | [61,62] | |
| SIRT1 | |||||
| Histone methylation | G9a | Transcriptional repression | NO downregulates expression | Anti-tumoral | [64,67,68] |
| SETDB2 | NO upregulates expression | Pro-tumoral | [64,69] | ||
| SUV39H2 | |||||
| SUV30H1 | NO indirectly targets SUV20H1 for proteasomal degradation | Anti-tumoral | [70,71,72] | ||
| MLL | Transcriptional activation | Not described | Pro-tumoral | [74] | |
| SET-1A | SET-1A trimethylates NOS2 promoter in response to IL-1 | Pro-tumoral | [75] | ||
| EZH2 | Transcriptional repression | EZH2 does not control NOS2 expression. Other mechanism should be involved | Pro-tumoral | [79,80] | |
| Histone demethylation | KDM3A | Transcriptional activation | NO inhibits KDM3A by forming a nitrosyl–iron complex | Anti-tumoral | [64,65,66] |
| KDM3B | NO upregulates expression. Compensatory mechanism in response to NO mediated KDM3A inhibition | Not described | [64] | ||
| KDM4A | |||||
| KDM4B | |||||
| KDM4C | |||||
| KDM4D | |||||
| KDM1 | |||||
| KDM7A | |||||
| KDMA | Transcriptional repression | Not described | Pro-tumoral | [76] | |
| KDMB | [77,78] | ||||
| KDM2A | Transcriptional repression | NO promotes the expression of Oct-4, which is related to reduced expression of demethylase KDM2A | Pro-tumoral | [81,82] |
Histone Phosphorylation
1.3.3. Non-Coding RNAs
Small RNAs
Long Non-Coding RNAs
| Type of Cancer | Expression | Molecular Mechanism | Interaction with NO | Impact of the Regulatory Mechanism in Carcinogenesis | References | ||||
|---|---|---|---|---|---|---|---|---|---|
| Control | Cancer | ||||||||
| miRNAs | miR-29b/c | Gastric cancer | − | + | Expression of miR-29b/c is regulated by NOS2 | Not specified | NOS2↑–miR-29b/c↑–PTEN↓-Migration↑–Apoptosis↓ | NOS2 regulates the expression of miR-29b/c, which in turns reduces PTEN and apoptosis, and increases migration | [92,93,94] |
| miR-335, miR-543 | Prostate cancer/ Liver cancer | + | − | Post-transcriptional regulation of NOS3 | NOS3 mRNA degradation (miRNA target) | miR-335, miR-543↓–NOS3↑–Metastatic potential↑ | miR-335 and miR-543 target NOS3 mRNA for degradation. In cancer, downregulation of these miRNAs, increases NOS3 expression leading to higher metastatic potential | [95,96] | |
| miR-193b | Breast cancer | + | − | Post-transcriptional regulation of NOS2 regulator DDHA1 | DDHA1 mRNA degradation (miRNA target) | miR-193↓–DDAH1↑–ADMA↓–NOS2↑–Angiogenesis↑ | Downregulation of miR-193b reduces DDAH1 mRNA degradation, which increases ADMA elimination and consequent increased NOS2 activity. This leads to increased angiogenesis | [97] | |
| miR-16 | Pan-cancer (macrophages) | + | − | NO production | Not specified | miR-16↓–NO production↓–Pro-tumoral microenvironment↑ and miR-16↓–PD-L1↑–Pro-tumoral microenvironment↑ | miR-16 in M1 macrophages is able to increase NO production, leading to an anti-tumoral microenvironment. Also, miR-16 targets PD-L1 mRNA for degradation, leading to reduced immunosuppression. In M2 macrophages, downregulation of miR-16 coincides in reduced NO production | [99] | |
| miR-155 | Pan-cancer (macrophages) | + | − | Post-transcriptional regulation of NOS2 | Not specified | miR-155↓–NOS2↓–FGF2↑–Proliferation↑ | Downregulation of miR-155 decreases NOS2 expression and increases FGF2, promoting tumor proliferation | [100] | |
| miR-155 | Liver cancer | − | + | Exogenous NO increases miR-155 expression | Not specified | miR-155↑–tumor suppressor gene P21WAF/CIP1↓ | In liver cancer, upregulation of miR-155 by exogenous NO donors, blocks tumor suppressor gene P21WAF/CIP1 | [102,103] | |
| miR-204 | Acute myeloid leukemia | + | − | Post-transcriptional regulation of SIRT1, NOS2 and COX2 | Not specified | miR-204↑–SIRT1↓/NOS2↓/COX2↓ | In AML cells, miR-204 reduces expression of SIRT1, COX2 and NOS2 exerting proapoptotic and antiproliferative properties | [118] | |
| miR-939-5p | Triple-negative breast cancer | + | − | Post-transcriptional regulation of NOS2 | Not specified | miR-939-5p↑–NOS2↑–NO↑ | miR-939-5p downregulates NOS2 expression in cultured human hepatocytes and in TNBC | [119] | |
| miR-148b-3p | Liver cancer (Hepatic sinusoidal endothelial cells) | + | − | Post-transcriptional regulation of NOS3 and NOX4 | NOX4 mRNA degradation (miRNA target) | miR-148b-3p↑–NOS3↑/NO↑–NOX4↓ | miR-148b-3p regulates negatively NOX4, it also enhances NOS3 expression and NO production in HSEC | [120] | |
| miR-122 | Liver cancer | + | − | Post-transcriptional regulation of SLC7A1 arginine transporter | SLC7A1 mRNA degradation (miRNA target) | miR-122↓–SLC7A1↑–Arginine↑–NO production↑–Cell proliferation↑ | Downregulation of miR-122 promotes cell proliferation in liver cancer through upregulation of NO production. In particular, miR-122 targets arginine transporter SLC7A1. Under circumstances of reduced expression of miR-122, SLC7A1 is not degraded and arginine availability increases | [104] | |
| lncRNAs | UCA1 | Acute myeloid leukemia | − | + | Post-transcriptional regulation | miR-204 mRNA degradation | UCA1↑–miR-204↓–SIRT1↑/NOS2↑/COX2↑ | UCA1 downregulates miR-204 expression and it enhances expression of SIRT1, NOS2 and COX2 | [118] |
| HEIH | Triple-negative breast cancer | − | + | Post-transcriptional regulation | miR-939-5p degradation | HEIH↑–miR-939-5p↓–NOS2↑–NO↑ | In TNBC HEIH decreases miR-939-5p expression, which consequently enhances NOS2 expression and NO production | [119] | |
| H19 | Liver cancer (Hepatic sinusoidal endothelial cells) | − | + | Post-transcriptional regulation | miR-148b-3p degradation | H19↑–miR-148b-3p ↓–NOS3↓/NO↓–NOX4↑ | H19 negatively regulates miR-148b-3p, so it turns to downregulate NOS3/NO and upregulates its direct target NOX4 in HSEC | [120] | |
2. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AID | Activation-induced cytidine deaminase |
| AML | acute myeloid leukemia |
| AFP | alpha-fetoprotein |
| ADMA | asymmetric dimethylarginine |
| AMPK | AMP-activated protein kinase |
| BE | Barrett’s esophagus |
| BCP-ALL | B-cell precursor-acute lymphoblastic leukemia |
| CAFs | cancer associated fibroblasts |
| COX2 | cyclooxygenase-2 |
| ceRNA | competing endogenous RNA |
| CBP | CREB-binding protein |
| cGMP | cyclic guanosine monophosphate |
| DDAH1 | dimethylarginine dimethylaminohydrolase 1 |
| DSBs | DNA double-strand breaks |
| DNMTs | DNA methyltransferases |
| DNA-PK | DNA-dependent protein kinase |
| ER | endoplasmic reticulum |
| eNOS/NOS3 | endothelial NOS |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial mesenchymal transition |
| EACs | esophageal adenocarcinoma cells |
| ERrα | estrogen receptor α |
| ERβ | estrogen receptor β |
| FGF2 | fibroblast growth factor 2 |
| FAD | flavin adenine dinucleotide |
| FMN | flavin adenine mononucleotide |
| Grx | glutaredoxin |
| HEIH | HCC upregulated EZH2-associated lncRNA |
| HSEC | hepatic sinusoidal endothelial cells |
| HCC | hepatocellular carcinoma |
| HGF | hepatocyte growth factor |
| HATs | histone acetyltransferases |
| HDACs | histone deacetylases |
| HMTs | histone methyltransferases |
| HOTAIR | HOX transcript antisense intergenic RNA |
| HIF1α | hypoxia inducible factor 1α |
| iNOS/NOS2 | inducible NOS |
| IFN-γ | interferon-γ |
| IL-1β | interleukin-1β |
| CSCs | cancer stem cells |
| KDM2A | lysine demethylase 2A |
| KDM3A | lysine demethylase 3A |
| lncRNAs | long non-coding RNAs |
| mTOR | mammalian target of rapamycin |
| MMP1 | matrix metalloproteinase 1 |
| MMP3 | matrix metalloproteinase 3 |
| METTL6 | methyltransferase-like protein 6 |
| miRNAs | microRNAs |
| NOX4 | NADPH oxidase 4 |
| nNOS/NOS1 | neuronal NOS |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NOS | nitric oxide synthases |
| NO | nitric oxide |
| NO-ASA | NO-releasing acetylsalicylic acid |
| ncRNAs | non-coding RNAs |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| PTEN | phosphatase and tensin homolog |
| PIKKs | phosphoinositide 3-kinase-related protein kinases |
| PPAT | phosphoribosyl pyrophosphate amidotransferase |
| piRNAs | Piwi-interacting RNAs |
| PTMs | post-translational modifications |
| rRNAs | ribosomal RNAs |
| POLR3G | RNA polymerase III subunit G |
| SETDB2 | SET domain bifurcated 2 |
| SNAP | S-nitroso-N-acetylpenicillamine |
| GSNO | S-nitrosoglutathione |
| siRNAs | small interfering RNAs |
| snRNAs | small nuclear RNAs |
| snoRNAs | small nucleolar RNAs |
| SNOC | S-nitrosoglutathione-oligosaccharide-chitosan |
| sdnRNAs | snRNA/snoRNA-derived nuclear RNAs |
| SNP | sodium nitroprusside |
| SUV30H1 | suppressor of variegation 3–9 homolog 1 |
| SUV39H2 | suppressor of variegation 3–9 homolog 2 |
| TNBC | triple-negative breast cancer |
| TAMs | tumor-associated macrophages |
| BH4 | tetrahydrobiopterin |
| Trx | thioredoxin |
| tRNAs | transfer RNAs |
| TGF-β | transforming growth factor β |
| TNF-α | tumor necrosis factor α |
| UCA1 | urothelial carcinoma-associated 1 |
References
- Somasundaram, V.; Basudhar, D.; Bharadwaj, G.; No, J.H.; Ridnour, L.A.; Cheng, R.Y.; Fujita, M.; Thomas, D.D.; Anderson, S.; McVicar, D.W.; et al. Molecular Mechanisms of Nitric Oxide in Cancer Progression, Signal Transduction, and Metabolism. Antioxid. Redox Signal. 2019, 30, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar] [CrossRef]
- Oswald, S.E.; Griepentrog, M.; Schirmer, M.; Balcke, G.U. Interplay between oxygen demand reactions and kinetic gas–water transfer in porous media. Water Res. 2008, 42, 3579–3590. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Koesling, D. Regulation of Nitric Oxide-Sensitive Guanylyl Cyclase. Circ. Res. 2003, 93, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 78, 931–936. [Google Scholar] [CrossRef]
- Muntané, J.; Angel, J.; Marín, L.M.; Padillo, F.J. Nitric oxide and cell death in liver cancer cells. Mitochondrion 2013, 13, 257–262. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, L.; Tian, Z.; Wang, J.; Yang, Y.; Liu, J.; Chen, Y.; Hu, C.; Chen, T.; Zhao, Y.; et al. Nitric oxide inhibits autophagy and promotes apoptosis in hepatocellular carcinoma. Cancer Sci. 2019, 110, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Li, L.; Zhang, Q.; Yang, X.; Zou, Z.; Hao, B.; Marincola, F.M.; Liu, Z.; Zhong, Z.; Wang, M.; et al. NOS1 S-nitrosylates PTEN and inhibits autophagy in nasopharyngeal carcinoma cells. Cell Death Discov. 2017, 3, 17011. [Google Scholar] [CrossRef]
- Augsten, M.; Sjöberg, E.; Frings, O.; Vorrink, S.U.; Frijhoff, J.; Olsson, E.; Borg, Å.; Östman, A. Cancer-Associated Fibroblasts Expressing CXCL14 Rely upon NOS1-Derived Nitric Oxide Signaling for Their Tumor-Supporting Properties. Cancer Res. 2014, 74, 2999–3010. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, X.; Xu, P.; Li, K.; Ye, S.; Zeng, S.; Huang, M.; Gao, W.; Chen, J.; Zhang, Q.; et al. Mitochondrial NOS1 suppresses apoptosis in colon cancer cells through increasing SIRT3 activity. Biochem. Biophys. Res. Commun. 2019, 515, 517–523. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Bardi, G.T.; Smith, M.A.; Hood, J.L. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine 2018, 105, 63–72. [Google Scholar] [CrossRef]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230. [Google Scholar] [CrossRef] [Green Version]
- Dávila-González, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.-F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.H.; et al. Pharmacological Inhibition of NOS Activates ASK1/JNK Pathway Augmenting Docetaxel-Mediated Apoptosis in Triple-Negative Breast Cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Dave, B.; Gonzalez, D.D.; Liu, Z.-B.; Li, X.; Wong, H.; Granados, S.; Ezzedine, N.E.; Sieglaff, D.H.; Ensor, J.; Miller, K.D.; et al. Role of RPL39 in Metaplastic Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw292. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; He, P.; Gaida, M.; Yang, S.; Schetter, A.J.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.; Lee, D.; et al. Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer. Oncotarget 2016, 7, 52993–53004. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Wang, Y.; Tian, D.-A.; Yang, J.; Yang, Y.-Z. Decreased levels of nitric oxide production and nitric oxide synthase-2 expression are associated with the development and metastasis of hepatocellular carcinoma. Mol. Med. Rep. 2012, 6, 1261–1266. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Tsung, A.; Huang, H.; Du, Q.; Yang, M.; Deng, M.; Xiong, S.; Wang, X.; Zhang, L.; et al. iNOS promotes CD24+CD133+liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10127–E10136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeguchi, M.; Ueta, T.; Yamane, Y.; Hirooka, Y.; Kaibara, N. Inducible nitric oxide synthase and survivin messenger RNA expression in hepatocellular carcinoma. Clin. Cancer Res. 2002, 8, 3131–3136. [Google Scholar] [PubMed]
- Oláh, G.; Módis, K.; Törö, G.; Hellmich, M.R.; Szczesny, B.; Szabo, C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem. Pharmacol. 2018, 149, 186–204. [Google Scholar] [CrossRef] [PubMed]
- González, R.; López-Grueso, M.J.; Muntané, J.; Bárcena, J.A.; Padilla, C.A. Redox regulation of metabolic and signaling pathways by thioredoxin and glutaredoxin in NOS-3 overexpressing hepatoblastoma cells. Redox Biol. 2015, 6, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Hernández, A.; Navarro-Villarán, E.; González, R.; Pereira, S.; Castro, L.S.-D.; Sarrias-Giménez, A.; Barrera-Pulido, L.; Álamo-Martínez, J.; Serrablo-Requejo, A.; Blanco, G.; et al. Regulation of cell death receptor S-nitrosylation and apoptotic signaling by Sorafenib in hepatoblastoma cells. Redox Biol. 2015, 6, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Socco, S.; Bovee, R.C.; Palczewski, M.B.; Hickok, J.R.; Thomas, D.D. Epigenetics: The third pillar of nitric oxide signaling. Pharmacol. Res. 2017, 121, 52–58. [Google Scholar] [CrossRef]
- Vasudevan, D.; Bovee, R.C.; Thomas, D.D. Nitric oxide, the new architect of epigenetic landscapes. Nitric Oxide 2016, 59, 54–62. [Google Scholar] [CrossRef]
- Hardy, T.; Mann, D.A. Epigenetics in liver disease: From biology to therapeutics. Gut 2016, 65, 1895–1905. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Baylin, S.B. Cancer epigenetics: Linking basic biology to clinical medicine. Cell Res. 2011, 21, 502–517. [Google Scholar] [CrossRef] [Green Version]
- Hmadcha, A.; Bedoya, F.J.; Sobrino, F.; Pintado, E. Methylation-dependent gene silencing induced by interleukin 1beta via nitric oxide production. J. Exp. Med. 1999, 190, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wu, W. DNA Methylation Alterations in Human Cancers. In Epigenetics in Human Disease; Academic Press: London, UK, 2018; pp. 109–139. [Google Scholar]
- Fan, G.; Tu, Y.; Chen, C.; Sun, H.; Wan, C.; Cai, X. DNA methylation biomarkers for hepatocellular carcinoma. Cancer Cell Int. 2018, 18, 1–13. [Google Scholar] [CrossRef]
- Takeshima, H.; Niwa, T.; Yamashita, S.; Takamura-Enya, T.; Iida, N.; Wakabayashi, M.; Nanjo, S.; Abe, M.; Sugiyama, T.; Kim, Y.-J.; et al. TET repression and increased DNMT activity synergistically induce aberrant DNA methylation. J. Clin. Investig. 2020, 130, 5370–5379. [Google Scholar] [CrossRef]
- Gao, W.; Kondo, Y.; Shen, L.; Shimizu, Y.; Sano, T.; Yamao, K.; Natsume, A.; Goto, Y.; Ito, M.; Murakami, H.; et al. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis 2008, 29, 1901–1910. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, H.J.; Kim, J.-H.; Lee, H.-S.; Jang, J.J.; Kang, G.H. Aberrant CpG Island Hypermethylation Along Multistep Hepatocarcinogenesis. Am. J. Pathol. 2003, 163, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Um, T.-H.; Kim, H.; Oh, B.-K.; Kim, M.S.; Kim, K.S.; Jung, G.; Park, Y.N. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J. Hepatol. 2011, 54, 939–947. [Google Scholar] [CrossRef]
- Masferrer, J.L.; Leahy, K.M.; Koki, A.T.; Zweifel, B.S.; Settle, S.L.; Woerner, B.M.; Edwards, D.A.; Flickinger, A.G.; Moore, R.J.; Seibert, K. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000, 60, 1306–1311. [Google Scholar]
- Cervello, M.; Foderà, D.; Florena, A.M.; Soresi, M.; Tripodo, C.; D’Alessandro, N.; Montalto, G. Correlation between expression of cyclooxygenase-2 and the presence of inflammatory cells in human primary hepatocellular carcinoma: Possible role in tumor promotion and angiogenesis. World. J. Gastroenterol. 2005, 11, 4638–4643. [Google Scholar] [CrossRef]
- Koga, H.; Sakisaka, S.; Ohishi, M.; Kawaguchi, T.; Taniguchi, E.; Sasatomi, K.; Harada, M.; Kusaba, T.; Tanaka, M.; Kimura, R.; et al. Expression of cyclooxygenase-2 in human hepatocellular carcinoma: Relevance to tumor dedifferentiation. Hepatology 1999, 29, 688–696. [Google Scholar] [CrossRef]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase Regulates Angiogenesis Induced by Colon Cancer Cells. Cell 1998, 93, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Cozma, A.; Fodor, A.; Vulturar, R.; Sitar-Tăut, A.-V.; Orăşan, O.H.; Mureşan, F.; Login, C.; Suharoschi, R. DNA Methylation and Micro-RNAs: The Most Recent and Relevant Biomarkers in the Early Diagnosis of Hepatocellular Carcinoma. Medicina 2019, 55, 607. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.H.; Jung, E.S.; Park, Y.M.; Kim, B.S.; Kim, D.G.; Ryu, W.S. Expression of cyclooxygenase-2 (COX-2) in hepatocellular carcinoma and growth inhibition of hepatoma cell lines by a COX-2 inhibitor, NS-398. Clin. Cancer Res. 2001, 7, 1410–1418. [Google Scholar]
- Rahman, M.A.; Dhar, D.K.; Yamaguchi, E.; Maruyama, S.; Sato, T.; Hayashi, H.; Ono, T.; Yamanoi, A.; Kohno, H.; Nagasue, N. Coexpression of inducible nitric oxide synthase and COX-2 in hepatocellular carcinoma and surrounding liver: Possible involvement of COX-2 in the angiogenesis of hepatitis C virus-positive cases. Clin. Cancer Res. 2001, 7, 1325–1332. [Google Scholar]
- Huang, F.Y.; Chan, A.O.; Rashid, A.; Wong, D.K.; Cho, C.H.; Yuen, M.F. Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1beta activation of nitric oxide production in gastric cancer cells. Cancer 2012, 118, 4969–4980. [Google Scholar] [CrossRef]
- Katayama, Y.; Takahashi, M.; Kuwayama, H. Helicobacter pylori causes runx3 gene methylation and its loss of expression in gastric epithelial cells, which is mediated by nitric oxide produced by macrophages. Biochem. Biophys. Res. Commun. 2009, 388, 496–500. [Google Scholar] [CrossRef]
- Tatemichi, M.; Hata, H.; Nakadate, T. Ectopic expression of activation-induced cytidine deaminase caused by epigenetics modification. Oncol. Rep. 2011, 25, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Vasudevan, D.; Hickok, J.R.; Bovee, R.C.; Pham, V.; Mantell, L.L.; Bahroos, N.; Kanabar, P.; Cao, X.J.; Maienschein-Cline, M.; Garcia, B.A.; et al. Nitric Oxide Regulates Gene Expression in Cancers by Controlling Histone Posttranslational Modifications. Cancer Res. 2015, 75, 5299–5308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; De Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Siddiqui, H.; Solomon, D.A.; Gunawardena, R.W.; Wang, Y.; Knudsen, E.S. Histone Deacetylation of RB-Responsive Promoters: Requisite for Specific Gene Repression but Dispensable for Cell Cycle Inhibition. Mol. Cell. Biol. 2003, 23, 7719–7731. [Google Scholar] [CrossRef] [Green Version]
- Godman, C.A.; Joshi, R.; Tierney, B.R.; Greenspan, E.; Rasmussen, T.; Wang, H.-W.; Shin, D.-G.; Rosenberg, D.W.; Giardina, C. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol. Ther. 2008, 7, 1570–1580. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Bae, H.J.; Chang, Y.G.; Kim, M.G.; Park, W.S.; Lee, J.Y.; Lee, S.-Y.; et al. Histone deacetylase 6 functions as a tumor suppressor by activating c-Jun NH2-terminal kinase-mediated beclin 1-dependent autophagic cell death in liver cancer. Hepatology 2012, 56, 644–657. [Google Scholar] [CrossRef]
- Wu, J.; Du, C.; Lv, Z.; Ding, C.; Cheng, J.; Xie, H.; Zhou, L.; Zheng, S. The Up-Regulation of Histone Deacetylase 8 Promotes Proliferation and Inhibits Apoptosis in Hepatocellular Carcinoma. Dig. Dis. Sci. 2013, 58, 3545–3553. [Google Scholar] [CrossRef]
- Nott, A.; Watson, P.M.; Robinson, J.D.; Crepaldi, L.; Riccio, A. S-nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 2008, 455, 411–415. [Google Scholar] [CrossRef]
- Nott, A.; Nitarska, J.; Veenvliet, J.V.; Schacke, S.; Derijck, A.A.H.A.; Sirko, P.; Muchardt, C.; Pasterkamp, J.; Smidt, M.P.; Riccio, A. S-nitrosylation of HDAC2 regulates the expression of the chromatin-remodeling factor Brm during radial neuron migration. Proc. Natl. Acad. Sci. USA 2013, 110, 3113–3118. [Google Scholar] [CrossRef] [Green Version]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 Is Essential for the Maintenance of Chromatin Structure and Genome Stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Okuda, K.; Ito, A.; Uehara, T. Regulation of Histone Deacetylase 6 Activity via S-Nitrosylation. Biol. Pharm. Bull. 2015, 38, 1434–1437. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Cao, Z.; Cheng, Y.; Wang, J.; Liu, Y.; Yang, R.; Li, H.; Jiang, W.; Li, G.; Zhao, W.; et al. Acetylation of alpha-fetoprotein promotes hepatocellular carcinoma progression. Cancer Lett. 2020, 471, 12–26. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, H.; Zhu, L.-H.; Wang, L.; Xu, L. Downregulation of CREB-binding protein expression sensitizes endothelial cells to serum-deprived apoptosis: Important role of nitric oxide. Mol. Cell. Biochem. 2010, 337, 159–166. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Hickok, J.R.; Vasudevan, D.; Antholine, W.E.; Thomas, D.D. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. J. Biol. Chem. 2013, 288, 16004–16015. [Google Scholar] [CrossRef] [Green Version]
- Yamada, D.; Kobayashi, S.; Yamamoto, H.; Tomimaru, Y.; Noda, T.; Uemura, M.; Wada, H.; Marubashi, S.; Eguchi, H.; Tanemura, M.; et al. Role of the Hypoxia-Related Gene, JMJD1A, in Hepatocellular Carcinoma: Clinical Impact on Recurrence after Hepatic Resection. Ann. Surg. Oncol. 2012, 19 (Suppl. 3), S355–S364. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, J.-G.; Son, T.G.; Yi, J.M.; Kim, N.D.; Yang, K.; Heo, K. The histone demethylase JMJD1A regulates adrenomedullin-mediated cell proliferation in hepatocellular carcinoma under hypoxia. Biochem. Biophys. Res. Commun. 2013, 434, 722–727. [Google Scholar] [CrossRef]
- Hu, Y.; Zheng, Y.; Dai, M.; Wang, X.; Wu, J.; Yu, B.; Zhang, H.; Cui, Y.; Kong, W.; Wu, H.; et al. G9a and histone deacetylases are crucial for Snail2-mediated E-cadherin repression and metastasis in hepatocellular carcinoma. Cancer Sci. 2019, 110, 3442–3452. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Chiu, D.K.-C.; Tsang, F.H.-C.; Law, C.-T.; Cheng, C.L.-H.; Au, S.L.-K.; Lee, J.M.-F.; Wong, C.C.-L.; Ng, I.O.-L.; Wong, C.-M. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Jiang, W.; Ma, L.; Sun, J.; Yan, X.; Qian, J.; Wang, Y.; Shi, Y.; Ni, S.; Yao, N. A metabolism-related gene signature for predicting the prognosis and therapeutic responses in patients with hepatocellular carcinoma. Ann. Transl. Med. 2021, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.N.-Y.; Tsang, F.H.-C.; Tam, A.H.-K.; Au, S.L.-K.; Wong, C.C.-L.; Wei, L.; Lee, J.M.-F.; He, X.; Ng, I.O.-L.; Wong, C.-M. Histone lysine methyltransferase, suppressor of variegation 3-9 homolog 1, promotes hepatocellular carcinoma progression and is negatively regulated by microRNA-125b. Hepatology 2013, 57, 637–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Higashi, S.; Kimura, H.; Yamamoto, H.; Mori, M.; Matsuura, S.; Matsuura, N. Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci. 2013, 104, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Snyder, S.H. Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc. Natl. Acad. Sci. USA 2011, 108, 20178–20183. [Google Scholar] [CrossRef] [Green Version]
- Accordi, B.; Galla, L.; Milani, G.; Curtarello, M.; Serafin, V.; Lissandron, V.; Viola, G.; Kronnie, G.T.; De Maria, R.; Petricoin, E.F.; et al. AMPK inhibition enhances apoptosis in MLL-rearranged pediatric B-acute lymphoblastic leukemia cells. Leukemia 2013, 27, 1019–1027. [Google Scholar] [CrossRef]
- Takeda, S.; Liu, H.; Sasagawa, S.; Dong, Y.; Trainor, P.A.; Cheng, E.H.; Hsieh, J.J. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J. Clin. Investig. 2013, 123, 3154–3165. [Google Scholar] [CrossRef] [Green Version]
- El Mansouri, F.E.; Chabane, N.; Zayed, N.; Kapoor, M.; Benderdour, M.; Martel-Pelletier, J.; Pelletier, J.P.; Duval, N.; Fahmi, H. Contribution of H3K4 methylation by SET-1A to interleukin-1-induced cyclooxygenase 2 and inducible nitric oxide synthase expression in human osteoarthritis chondrocytes. Arthritis Rheum 2011, 63, 168–179. [Google Scholar] [CrossRef]
- Gale, M.; Sayegh, J.; Cao, J.; Norcia, M.; Gareiss, P.; Hoyer, D.; Merkel, J.S.; Yan, Q. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget 2016, 7, 39931–39944. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Han, S.; Peng, R.; Jiao, C.; Wang, X.; Yang, X.; Yang, R.; Li, X. Depletion of histone demethylase KDM5B inhibits cell proliferation of hepatocellular carcinoma by regulation of cell cycle checkpoint proteins p15 and Pj. Exp. Clin. Cancer Res. 2016, 35, 37. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Qi, G.; Tang, F.; Yuan, S.; Wang, Z.; Liang, X.; Li, B.; Yu, S.; Liu, J.; Huang, Q.; et al. JARID1B promotes metastasis and epithelial-mesenchymal transition via PTEN/AKT signaling in hepatocellular carcinoma cells. Oncotarget 2015, 6, 12723–12739. [Google Scholar] [CrossRef] [Green Version]
- Pediconi, N.; Salerno, D.; Lupacchini, L.; Angrisani, A.; Peruzzi, G.; De Smaele, E.; Levrero, M.; Belloni, L. EZH2, JMJD3, and UTX epigenetically regulate hepatic plasticity inducing retro-differentiation and proliferation of liver cells. Cell Death Dis. 2019, 10, 518. [Google Scholar] [CrossRef] [Green Version]
- Dreger, H.; Ludwig, A.; Weller, A.; Baumann, G.; Stangl, V.; Stangl, K. Epigenetic suppression of iNOS expression in human endothelial cells: A potential role of Ezh2-mediated H3K27me3. Genomics 2016, 107, 145–149. [Google Scholar] [CrossRef]
- Maiuthed, A.; Bhummaphan, N.; Luanpitpong, S.; Mutirangura, A.; Aporntewan, C.; Meeprasert, A.; Rungrotmongkol, T.; Rojanasakul, Y.; Chanvorachote, P. Nitric oxide promotes cancer cell dedifferentiation by disrupting an Oct4:caveolin-1 complex: A new regulatory mechanism for cancer stem cell formation. J. Biol. Chem. 2018, 293, 13534–13552. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Wu, Z.; Yue, X.; Yu, X.; Wang, Z.; Song, X.; Xu, L.; He, Y.; Ge, Y.; Tan, S.; et al. ZHX2 restricts hepatocellular carcinoma by suppressing stem cell-like traits through KDM2A-mediated H3K36 demethylation. EBioMedicine 2020, 53, 102676. [Google Scholar] [CrossRef]
- Tanaka, T.; Huang, X.; Halicka, H.D.; Zhao, H.; Traganos, F.; Albino, A.P.; Dai, W.; Darzynkiewicz, Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytom. Part A 2007, 71, 648–661. [Google Scholar] [CrossRef]
- Tanaka, T.; Kurose, A.; Halicka, H.D.; Huang, X.; Traganos, F.; Darzynkiewicz, Z. Nitrogen Oxide-Releasing Aspirin Induces Histone H2AX Phosphorylation, ATM Activation and Apoptosis Preferentially in S-Phase Cells: Involvement of Reactive Oxygen Species. Cell Cycle 2006, 5, 1669–1674. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Kodela, R.; Olson, K.R.; Kashfi, K. NOSH–aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model. Biochem. Biophys. Res. Commun. 2012, 419, 523–528. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Kodela, R.; Santiago, G.; Le, T.T.C.; Nath, N.; Kashfi, K. NOSH-aspirin (NBS-1120) inhibits pancreatic cancer cell growth in a xenograft mouse model: Modulation of FoxM1, p53, NF-κB, iNOS, caspase-3 and ROS. Biochem. Pharmacol. 2020, 176, 113857. [Google Scholar] [CrossRef]
- Clemons, N.J.; McColl, K.E.; Fitzgerald, R.C. Nitric Oxide and Acid Induce Double-Strand DNA Breaks in Barrett’s Esophagus Carcinogenesis via Distinct Mechanisms. Gastroenterology 2007, 133, 1198–1209. [Google Scholar] [CrossRef]
- Xiao, H.; Tong, R.; Ding, C.; Lv, Z.; Du, C.; Peng, C.; Cheng, S.; Xie, H.; Zhou, L.; Wu, J.; et al. γ-H2AX promotes hepatocellular carcinoma angiogenesis via EGFR/HIF-1alpha/VEGF pathways under hypoxic condition. Oncotarget 2015, 6, 2180–2192. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef] [PubMed]
- Ambs, S.; Ogunfusika, M.O.; Merriam, W.G.; Bennett, W.P.; Billiar, T.R.; Harris, C.C. Up-regulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc. Natl. Acad. Sci. USA 1998, 95, 8823–8828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathé, E.; Nguyen, G.H.; Funamizu, N.; He, P.; Moake, M.; Croce, C.M.; Hussain, S.P. Inflammation regulates microRNA expression in cooperation with p53 and nitric oxide. Int. J. Cancer 2012, 131, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Bian, Z.; Wei, D.; Zhang, J.-G. miR-29b regulates migration of human breast cancer cells. Mol. Cell. Biochem. 2011, 352, 197–207. [Google Scholar] [CrossRef]
- Cui, H.; Wang, L.; Gong, P.; Zhao, C.; Zhang, S.; Zhang, K.; Zhou, R.; Zhao, Z.; Fan, H. Deregulation between miR-29b/c and DNMT3A Is Associated with Epigenetic Silencing of the CDH1 Gene, Affecting Cell Migration and Invasion in Gastric Cancer. PLoS ONE 2015, 10, e0123926. [Google Scholar] [CrossRef]
- Fu, Q.; Liu, X.; Liu, Y.; Yang, J.; Lv, G.; Dong, S. MicroRNA-335 and -543 suppress bone metastasis in prostate cancer via targeting endothelial nitric oxide synthase. Int. J. Mol. Med. 2015, 36, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Li, L.; Piontek, K.; Sakaguchi, M.; Selaru, F.M. Exosome miR-335 as a novel therapeutic strategy in hepatocellular carcinoma. Hepatology 2018, 67, 940–954. [Google Scholar] [CrossRef]
- Hulin, J.-A.; Tommasi, S.; Elliot, D.; Hu, D.G.; Lewis, B.C.; Mangoni, A.A. MiR-193b regulates breast cancer cell migration and vasculogenic mimicry by targeting dimethylarginine dimethylaminohydrolase 1. Sci. Rep. 2017, 7, 13996. [Google Scholar] [CrossRef]
- Roy, S.; Benz, F.; Vargas Cardenas, D.; Vucur, M.; Gautheron, J.; Schneider, A.; Hellerbrand, C.; Pottier, N.; Alder, J.; Tacke, F.; et al. miR-30c and miR-193 are a part of the TGF-beta-dependent regulatory network controlling extracellular matrix genes in liver fibrosis. J. Dig. Dis. 2015, 16, 513–524. [Google Scholar] [CrossRef]
- Jia, X.; Li, X.; Shen, Y.; Miao, J.; Liu, H.; Li, G.; Wang, Z. MiR-16 regulates mouse peritoneal macrophage polarization and affects T-cell activation. J. Cell. Mol. Med. 2016, 20, 1898–1907. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xu, L.-J.; Qin, J.-J.; Zhang, L.; Zhuang, G.-H. MicroRNA-155 inversely correlates with esophageal cancer progression through regulating tumor-associated macrophage FGF2 expression. Biochem. Biophys. Res. Commun. 2018, 503, 452–458. [Google Scholar] [CrossRef]
- Shi, Y.; Shi, Q.; Shen, Q.; Zhang, Q.; Cao, X. Dicer-independent snRNA/snoRNA-derived nuclear RNA 3 regulates tumor-associated macrophage function by epigenetically repressing inducible nitric oxide synthase transcription. Cancer Commun. 2021, 41, 140–153. [Google Scholar] [CrossRef]
- Yuhas, Y.; Berent, E.; Ashkenazi, S. Effect of nitric oxide on microRNA-155 expression in human hepatic epithelial cells. Inflamm. Res. 2014, 63, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Lu, Y.; Xie, S.; Chen, Y.; Jiang, X.; Song, S.; Wang, L.; Pu, H.; Gui, X.; Li, T.; et al. miR-155 Accelerates the Growth of Human Liver Cancer Cells by Activating CDK2 via Targeting H3F3A. Mol. Ther. Oncolytics 2020, 17, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, T.; Otsuka, M.; Tan, P.S.; Ohno, M.; Sun, X.; Yoshikawa, T.; Shibata, C.; Takata, A.; Kojima, K.; Takehana, K.; et al. Decreased miR122 in hepatocellular carcinoma leads to chemoresistance with increased arginine. Oncotarget 2015, 6, 8339–8352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Han, S.; Feng, B.; Chu, X.; Chen, L.; Wang, R. Hepatitis B virus X protein-mediated non-coding RNA aberrations in the development of human hepatocellular carcinoma. Exp. Mol. Med. 2017, 49, e293. [Google Scholar] [CrossRef] [PubMed]
- Youness, R.A.; Gad, M.Z. Long non-coding RNAs: Functional regulatory players in breast cancer. Non-Coding RNA Res. 2019, 4, 36–44. [Google Scholar] [CrossRef] [PubMed]
- García-Guede, Á.; Vera, O.; Ibáñez-de-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. [Google Scholar] [CrossRef]
- Mahpour, A.; Mullen, A.C. Our emerging understanding of the roles of long non-coding RNAs in normal liver function, disease, and malignancy. JHEP Rep. 2021, 3, 100177. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Mozdarani, H.; Ezzatizadeh, V.; Parvaneh, R.R. The emerging role of the long non-coding RNA HOTAIR in breast cancer development and treatment. J. Transl. Med. 2020, 18, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhou, L.; Wu, L.-M.; Lai, M.-C.; Xie, H.-Y.; Zhang, F.; Zheng, S.-S. Overexpression of Long Non-coding RNA HOTAIR Predicts Tumor Recurrence in Hepatocellular Carcinoma Patients Following Liver Transplantation. Ann. Surg. Oncol. 2011, 18, 1243–1250. [Google Scholar] [CrossRef]
- Aiello, A.; Bacci, L.; Re, A.; Ripoli, C.; Pierconti, F.; Pinto, F.; Masetti, R.; Grassi, C.; Gaetano, C.; Bassi, P.F.; et al. MALAT1 and HOTAIR Long Non-Coding RNAs Play Opposite Role in Estrogen-Mediated Transcriptional Regulation in Prostate Cancer Cells. Sci. Rep. 2016, 6, 38414. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, L.; Huo, X.-S.; Yuan, J.-H.; Xu, D.; Yuan, S.-X.; Zhu, N.; Zhou, W.-P.; Yang, G.-S.; Wang, Y.-Z.; et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology 2011, 54, 1679–1689. [Google Scholar] [CrossRef]
- Nafea, H.; Youness, R.A.; Abou-Aisha, K.; Gad, M.Z. LncRNA HEIH/miR-939-5p interplay modulates triple-negative breast cancer progression through NOS2-induced nitric oxide production. J. Cell Physiol. 2021, 236, 5362–5372. [Google Scholar] [CrossRef]
- Guo, Z.; Shao, L.; Zheng, L.; Du, Q.; Li, P.; John, B.; Geller, D.A. miRNA-939 regulates human inducible nitric oxide synthase posttranscriptional gene expression in human hepatocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 5826–5831. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Li, C.; Xu, M.; Liu, Y.; Liu, S. LncRNA UCA1 attenuates autophagy-dependent cell death through blocking autophagic flux under arsenic stress. Toxicol. Lett. 2018, 284, 195–204. [Google Scholar] [CrossRef]
- Yang, L.; Tang, Y.; Xiong, F.; He, Y.; Wei, F.; Zhang, S.; Guo, C.; Xiang, B.; Zhou, M.; Xie, N.; et al. LncRNAs regulate cancer metastasis via binding to functional proteins. Oncotarget 2017, 9, 1426–1443. [Google Scholar] [CrossRef]
- Liang, Y.; Li, E.; Zhang, H.; Zhang, L.; Tang, Y.; Wanyan, Y. Silencing of lncRNA UCA1 curbs proliferation and accelerates apoptosis by repressing SIRT1 signals by targeting miR-204 in pediatric AML. J. Biochem. Mol. Toxicol. 2020, 34, e22435. [Google Scholar] [CrossRef]
- Bao, Y.; Deng, L.; Su, D.; Xiao, J.; Ge, D.; Geng, Y.; Jing, H. Identification of crucial microRNAs and genes in hypoxia-induced human lung adenocarcinoma cells. OncoTargets Ther. 2016, 9, 4605–4616. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Ni, T.; Lin, J.; Zhang, C.; Zheng, L.; Luo, M. Long non-coding RNA H19, a negative regulator of microRNA-148b-3p, participates in hypoxia stress in human hepatic sinusoidal endothelial cells via NOX4 and eNOS/NO signaling. Biochimie 2019, 163, 128–136. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Cruz-Ojeda, P.; Flores-Campos, R.; Dios-Barbeito, S.; Navarro-Villarán, E.; Muntané, J. Role of Nitric Oxide in Gene Expression Regulation during Cancer: Epigenetic Modifications and Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 6264. https://doi.org/10.3390/ijms22126264
de la Cruz-Ojeda P, Flores-Campos R, Dios-Barbeito S, Navarro-Villarán E, Muntané J. Role of Nitric Oxide in Gene Expression Regulation during Cancer: Epigenetic Modifications and Non-Coding RNAs. International Journal of Molecular Sciences. 2021; 22(12):6264. https://doi.org/10.3390/ijms22126264
Chicago/Turabian Stylede la Cruz-Ojeda, Patricia, Rocío Flores-Campos, Sandra Dios-Barbeito, Elena Navarro-Villarán, and Jordi Muntané. 2021. "Role of Nitric Oxide in Gene Expression Regulation during Cancer: Epigenetic Modifications and Non-Coding RNAs" International Journal of Molecular Sciences 22, no. 12: 6264. https://doi.org/10.3390/ijms22126264
APA Stylede la Cruz-Ojeda, P., Flores-Campos, R., Dios-Barbeito, S., Navarro-Villarán, E., & Muntané, J. (2021). Role of Nitric Oxide in Gene Expression Regulation during Cancer: Epigenetic Modifications and Non-Coding RNAs. International Journal of Molecular Sciences, 22(12), 6264. https://doi.org/10.3390/ijms22126264