
Amyloid Beta-Mediated Changes in Synaptic Function and Spine Number of Neocortical Neurons Depend on NMDA Receptors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
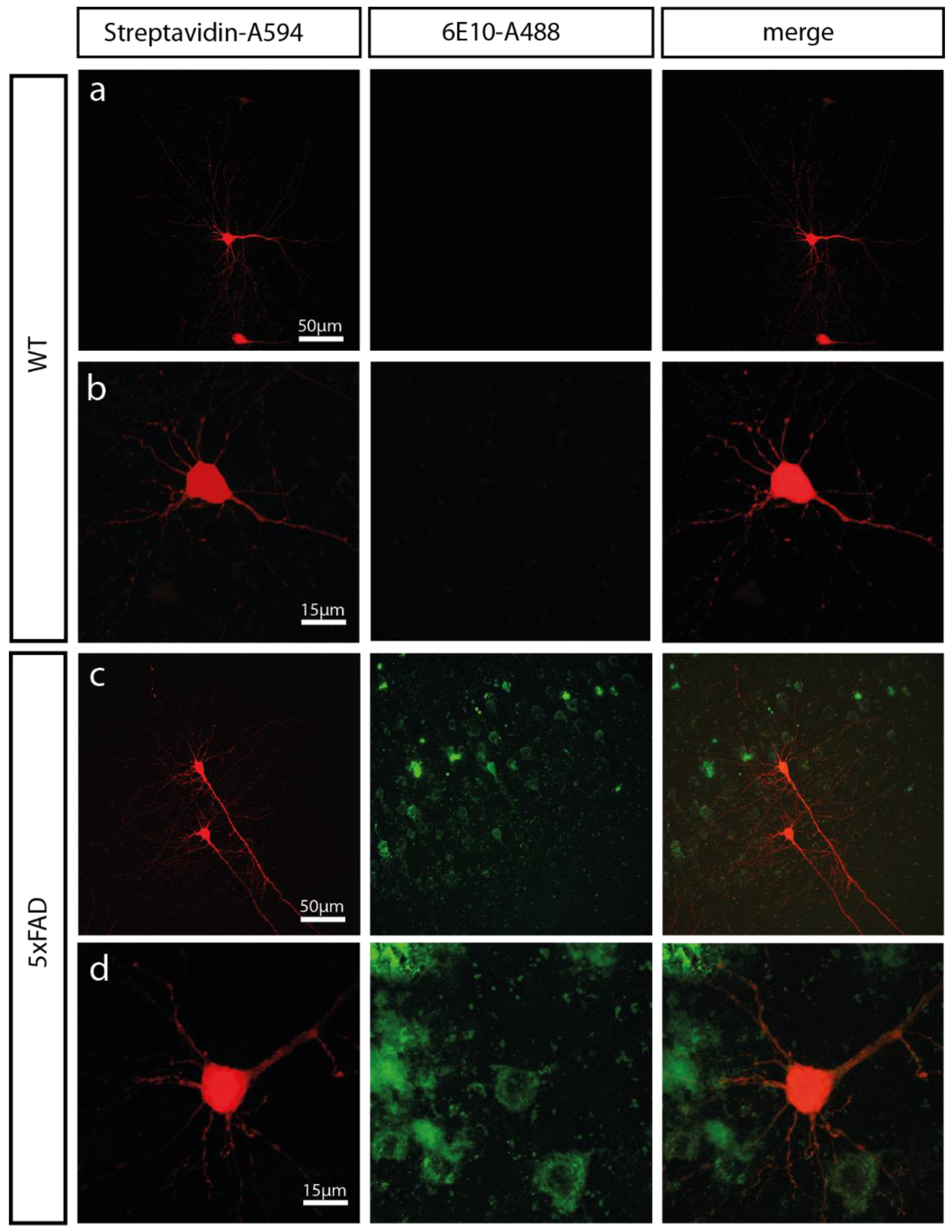
2.1. 5xFAD Mice Show a High Intracellular Aβ Burden and Aβ Plaques in the Somatosensory Cortex
2.2. NMDAR Subunit Expression Is Regulated by Aβ
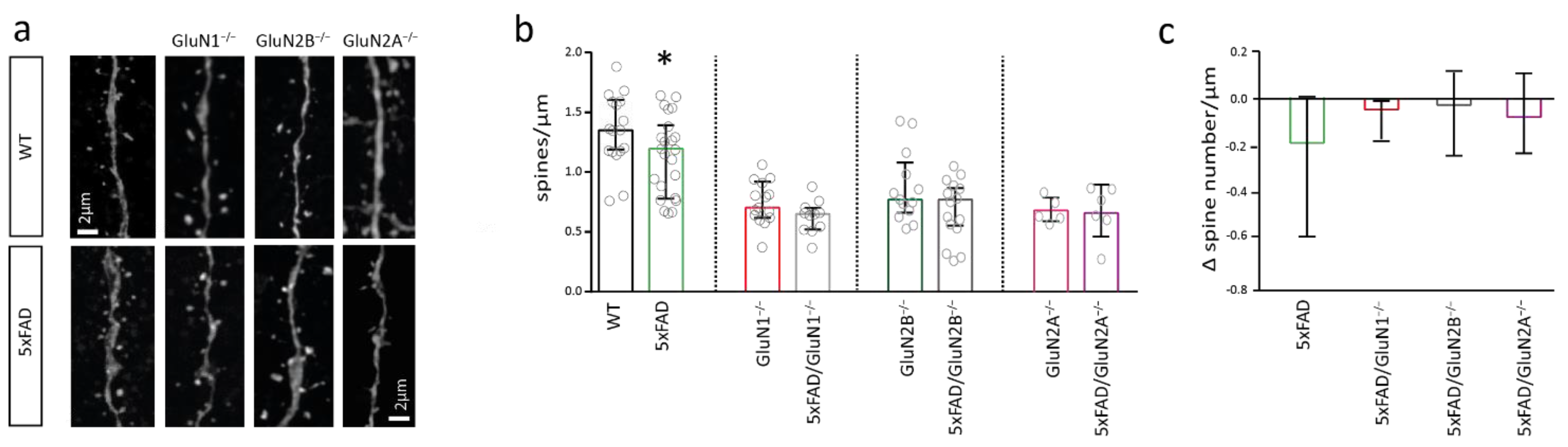
2.3. NMDAR Subunits Are Involved in Mediating Spine Loss in Somatosensory Cortex Pyramidal Cells of 5xFAD Mice
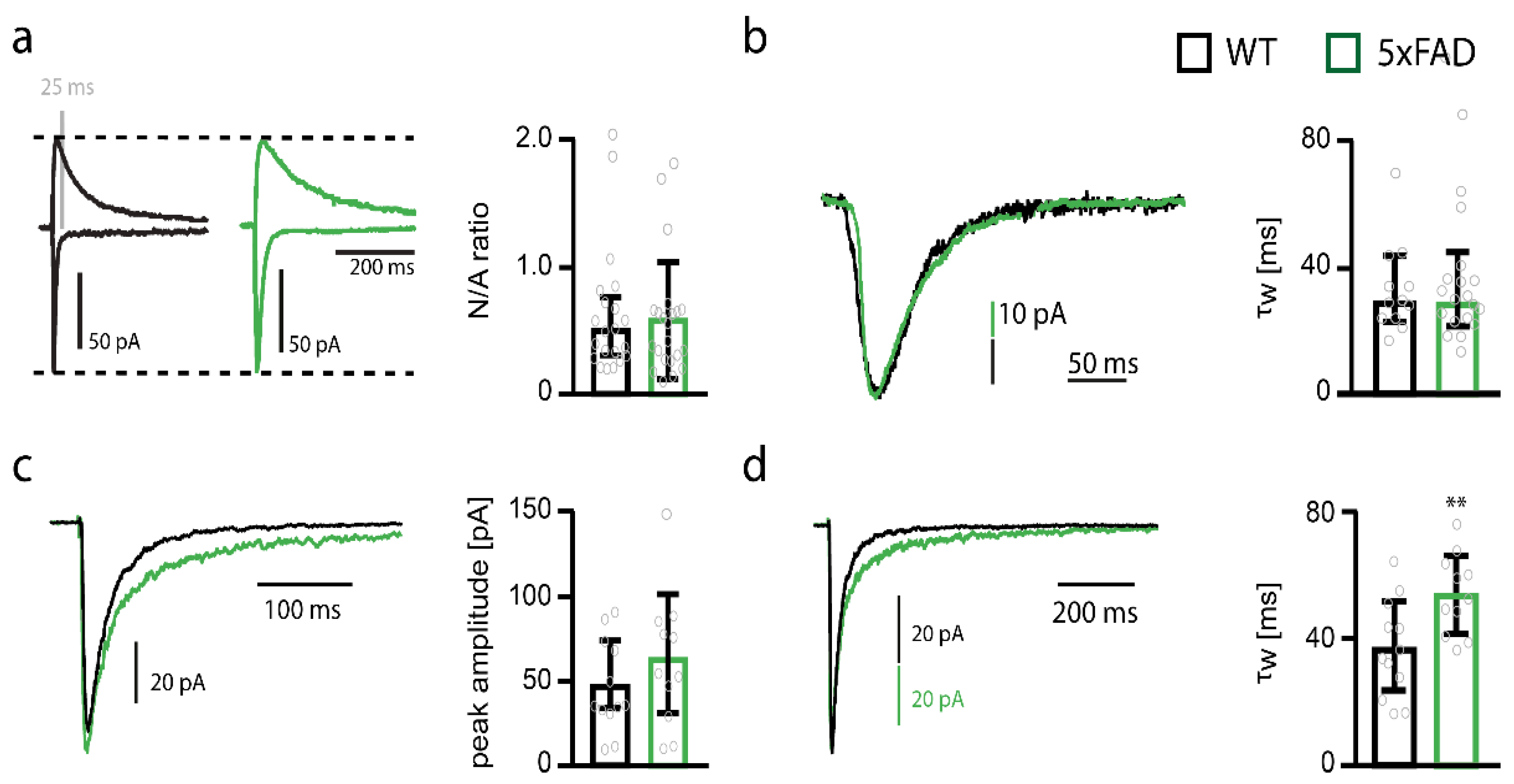
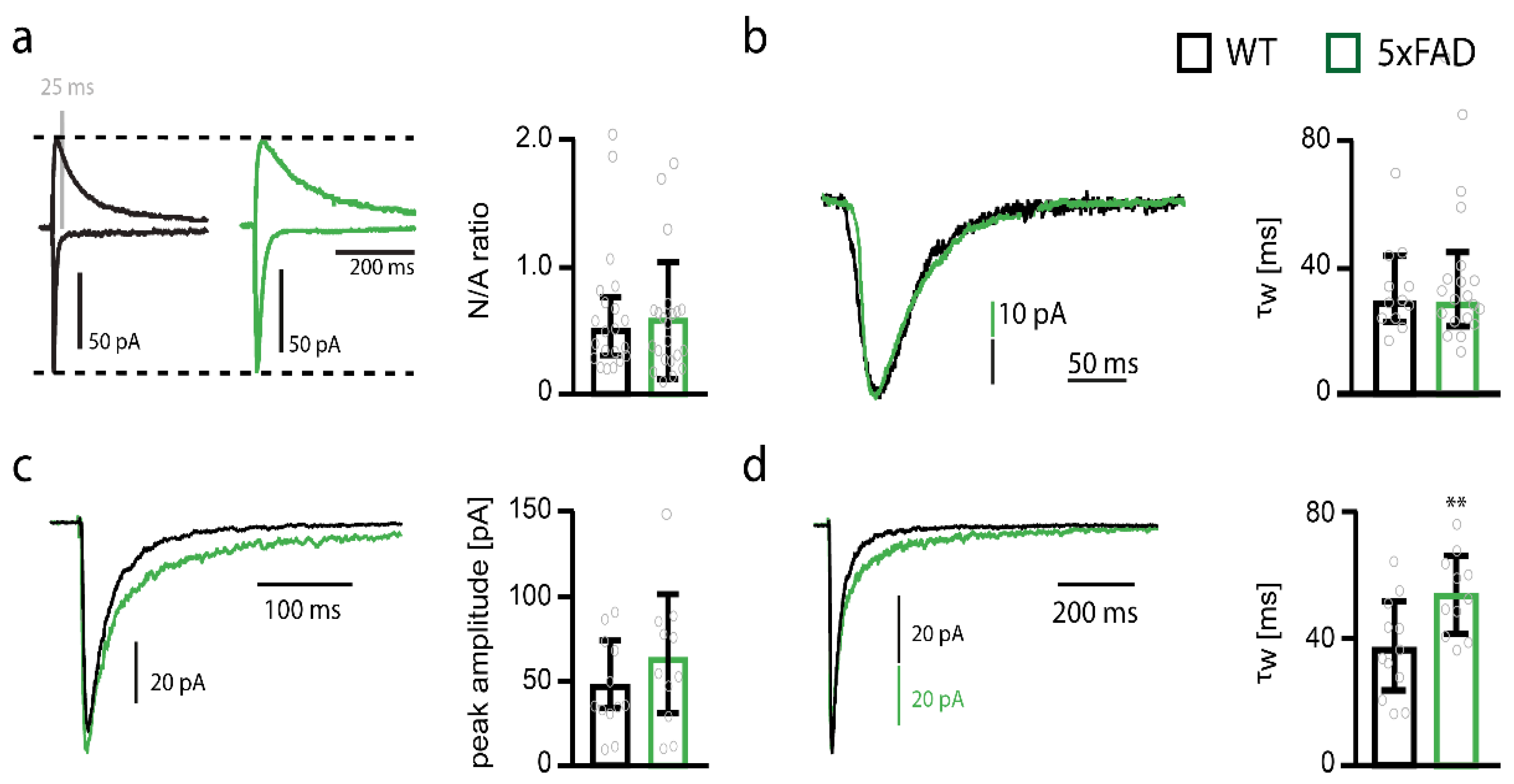
2.4. NMDARs Are Involved in Mediating the Decrease in Functional Synapse Number in Pyramidal Cells of the Somatosensory Cortex of 5xFAD Mice
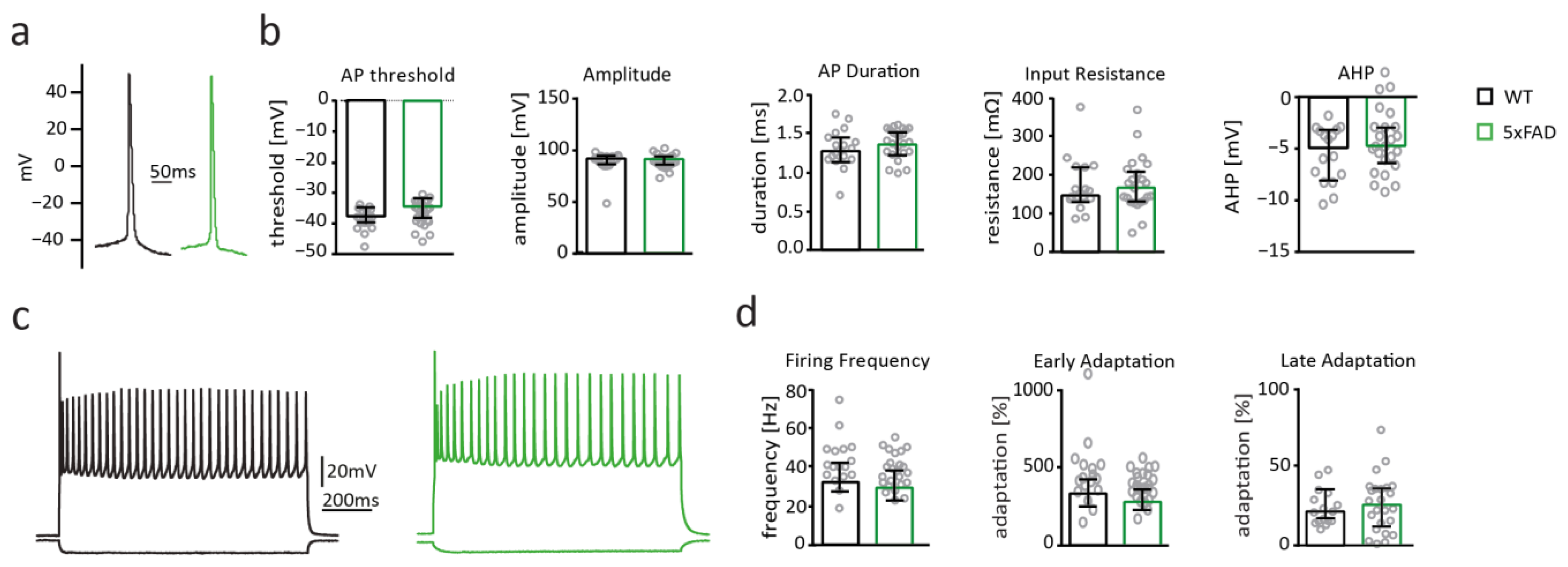
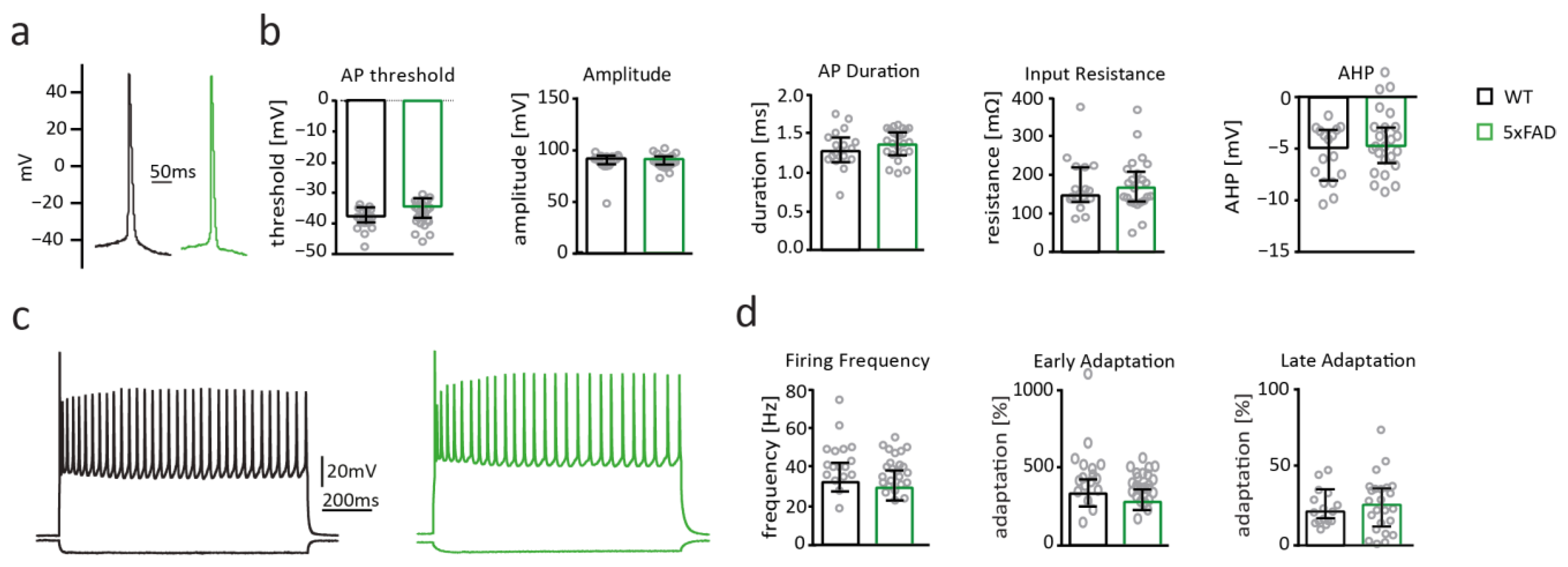
2.5. Neuronal Excitability of Somatosensory Cortex Pyramidal Cells Is Not Altered in 5xFAD Mice
2.6. Spine Number Is Increased after Short-Term Overexpression of Aβ in the Somatosensory Cortex
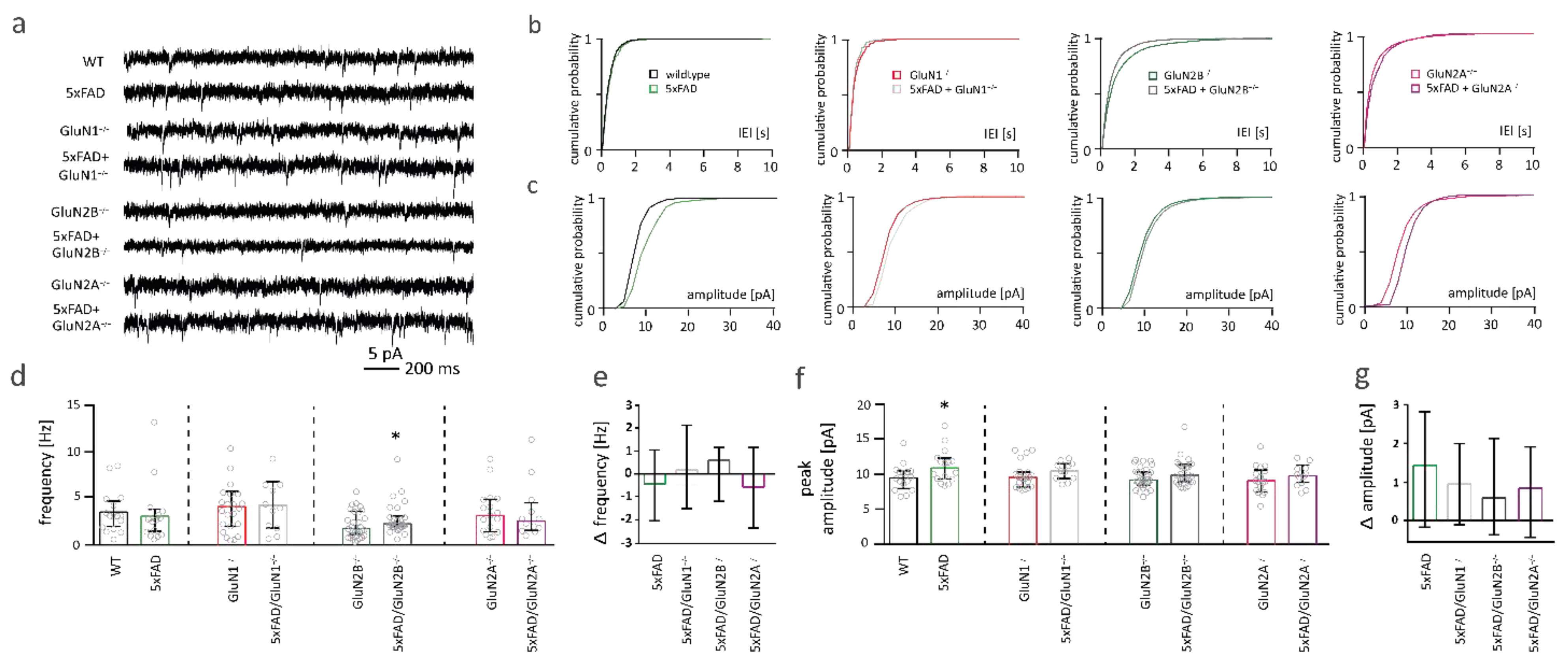
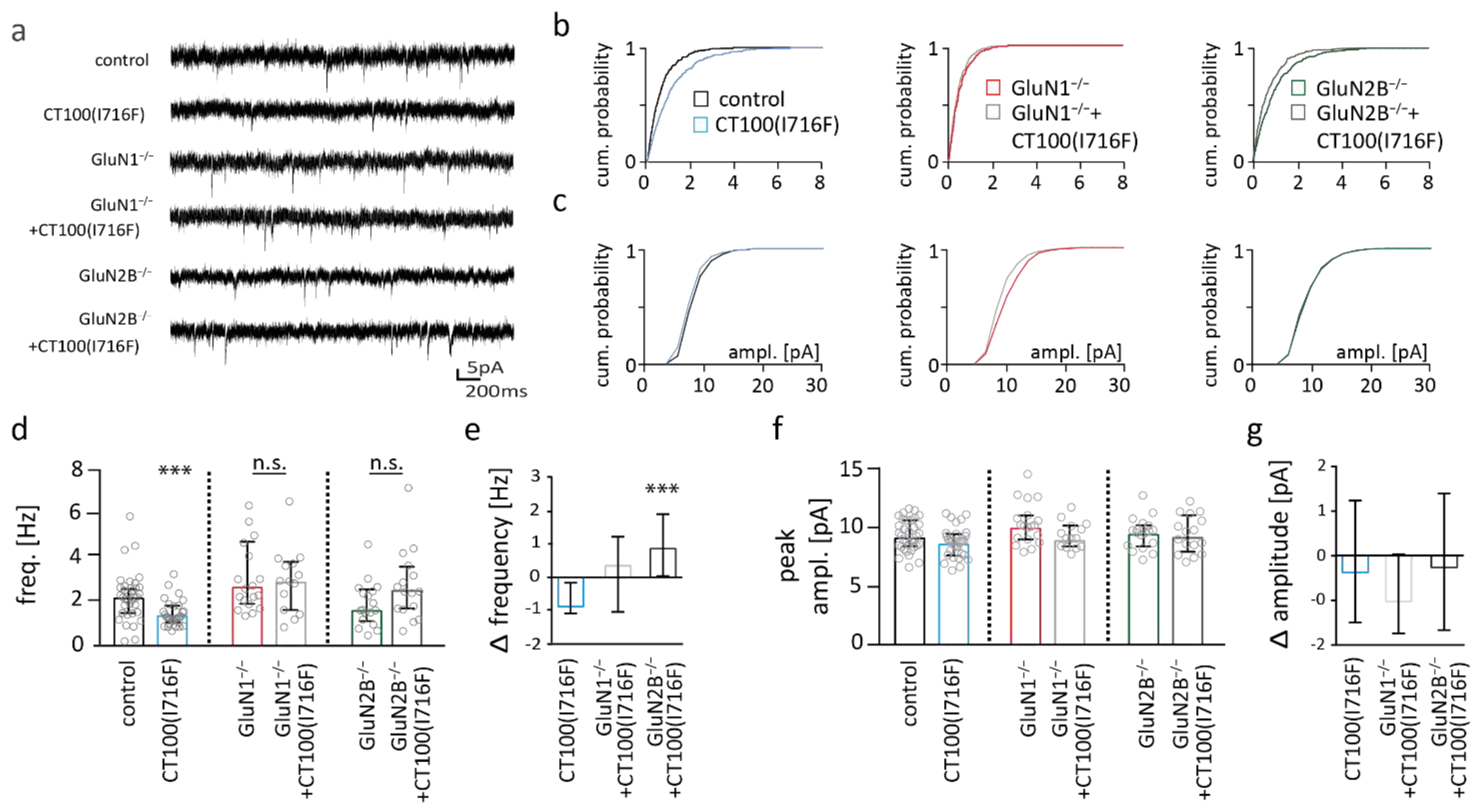
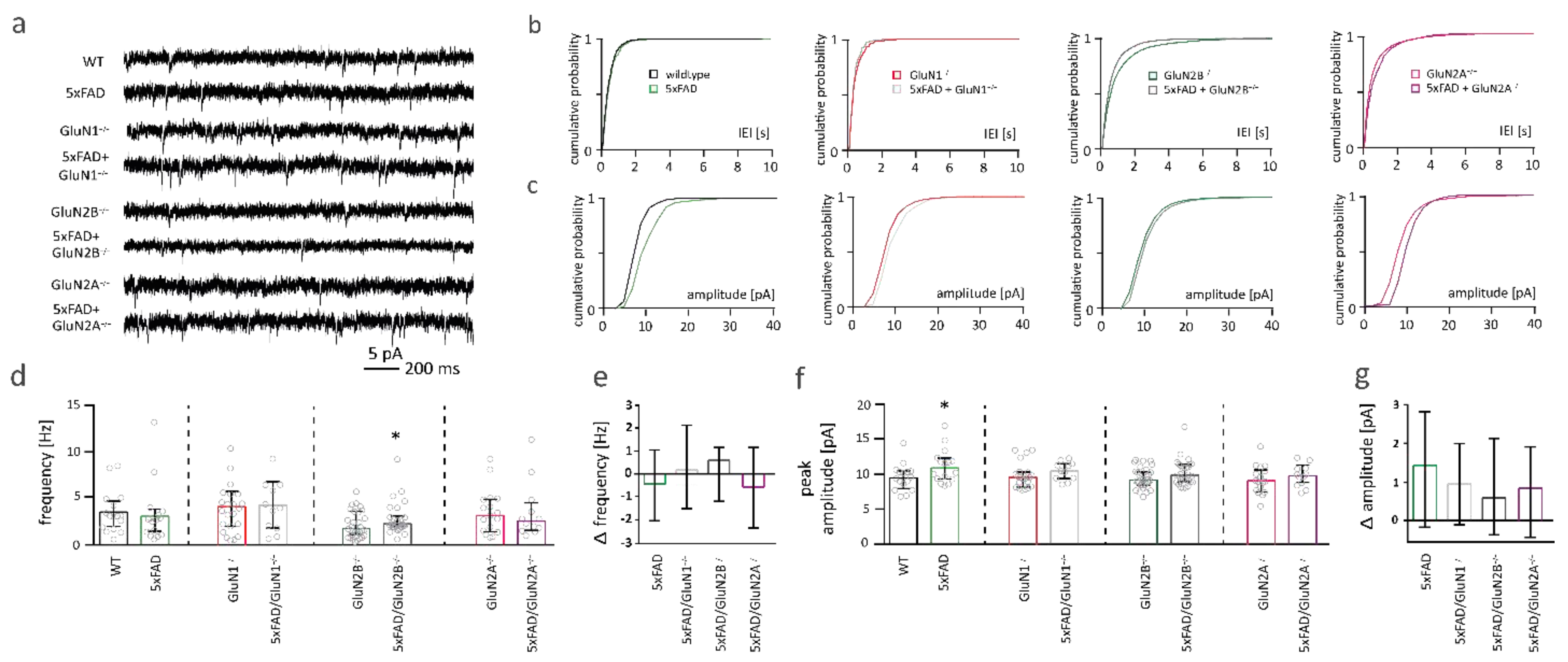
2.7. NMDAR Subunits Are Involved in Aβ-Mediated Changes in Functional Synapse Number
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Stereotactic Injection of rAAVs
4.3. Preparation of Acute Slices
4.4. Electrophysiology
4.5. Morphological Analysis
4.6. Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Näslund, J.; Haroutunian, V.; Mohs, R.C.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J. Correlation Between Elevated Levels of Amyloid β-Peptide in the Brain and Cognitive Decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, J.D.; Buckley, A.R.; Knox, J.E.; Kuan, L.; Graddis, N.; Pelos, A.; Mukora, A.; Wakeman, W.; Bohn, P.; Ho, A.; et al. Whole brain imaging reveals distinct spatial patterns of amyloid beta deposition in three mouse models of Alzheimer’s disease. J. Comp. Neurol. 2019, 527, 2122–2145. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Pimplikar, S.W. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2009, 41, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Koychev, I.; Hofer, M.; Friedman, N.C. Correlation of Alzheimer Disease Neuropathologic Staging with Amyloid and Tau Scintigraphic Imaging Biomarkers. J. Nucl. Med. 2020, 61, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Crowe, S.E.; Ellis-Davies, G.C.R. Spine pruning in 5xFAD mice starts on basal dendrites of layer 5 pyramidal neurons. Brain Struct. Funct. 2013, 219, 571–580. [Google Scholar] [CrossRef]
- Winblad, B.; Poritis, N. Memantine in severe dementia: Results of the 9M-best study (benefit and efficacy in severly demented patients during treatment with memantine). Int. J. Geriatr. Psychiatry 1999, 14, 135–146. [Google Scholar] [CrossRef]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine in Moderate-to-Severe Alzheimer’s Disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.-Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.; Salter, M.W.; Lombroso, P.J.; Gouras, G.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.; Nabavi, S.; Malinow, R. Metabotropic NMDA receptor function is required for -amyloid-induced synaptic depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4033–4038. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR Removal Underlies Aβ-Induced Synaptic Depression and Dendritic Spine Loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Bloodgood, B.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural Oligomers of the Alzheimer Amyloid-β Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Calabrese, B.; Shaked, G.M.; Tabarean, I.V.; Braga, J.; Koo, E.H.; Halpain, S. Rapid, concurrent alterations in pre- and postsynaptic structure induced by naturally-secreted amyloid-β protein. Mol. Cell. Neurosci. 2007, 35, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, N.-W.; Klyubin, I.; Anwyl, R.; Rowan, M.J. GluN2B subunit-containing NMDA receptor antagonists prevent Aβ-mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20504–20509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rammes, G.; Hasenjäger, A.; Sroka-Saidi, K.; Deussing, J.; Parsons, C.G. Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of β-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology 2011, 60, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Rammes, G.; Mattusch, C.; Wulff, M.; Seeser, F.; Kreuzer, M.; Zhu, K.; Deussing, J.M.; Herms, J.; Parsons, C.G. Involvement of GluN2B subunit containing N-methyl- d -aspartate (NMDA) receptors in mediating the acute and chronic synaptotoxic effects of oligomeric amyloid-beta (Aβ) in murine models of Alzheimer’s disease (AD). Neuropharmacology 2017, 123, 100–115. [Google Scholar] [CrossRef]
- Rammes, G.; Seeser, F.; Mattusch, K.; Zhu, K.; Haas, L.; Kummer, M.; Heneka, M.; Herms, J.; Parsons, C.G. The NMDA receptor antagonist Radiprodil reverses the synaptotoxic effects of different amyloid-beta (Aβ) species on long-term potentiation (LTP). Neuropharmacology 2018, 140, 184–192. [Google Scholar] [CrossRef]
- Rönicke, R.; Mikhaylova, M.; Rönicke, S.; Meinhardt, J.; Schröder, U.H.; Fändrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid β oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar] [CrossRef]
- Gambrill, A.C.; Barria, A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc. Natl. Acad. Sci. USA 2011, 108, 5855–5860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutsuwada, T.; Kashiwabuchi, N.; Mori, H.; Sakimura, K.; Kushiya, E.; Araki, K.; Meguro, H.; Masaki, H.; Kumanishi, T.; Arakawa, M.; et al. Molecular diversity of the NMDA receptor channel. Nature 1992, 358, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Monyer, H.; Sprengel, R.; Schoepfer, R.; Herb, A.; Higuchi, M.; Lomeli, H.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Heteromeric NMDA Receptors: Molecular and Functional Distinction of Subtypes. Science 1992, 256, 1217–1221. [Google Scholar] [CrossRef]
- Chazot, P.L.; Stephenson, F.A. Molecular dissection of native mammalian forebrain NMDA receptors containing the NR1 C2 exon: Direct demonstration of NMDA receptors comprising NR1, NR2A, and NR2B subunits within the same complex. J. Neurochem. 2002, 69, 2138–2144. [Google Scholar] [CrossRef]
- Rauner, C.; Köhr, G. Triheteromeric NR1/NR2A/NR2B Receptors Constitute the Major N-Methyl-d-aspartate Receptor Population in Adult Hippocampal Synapses. J. Biol. Chem. 2011, 286, 7558–7566. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Gascón, S.; Sobrado, M.; Roda, J.M.; Rodríguez-Peña, Á.; Díaz-Guerra, M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol. Psychiatry 2007, 13, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Von Engelhardt, J.; Bocklisch, C.; Tönges, L.; Herb, A.; Mishina, M.; Monyer, H. GluN2D-containing NMDA receptors-mediate synaptic currents in hippocampal interneurons and pyramidal cells in juvenile mice. Front. Cell. Neurosci. 2015, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Wang, Y.; Yasuda, R.P.; Dunah, A.W.; Wolfe, B.B. The Majority of N-Methyl-d-Aspartate Receptor Complexes in Adult Rat Cerebral Cortex Contain at Least Three Different Subunits (NR1/NR2A/NR2B). Mol. Pharmacol. 1997, 51, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Tovar, K.R.; McGinley, M.J.; Westbrook, G.L. Triheteromeric NMDA Receptors at Hippocampal Synapses. J. Neurosci. 2013, 33, 9150–9160. [Google Scholar] [CrossRef] [PubMed]
- Vicini, S.; Wang, J.F.; Li, J.H.; Zhu, W.J.; Wang, Y.H.; Luo, J.H.; Wolfe, B.B.; Grayson, D.R. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 1998, 79, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Ferreira, I.; Bajouco, L.; Mota, S.; Auberson, Y.; Oliveira, C.; Rego, A. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012, 51, 95–106. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Von Engelhardt, J.; Doganci, B.; Seeburg, P.H.; Monyer, H. Synaptic NR2A-but not NR2B-containing NMDA receptors increase with blockade of ionotropic glutamate receptors. Front. Mol. Neurosci. 2009, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA Receptor Involvement in Central Nervous System Disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, M.; Augusto, E.; Oliveira, C.; Agostinho, P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 2008, 156, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble Oligomers of Amyloid β Protein Facilitate Hippocampal Long-Term Depression by Disrupting Neuronal Glutamate Uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. GlutamateNMDA receptor NR1 subunit mRNA expression in Alzheimer’s disease. J. Neurochem. 2001, 78, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.; et al. Early Increase in Extrasynaptic NMDA Receptor Signaling and Expression Contributes to Phenotype Onset in Huntington’s Disease Mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Léveillé, F.; El Gaamouch, F.; Gouix, E.; Lecocq, M.; Lobner, D.; Nicole, O.; Buisson, A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 2008, 22, 4258–4271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Hollern, D.; Liao, J.; Andrechek, E.; Wang, H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013, 4, e560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.K.; Jacobi, E.; Sakimura, K.; Malinow, R.; Von Engelhardt, J. NMDA receptors mediate synaptic depression, but not spine loss in the dentate gyrus of adult amyloid Beta (Aβ) overexpressing mice. Acta Neuropathol. Commun. 2018, 6, 110. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Engelhardt, J.; Coserea, I.; Pawlak, V.; Fuchs, E.C.; Köhr, G.; Seeburg, P.H.; Monyer, H. Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology 2007, 53, 10–17. [Google Scholar] [CrossRef]
- Sattler, R.; Xiong, Z.; Lu, W.-Y.; Macdonald, J.F.; Tymianski, M. Distinct Roles of Synaptic and Extrasynaptic NMDA Receptors in Excitotoxicity. J. Neurosci. 2000, 20, 22–33. [Google Scholar] [CrossRef]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.-P.; Oliet, S.H. Synaptic and Extrasynaptic NMDA Receptors Are Gated by Different Endogenous Coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wroge, C.M.; Hogins, J.; Eisenman, L.; Mennerick, S. Synaptic NMDA Receptors Mediate Hypoxic Excitotoxic Death. J. Neurosci. 2012, 32, 6732–6742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Ding, Q.; Chen, Z.; Yun, H.; Wang, H. Involvement of the GluN2A and GluN2B Subunits in Synaptic and Extrasynaptic N-methyl-d-aspartate Receptor Function and Neuronal Excitotoxicity. J. Biol. Chem. 2013, 288, 24151–24159. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.B.; Ogden, K.K.; Yuan, H.; Traynelis, S.F. Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 2014, 81, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Queenan, B.N.; Lee, K.J.; Pak, D.T.S. Wherefore Art Thou, Homeo(stasis)? Functional Diversity in Homeostatic Synaptic Plasticity. Neural Plast. 2012, 2012, 718203. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.T.; Chin, J.; Leiser, S.C.; Pangalos, M.N.; Randall, A.D. Altered intrinsic neuronal excitability and reduced Na+ currents in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 2109.e1–2109.e14. [Google Scholar] [CrossRef]
- Kerrigan, T.; Brown, J.T.; Randall, A. Characterization of altered intrinsic excitability in hippocampal CA1 pyramidal cells of the Aβ-overproducing PDAPP mouse. Neuropharmacology 2014, 79, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Tamagnini, F.; Scullion, S.; Brown, J.T.; Randall, A.D. Intrinsic excitability changes induced by acute treatment of hippocampal CA1 pyramidal neurons with exogenous amyloid β peptide. Hippocampus 2015, 25, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Born, H. Seizures in Alzheimer’s disease. Neuroscience 2015, 286, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.A.; Shi, Y.; Usui, H.; During, M.J.; Sakimura, K.; Nicoll, R.A. Distinct Modes of AMPA Receptor Suppression at Developing Synapses by GluN2A and GluN2B: Single-Cell NMDA Receptor Subunit Deletion In Vivo. Neuron 2011, 71, 1085–1101. [Google Scholar] [CrossRef] [Green Version]
- Guardia-Laguarta, C.; Pera, M.; Clarimón, J.; Molinuevo, J.L.; Sànchez-Valle, R.; Lladó, A.; Coma, M.; Gómez-Isla, T.; Blesa, R.; Ferrer, I.; et al. Clinical, Neuropathologic, and Biochemical Profile of the Amyloid Precursor Protein I716F Mutation. J. Neuropathol. Exp. Neurol. 2010, 69, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Crouzin, N.; Baranger, K.; Cavalier, M.; Marchalant, Y.; Cohen-Solal, C.; Roman, F.S.; Khrestchatisky, M.; Rivera, S.; Féron, F.; Vignes, M. Area-Specific Alterations of Synaptic Plasticity in the 5XFAD Mouse Model of Alzheimer’s Disease: Dissociation between Somatosensory Cortex and Hippocampus. PLoS ONE 2013, 8, e74667. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ Oligomers Inhibit Long-Term Potentiation through a Mechanism Involving Excessive Activation of Extrasynaptic NR2B-Containing NMDA Receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Sun, W.; Hansen, K.B.; Jahr, C.E. Allosteric Interactions between NMDA Receptor Subunits Shape the Developmental Shift in Channel Properties. Neuron 2017, 94, 58–64.e3. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.-I.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollmann, M.; Boulter, J.; Maron, C.; Beasley, L.; Sullivan, J.; Pecht, G.; Heinemann, S. Zinc potentiates agonist-induced currents at certain splice variants of the NMDA receptor. Neuron 1993, 10, 943–954. [Google Scholar] [CrossRef]
- Traynelis, S.; Hartley, M.; Heinemann, S. Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science 1995, 268, 873–876. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Burgess, M.F.; Zheng, F.; Lyuboslavsky, P.; Powers, J.L. Control of Voltage-Independent Zinc Inhibition of NMDA Receptors by the NR1 Subunit. J. Neurosci. 1998, 18, 6163–6175. [Google Scholar] [CrossRef]
- Rumbaugh, G.; Prybylowski, K.; Wang, J.F.; Vicini, S. Exon 5 and spermine regulate deactivation of NMDA receptor subtypes. J. Neurophysiol. 2000, 83, 1300–1306. [Google Scholar] [CrossRef]
- Laurie, D.J.; Seeburg, P.H. Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J. Neurosci. 1994, 14, 3180–3194. [Google Scholar] [CrossRef] [Green Version]
- Standaert, D.G.; Testa, C.M.; Penney, J.B.; Young, A.B. Alternatively spliced isoforms of the NMDAR1 glutamate receptor subunit: Differential expression in the basal ganglia of the rat. Neurosci. Lett. 1993, 152, 161–164. [Google Scholar] [CrossRef]
- Standaert, D.; Testa, C.M.; Young, A.B.; Penney, J.B. Organization of N-methyl-D-aspartate glutamate receptor gene expression in the basal ganglia of the rat. J. Comp. Neurol. 1994, 343, 1–16. [Google Scholar] [CrossRef]
- Paupard, M.-C.; Friedman, L.; Zukin, R. Developmental regulation and cell-specific expression of N-methyl-d-aspartate receptor splice variants in rat hippocampus. Neuroscience 1997, 79, 399–409. [Google Scholar] [CrossRef]
- Zhong, J.; Carrozza, D.P.; Williams, K.; Pritchett, D.B.; Molinoff, P.B. Expression of mRNAs Encoding Subunits of the NMDA Receptor in Developing Rat Brain. J. Neurochem. 2002, 64, 531–539. [Google Scholar] [CrossRef]
- Lee, K.; Moussa, C.; Lee, Y.; Sung, Y.; Howell, B.; Turner, R.; Pak, D.; Hoe, H. Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience 2010, 169, 344–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Abeta Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Koistinaho, M.; Ort, M.; Cimadevilla, J.M.; Vondrous, R.; Cordell, B.; Bures, J.; Higgins, L.S. Specific spatial learning deficits become severe with age in β-amyloid precursor protein transgenic mice that harbor diffuse β-amyloid deposits but do not form plaques. Proc. Natl. Acad. Sci. USA 2001, 98, 14675–14680. [Google Scholar] [CrossRef] [Green Version]
- Preda, S.; Govoni, S.; Lanni, C.; Racchi, M.; Mura, E.; Grilli, M.; Marchi, M. Acute β-Amyloid Administration Disrupts the Cholinergic Control of Dopamine Release in the Nucleus Accumbens. Neuropsychopharmacology 2007, 33, 1062–1070. [Google Scholar] [CrossRef] [Green Version]
- Puzzo, D.; Privitera, L.; Fa’, M.; Staniszewski, A.; Hashimoto, G.; Aziz, F.; Sakurai, M.; Ribe, E.M.; Troy, C.M.; Mercken, M.; et al. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 2010, 69, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar Amyloid-β Positively Modulates Synaptic Plasticity and Memory in Hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef]
- Grilli, M.; Lagomarsino, F.; Zappettini, S.; Preda, S.; Mura, E.; Govoni, S.; Marchi, M. Specific inhibitory effect of amyloid-β on presynaptic muscarinic receptor subtypes modulating neurotransmitter release in the rat nucleus accumbens. Neuroscience 2010, 167, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Pardossi-Piquard, R.; Checler, F. The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 2011, 120, 109–124. [Google Scholar] [CrossRef]
- Montagna, E.; Dorostkar, M.M.; Herms, J. The Role of APP in Structural Spine Plasticity. Front. Mol. Neurosci. 2017, 10, 136. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Meyer, H.E.; Egensperger, R.; Marcus, K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics—Relevance for Alzheimer’s disease. Prog. Neurobiol. 2008, 85, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Schettini, G.; Govoni, S.; Racchi, M.; Rodriguez, G. Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: Signal transduction and/or transcriptional role—Relevance for Alzheimer pathology. J. Neurochem. 2010, 115, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Mehr, A.; Hick, M.; Ludewig, S.; Müller, M.; Herrmann, U.; Von Engelhardt, J.; Wolfer, D.P.; Korte, M.; Müller, U.C. Lack of APP and APLP2 in GABAergic Forebrain Neurons Impairs Synaptic Plasticity and Cognition. Cereb. Cortex 2020, 30, 4044–4063. [Google Scholar] [CrossRef] [PubMed]
- Tyan, S.-H.; Shih, A.Y.-J.; Walsh, J.; Maruyama, H.; Sarsoza, F.; Ku, L.; Eggert, S.; Hof, P.R.; Koo, E.H.; Dickstein, D.L. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell. Neurosci. 2012, 51, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Weyer, S.W.; Zagrebelsky, M.; Herrmann, U.; Hick, M.; Ganss, L.; Gobbert, J.; Gruber, M.; Altmann, C.; Korte, M.; Deller, T.; et al. Comparative analysis of single and combined APP/APLP knockouts reveals reduced spine density in APP-KO mice that is prevented by APPsα expression. Acta Neuropathol. Commun. 2014, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Steubler, V.; Erdinger, S.; Back, M.K.; Ludewig, S.; Fässler, D.; Richter, M.; Han, K.; Slomianka, L.; Amrein, I.; von Engelhardt, J.; et al. Loss of all three APP family members during development impairs synaptic function and plasticity, disrupts learning, and causes an autism-like phenotype. EMBO J. 2021, e107471. [Google Scholar] [CrossRef]
- Holtmaat, A.J.; Trachtenberg, J.T.; Wilbrecht, L.; Shepherd, G.M.; Zhang, X.; Knott, G.W.; Svoboda, K. Transient and Persistent Dendritic Spines in the Neocortex In Vivo. Neuron 2005, 45, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Berry, K.P.; Nedivi, E. Spine Dynamics: Are They All the Same? Neuron 2017, 96, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grutzendler, J.; Kasthuri, N.; Gan, W.-B. Long-term dendritic spine stability in the adult cortex. Nature 2002, 420, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Akashi, K.; Kakizaki, T.; Kamiya, H.; Fukaya, M.; Yamasaki, M.; Abe, M.; Natsume, R.; Watanabe, M.; Sakimura, K. NMDA Receptor GluN2B (GluRε2/NR2B) Subunit Is Crucial for Channel Function, Postsynaptic Macromolecular Organization, and Actin Cytoskeleton at Hippocampal CA3 Synapses. J. Neurosci. 2009, 29, 10869–10882. [Google Scholar] [CrossRef]
- Alvarez, V.A.; Ridenour, D.A.; Sabatini, B.L. Distinct Structural and Ionotropic Roles of NMDA Receptors in Controlling Spine and Synapse Stability. J. Neurosci. 2007, 27, 7365–7376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papouin, T.; Oliet, S.H.R. Organization, control and function of extrasynaptic NMDA receptors. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130601. [Google Scholar] [CrossRef]
- Papadia, S.; Stevenson, P.; Hardingham, N.R.; Bading, H.; Hardingham, G.E. Nuclear Ca2+ and the cAMP Response Element-Binding Protein Family Mediate a Late Phase of Activity-Dependent Neuroprotection. J. Neurosci. 2005, 25, 4279–4287. [Google Scholar] [CrossRef]
- Jiang, X.; Tian, F.; Mearow, K.; Okagaki, P.; Lipsky, R. The excitoprotective effect of N-methyl-d-aspartate receptors is mediated by a brain-derived neurotrophic factor autocrine loop in cultured hippocampal neurons. J. Neurochem. 2005, 94, 713–722. [Google Scholar] [CrossRef]
- Lau, D.; Bading, H. Synaptic Activity-Mediated Suppression of p53 and Induction of Nuclear Calcium-Regulated Neuroprotective Genes Promote Survival through Inhibition of Mitochondrial Permeability Transition. J. Neurosci. 2009, 29, 4420–4429. [Google Scholar] [CrossRef]
- Dieterich, D.C.; Karpova, A.; Mikhaylova, M.; Zdobnova, I.; König, I.; Landwehr, M.; Kreutz, M.; Smalla, K.-H.; Richter, K.; Landgraf, P.; et al. Caldendrin–Jacob: A Protein Liaison That Couples NMDA Receptor Signalling to the Nucleus. PLoS Biol. 2008, 6, e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Kurup, P.; Zhang, Y.; Goebel-Goody, S.M.; Wu, P.H.; Hawasli, A.H.; Baum, M.L.; Bibb, J.A.; Lombroso, P.J. Extrasynaptic NMDA Receptors Couple Preferentially to Excitotoxicity via Calpain-Mediated Cleavage of STEP. J. Neurosci. 2009, 29, 9330–9343. [Google Scholar] [CrossRef]
- Zhang, S.-J.; Steijaert, M.N.; Lau, D.; Schütz, G.; Delucinge-Vivier, C.; Descombes, P.; Bading, H. Decoding NMDA Receptor Signaling: Identification of Genomic Programs Specifying Neuronal Survival and Death. Neuron 2007, 53, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Voronin, L.L.; Cherubini, E. ‘Deaf, mute and whispering’ silent synapses: Their role in synaptic plasticity. J. Physiol. 2004, 557, 3–12. [Google Scholar] [CrossRef]
- Niewoehner, B.; Single, F.N.; Hvalby, Ø.; Jensen, V.; Borgloh, S.M.Z.A.; Seeburg, P.H.; Rawlins, J.N.P.; Sprengel, R.; Bannerman, D.M. Impaired spatial working memory but spared spatial reference memory following functional loss of NMDA receptors in the dentate gyrus. Eur. J. Neurosci. 2007, 25, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Engelhardt, J.; Doganci, B.; Jensen, V.; Hvalby, Ø.; Göngrich, C.; Taylor, A.; Barkus, C.; Sanderson, D.J.; Rawlins, J.N.P.; Seeburg, P.H.; et al. Contribution of Hippocampal and Extra-Hippocampal NR2B-Containing NMDA Receptors to Performance on Spatial Learning Tasks. Neuron 2008, 60, 846–860. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Haertel, C.; Maelicke, A.; Montag, D. Galantamine Slows Down Plaque Formation and Behavioral Decline in the 5XFAD Mouse Model of Alzheimer’s Disease. PLoS ONE 2014, 9, e89454. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Aslam, M.; Gollisch, T.; Allen, K.; Von Engelhardt, J. CKAMP44 modulates integration of visual inputs in the lateral geniculate nucleus. Nat. Commun. 2018, 9, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothman, J.S.; Silver, R.A. NeuroMatic: An Integrated Open-Source Software Toolkit for Acquisition, Analysis and Simulation of Electrophysiological Data. Front. Neuroinform. 2018, 12, 14. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Back, M.K.; Ruggieri, S.; Jacobi, E.; von Engelhardt, J. Amyloid Beta-Mediated Changes in Synaptic Function and Spine Number of Neocortical Neurons Depend on NMDA Receptors. Int. J. Mol. Sci. 2021, 22, 6298. https://doi.org/10.3390/ijms22126298
Back MK, Ruggieri S, Jacobi E, von Engelhardt J. Amyloid Beta-Mediated Changes in Synaptic Function and Spine Number of Neocortical Neurons Depend on NMDA Receptors. International Journal of Molecular Sciences. 2021; 22(12):6298. https://doi.org/10.3390/ijms22126298
Chicago/Turabian StyleBack, Michaela K., Sonia Ruggieri, Eric Jacobi, and Jakob von Engelhardt. 2021. "Amyloid Beta-Mediated Changes in Synaptic Function and Spine Number of Neocortical Neurons Depend on NMDA Receptors" International Journal of Molecular Sciences 22, no. 12: 6298. https://doi.org/10.3390/ijms22126298
APA StyleBack, M. K., Ruggieri, S., Jacobi, E., & von Engelhardt, J. (2021). Amyloid Beta-Mediated Changes in Synaptic Function and Spine Number of Neocortical Neurons Depend on NMDA Receptors. International Journal of Molecular Sciences, 22(12), 6298. https://doi.org/10.3390/ijms22126298