
n-Butylidenephthalide Modulates Autophagy to Ameliorate Neuropathological Progress of Spinocerebellar Ataxia Type 3 through mTOR Pathway

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
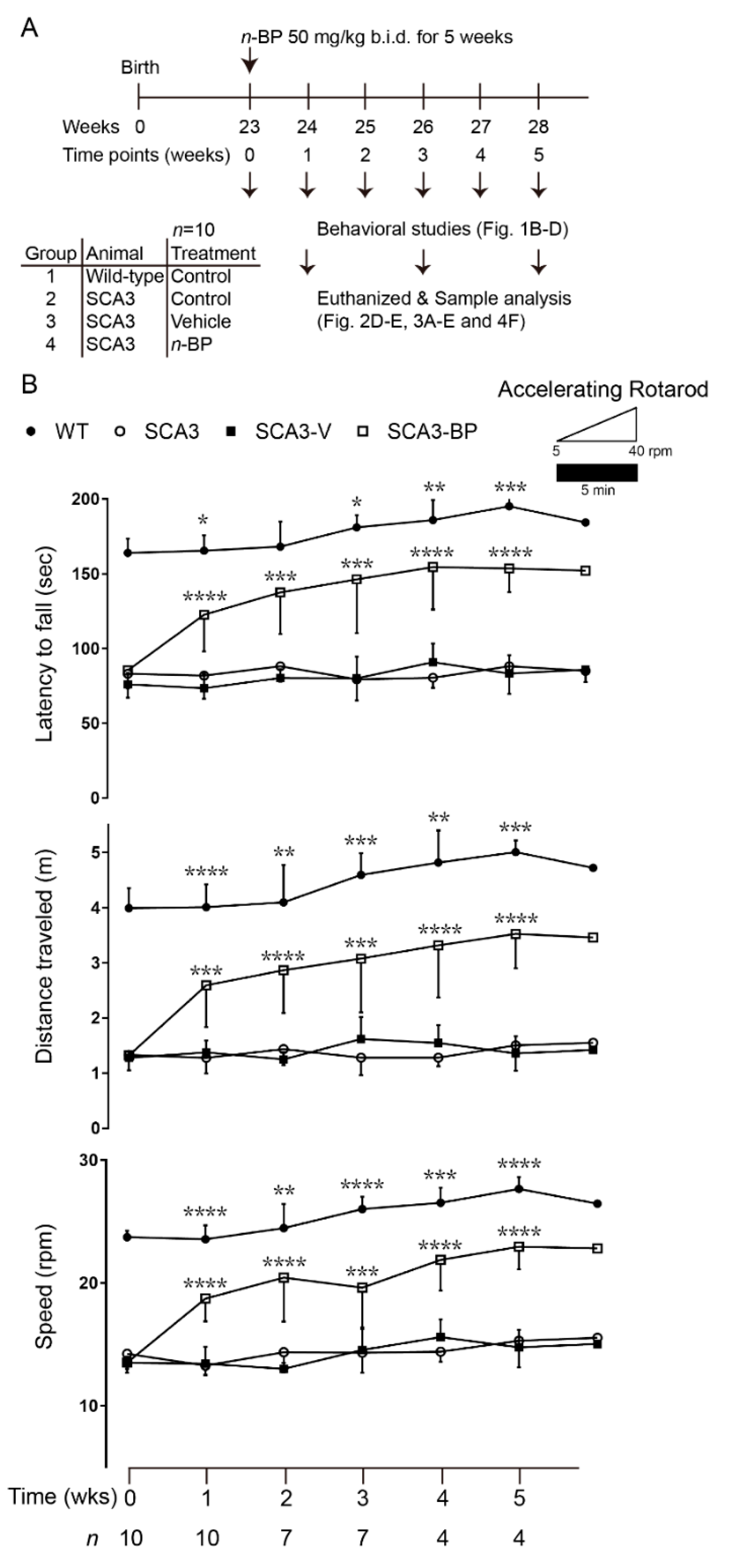
2.1. n-BP Effectively Improved Motor Function in SCA3 Animals
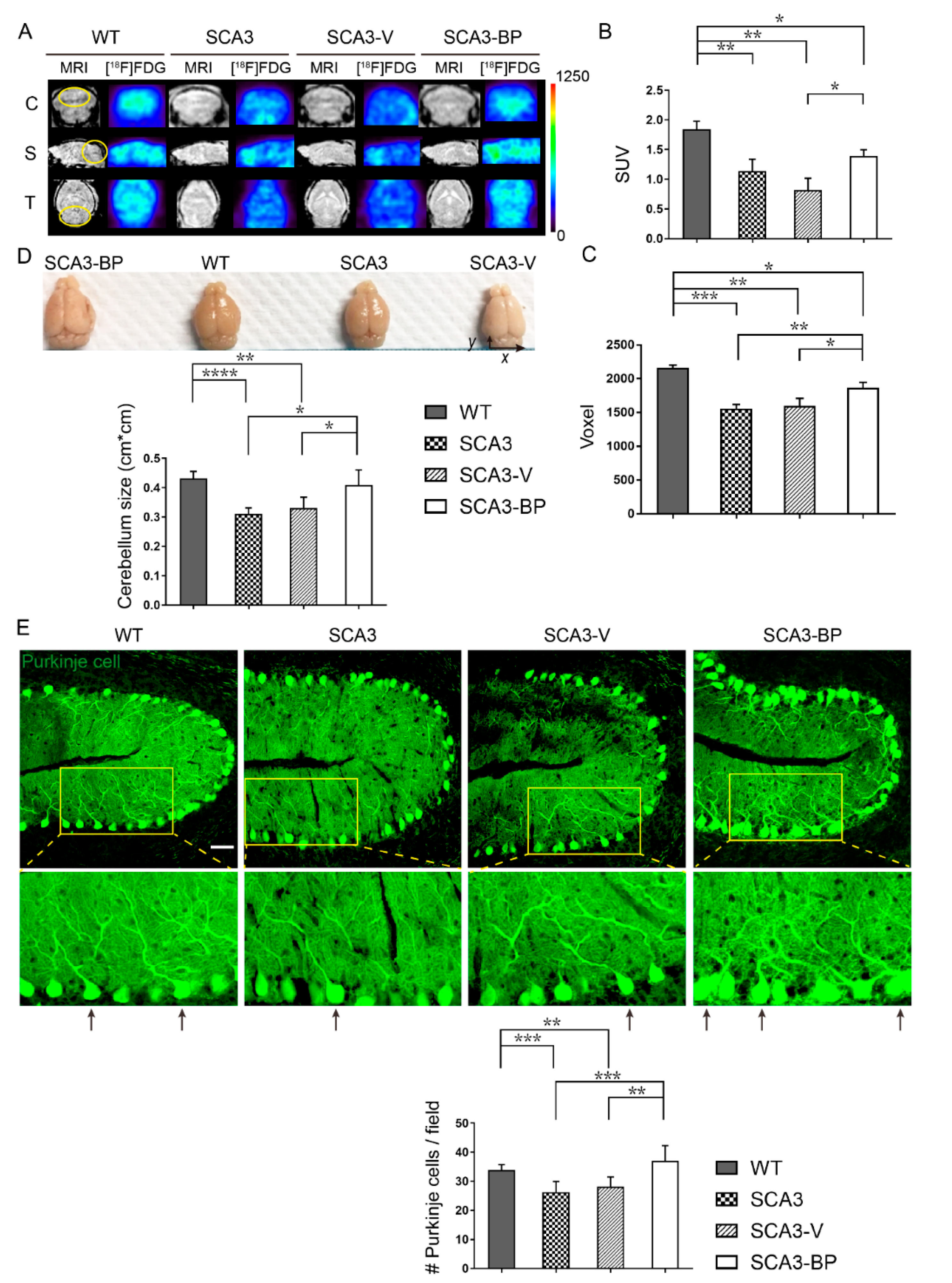
2.2. n-BP Ameliorated Cerebellar Neuropathology of SCA3 Mice
2.3. n-BP Promoted Autophagy to Eliminate the Aggregates In Vivo/In Vitro
2.4. n-BP Could Be an Autophagy Enhancer through Modulating AMPK/AKT/ERK1/2 to Inhibit mTOR Pathway In Vitro/In Vivo
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Behavior Assays
4.3. Animal Imaging
4.4. Immunofluorescence (IF) Staining
4.5. Western Blotting Analysis
4.6. Cell Culture and Treatment
4.7. Quantitative Real-Time PCR
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| AMPK | AMP-dependent protein kinase |
| ATG | Autophagy-related protein |
| BafA1 | Bafilomycin A1 |
| ERK | Extracellular signal-regulated protein kinase |
| HEK-293 | Human embryonic kidney-293 |
| LC3B | Light-chain-3 B subunit |
| MJD | Machado-Joseph disease |
| mTOR | Mammalian target of rapamycin |
| MRI | Magnetic resonance imaging |
| PET | Positron emission tomography |
| SCA | Spinocerebellar ataxia |
| WT | Wild type |
References
- Strong, T.V.; Tagle, D.A.; Valdes, J.M.; Elmer, L.W.; Boehm, K.; Swaroop, M.; Kaatz, K.W.; Collins, F.S.; Albin, R.L. Widespread expression of the human and rat Huntington′s disease gene in brain and nonneural tissues. Nat. Genet. 1993, 5, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32. 1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Koide, R.; Ikeuchi, T.; Onodera, O.; Tanaka, H.; Igarashi, S.; Endo, K.; Takahashi, H.; Kondo, R.; Ishikawa, A.; Hayashi, T. Unstable expansion of CAG repeat in hereditary dentatorubral—Pallidoluysian atrophy (DRPLA). Nat. Genet. 1994, 6, 9–13. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Abbas, N.; Stevanin, G.; Dürr, A.; Yvert, G.; Cancel, G.; Weber, C.; Imbert, G.; Saudou, F.; Antoniou, E. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997, 17, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α 1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Liu, C.; Shen, L.; Dai, H.; Pan, Q.; Jing, L.; Ouyang, S.; Xia, J. Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch. Neurol. 2000, 57, 540–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, B.; Li, F.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-K.; Chen, M.-H.; Chiang, Y.-H.; Chen, Y.-F.; Ma, W.-H.; Tseng, C.-Y.; Soong, B.-W.; Ho, J.H.; Lee, O.K. Mesenchymal stem cell transplantation ameliorates motor function deterioration of spinocerebellar ataxia by rescuing cerebellar Purkinje cells. J. Biomed. Sci. 2011, 18, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Kasumu, A.; Bezprozvanny, I. Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum 2012, 11, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Becher, M.; Rubinsztein, D.; Leggo, J.; Wagster, M.; Stine, O.; Ranen, N.; Franz, M.; Abbott, M.; Sherr, M.; MacMillan, J. Dentatorubral and pallidoluysian atrophy (DRPLA) Clinical and neuropathological findings in genetically confirmed north american and european pedigrees. Mov. Disord. 1997, 12, 519–530. [Google Scholar] [CrossRef]
- Durr, A.; Stevanin, G.; Cancel, G.; Duyckaerts, C.; Abbas, N.; Didierjean, O.; Chneiweiss, H.; Benomar, A.; Lyon-Caen, O.; Julien, J. Spinocerebellar ataxia 3 and Machado-Joseph disease: Clinical, molecular, and neuropathological features. Ann. Neurol. 1996, 39, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Van Alfen, N.; Sinke, R.J.; Zwarts, M.J.; Gabreëls-Festen, A.; Praamstra, P.; Kremer, B.P.; Horstink, M.W. Intermediate CAG repeat lengths (53, 54) for MJD/SCA3 are associated with an abnormal phenotype. Ann. Neurol. 2001, 49, 805–808. [Google Scholar] [CrossRef]
- Fan, H.-C.; Ho, L.-I.; Chi, C.-S.; Chen, S.-J.; Peng, G.-S.; Chan, T.-M.; Lin, S.-Z.; Harn, H.-J. Polyglutamine (PolyQ) diseases: Genetics to treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tüting, T. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado–Joseph disease. Nature 2011, 480, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Leotti, V.B.; de Vries, J.J.; Oliveira, C.M.; de Mattos, E.P.; Te Meerman, G.J.; Brunt, E.R.; Kampinga, H.H.; Jardim, L.B.; Verbeek, D.S. CAG repeat size influences the progression rate of spinocerebellar ataxia type 3. Ann. Neurol. 2021, 89, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Haacke, A.; Hartl, F.U.; Breuer, P. Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded ataxin-3. J. Biol. Chem. 2007, 282, 18851–18856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, A.T.; Gonçalves, N.; Koeppen, A.; Déglon, N.; Kügler, S.; Duarte, C.B.; de Almeida, L.P. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado–Joseph disease. Brain 2012, 135, 2428–2439. [Google Scholar] [CrossRef]
- Rajamani, K.; Liu, J.-W.; Wu, C.-H.; Chiang, I.-T.; You, D.-H.; Lin, S.-Y.; Hsieh, D.-K.; Lin, S.-Z.; Harn, H.-J.; Chiou, T.-W. n-Butylidenephthalide exhibits protection against neurotoxicity through regulation of tryptophan 2, 3 dioxygenase in spinocerebellar ataxia type 3. Neuropharmacology 2017, 117, 434–446. [Google Scholar] [CrossRef]
- Onofre, I.; Mendonça, N.; Lopes, S.; Nobre, R.; De Melo, J.B.; Carreira, I.M.; Januário, C.; Gonçalves, A.F.; De Almeida, L.P. Fibroblasts of Machado Joseph Disease patients reveal autophagy impairment. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Burnett, B.G.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. USA 2005, 102, 4330–4335. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Hong, S.; Ikeda, T.; Mori, H.; MacDougald, O.A.; Nada, S.; Okada, M.; Inoki, K. Amino acids enhance polyubiquitination of Rheb and its binding to mTORC1 by blocking lysosomal ATXN3 deubiquitinase activity. Mol. Cell 2020, 80, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.-J.; do Carmo Costa, M.; Silva, T.-L.; Ferreira, D.; Bajanca, F.; Logarinho, E.; Maciel, P. Absence of ataxin-3 leads to cytoskeletal disorganization and increased cell death. BBA-Mol. Cell Res. 2010, 1803, 1154–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warrick, J.M.; Morabito, L.M.; Bilen, J.; Gordesky-Gold, B.; Faust, L.Z.; Paulson, H.L.; Bonini, N.M. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell 2005, 18, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, G.; Menon, R.P.; Masino, L.; Knowles, P.P.; McDonald, N.Q.; Pastore, A. The solution structure of the Josephin domain of ataxin-3: Structural determinants for molecular recognition. Proc. Natl. Acad. Sci. USA 2005, 102, 10493–10498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Senic-Matuglia, F.; Di Fiore, P.P.; Polo, S.; Hodsdon, M.E.; De Camilli, P. Deubiquitinating function of ataxin-3: Insights from the solution structure of the Josephin domain. Proc. Natl. Acad. Sci. USA 2005, 102, 12700–12705. [Google Scholar] [CrossRef] [Green Version]
- Nascimento-Ferreira, I.; Nobrega, C.; Vasconcelos-Ferreira, A.; Onofre, I.; Albuquerque, D.; Aveleira, C.; Hirai, H.; Deglon, N.; de Almeida, L.P. Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado–Joseph disease. Brain 2013, 136, 2173–2188. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Herzog, L.K.; Kevei, É.; Marchante, R.; Böttcher, C.; Bindesbøll, C.; Lystad, A.H.; Pfeiffer, A.; Gierisch, M.E.; Salomons, F.A.; Simonsen, A. The Machado–Joseph disease deubiquitylase ataxin-3 interacts with LC3C/GABARAP and promotes autophagy. Aging Cell 2020, 19, 1–15. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Chiu, S.-C.; Chen, S.-P.; Huang, S.-Y.; Wang, M.-J.; Lin, S.-Z.; Harn, H.-J.; Pang, C.-Y. Induction of apoptosis coupled to endoplasmic reticulum stress in human prostate cancer cells by n-butylidenephthalide. PLoS ONE 2012, 7, e33742–e33755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, N.-M.; Lin, S.-Z.; Lee, C.-C.; Chen, S.-P.; Su, H.-C.; Chang, W.-L.; Harn, H.-J. The antitumor effects of Angelica sinensis on malignant brain tumors in vitro and in vivo. Clin. Cancer Res. 2005, 11, 3475–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-F.; Chang, J.T.; Huang, X.-F.; Lin, Y.-L.; Liao, K.-W.; Huang, C.-W.; Tsai, N.-M. Antitumor effects of n-butylidenephthalide encapsulated in lipopolyplexs in colorectal cancer cells. Molecules 2020, 25, 2394. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.-M.; Chen, W.-Y.; Ko, W.-C.; Ouyang, C. Antiplatelet effect of butylidenephthalide. BBA-Gen. Subj. 1987, 924, 375–382. [Google Scholar] [CrossRef]
- Zhou, Q.M.; Zhang, J.J.; Li, S.; Chen, S.; Le, W.D. n-Butylidenephthalide treatment prolongs life span and attenuates motor neuron loss in SOD 1G93A mouse model of amyotrophic lateral sclerosis. CNS Neurosci. Ther. 2017, 23, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, K.-W.; Chiou, T.-W.; Chiang, S.-F.; Yamashita, T.; Abe, K.; Borlongan, C.V.; Sanberg, P.R.; Lin, S.-Z.; Harn, H.-J. Autophagic down-regulation in motor neurons remarkably prolongs the survival of ALS mice. Neuropharmacology 2016, 108, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cueto, C.; Hernández-Gálvez, M.; Hillard, C.J.; Maciel, P.; García-García, L.; Valdeolivas, S.; Pozo, M.A.; Ramos, J.A.; Gómez-Ruiz, M.; Fernández-Ruiz, J. Dysregulation of the endocannabinoid signaling system in the cerebellum and brainstem in a transgenic mouse model of spinocerebellar ataxia type-3. Neuroscience 2016, 339, 191–209. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tang, T.-S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef] [Green Version]
- Trzesniewski, J.; Altmann, S.; Jäger, L.; Kapfhammer, J.P. Reduced Purkinje cell size is compatible with near normal morphology and function of the cerebellar cortex in a mouse model of spinocerebellar ataxia. Exp. Neurol. 2019, 311, 205–212. [Google Scholar] [CrossRef]
- Ishikawa, K.; Mizusawa, H.; Fujita, T.; Ohkoshi, N.; Doi, M.; Komatsuzaki, Y.; Iwamoto, H.; Ogata, T. Calbindin-D 28k immunoreactivity in the cerebellum of spinocerebellar degeneration. J. Neurol. Sci. 1995, 129, 179–185. [Google Scholar] [CrossRef]
- Musiwaro, P.; Smith, M.; Manifava, M.; Walker, S.A.; Ktistakis, N.T. Characteristics and requirements of basal autophagy in HEK 293 cells. Autophagy 2013, 9, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelárová, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Ktistakis, N.T.; Manifava, M.; Schoenfelder, P.; Rotondo, S. How phosphoinositide 3-phosphate controls growth downstream of amino acids and autophagy downstream of amino acid withdrawal. Biochem. Soc. Trans. 2012, 40, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cemal, C.K.; Carroll, C.J.; Lawrence, L.; Lowrie, M.B.; Ruddle, P.; Al-Mahdawi, S.; King, R.H.; Pook, M.A.; Huxley, C.; Chamberlain, S. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum. Mol. Genet. 2002, 11, 1075–1094. [Google Scholar] [CrossRef]
- Fu, R.-H.; Harn, H.-J.; Liu, S.-P.; Chen, C.-S.; Chang, W.-L.; Chen, Y.-M.; Huang, J.-E.; Li, R.-J.; Tsai, S.-Y.; Hung, H.-S. n-butylidenephthalide protects against dopaminergic neuron degeneration and α-synuclein accumulation in Caenorhabditis elegans models of Parkinson's disease. PLoS ONE 2014, 9, e85305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, K.; Fu, R.-H.; Huang, Y.-C.; Chen, S.-Y.; Hsu, C.-J.; Lin, S.-Z.; Tu, C.-T.; Chang, L.-H.; Wu, P.-A.; Liu, S.-P. Adipose-derived stem cells stimulated with n-butylidenephthalide exhibit therapeutic effects in a mouse model of Parkinson′s disease. Cell Transplant. 2018, 27, 456–470. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Liu, J.-W.; Lin, S.-Z.; Harn, H.-J.; Chiou, T.-W. Advances in patient-specific induced pluripotent stem cells shed light on drug discovery for amyotrophic lateral sclerosis. Cell Transplant. 2018, 27, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Hilbich, C.; Kisters-Woike, B.; Reed, J.; Masters, C.L.; Beyreuther, K. Aggregation and secondary structure of synthetic amyloid βA4 peptides of Alzheimer′s disease. J. Mol. Biol. 1991, 218, 149–163. [Google Scholar] [CrossRef]
- Levine, H., III. Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Baralle, M.; Buratti, E.; Baralle, F.E. The role of TDP-43 in the pathogenesis of ALS and FTLD. Curr. Opin. Neurol. 2013, 21, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Santos, J.; Duarte-Neves, J.; Carmona, V.; Guarente, L.; De Almeida, L.P.; Cavadas, C. Caloric restriction blocks neuropathology and motor deficits in Machado–Joseph disease mouse models through SIRT1 pathway. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Noorasyikin, M.; Elena, A.; Teh, P.; Waheeda, T.F.; Hajar, M.S.; Long, K.; Norlinah, M. Oral trehalose maybe helpful for patients with spinocerebellar ataxia 3 and should be better evaluated. Parkinsonism Relat. Disord. 2020, 70, 42–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Lebrón, E.; do Carmo Costa, M.; Luna-Cancalon, K.; Peron, T.M.; Fischer, S.; Boudreau, R.L.; Davidson, B.L.; Paulson, H.L. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther. 2013, 21, 1909–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotowska-Zimmer, A.; Ostrovska, Y.; Olejniczak, M. Universal RNAi triggers for the specific inhibition of mutant huntingtin, atrophin-1, ataxin-3, and ataxin-7 expression. Mol. Ther.-Nucl. Acids 2020, 19, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.-F.; Yang, G.-P.; Wang, J.-L.; Chuang, D.-M.; Song, W.-H.; Tang, B.-S.; Jiang, H. Safety and efficacy of valproic acid treatment in SCA3/MJD patients. Parkinsonism Relat. Disord. 2016, 26, 55–61. [Google Scholar] [CrossRef]
- Zesiewicz, T.A.; Greenstein, P.; Sullivan, K.L.; Wecker, L.; Miller, A.; Jahan, I.; Chen, R.; Perlman, S. A randomized trial of varenicline (Chantix) for the treatment of spinocerebellar ataxia type 3. Neurology 2012, 78, 545–550. [Google Scholar] [CrossRef]
- Saute, J.A.M.; de Castilhos, R.M.; Monte, T.L.; Schumacher-Schuh, A.F.; Donis, K.C.; D′Ávila, R.; Souza, G.N.; Russo, A.D.; Furtado, G.V.; Gheno, T.C. A randomized, phase 2 clinical trial of lithium carbonate in Machado-Joseph disease. Mov. Disord. 2014, 29, 568–573. [Google Scholar] [CrossRef]
- Jin, J.-L.; Liu, Z.; Lu, Z.-J.; Guan, D.-N.; Wang, C.; Chen, Z.-B.; Zhang, J.; Zhang, W.-Y.; Wu, J.-Y.; Xu, Y. Safety and efficacy of umbilical cord mesenchymal stem cell therapy in hereditary spinocerebellar ataxia. Curr. Neurovasc. Res. 2013, 10, 11–20. [Google Scholar] [CrossRef]
- Tsai, Y.-A.; Liu, R.-S.; Lirng, J.-F.; Yang, B.-H.; Chang, C.-H.; Wang, Y.-C.; Wu, Y.-S.; Ho, J.H.-C.; Lee, O.K.; Soong, B.-W. Treatment of spinocerebellar ataxia with mesenchymal stem cells: A phase I/IIa clinical study. Cell Transplant. 2017, 26, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Nascimento-Ferreira, I.; Santos-Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Colomer Gould, V.F.; Koeppen, A.; Déglon, N. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado–Joseph disease. Brain 2011, 134, 1400–1415. [Google Scholar] [CrossRef]
- De Carvalho, L.P.; Bochet, P.; Rossier, J. The endogeneous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem. Int. 1996, 28, 445–452. [Google Scholar] [CrossRef]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropath. Exp. Neur. 2011, 70, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhou, B.; Lin, M.-Y.; Wang, S.; Foust, K.D.; Sheng, Z.-H. Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS mice. Neuron 2015, 87, 355–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciel, P.; do Carmo Costa, M.; Ferro, A.; Rousseau, M.; Santos, C.S.; Gaspar, C.; Barros, J.; Rouleau, G.A.; Coutinho, P.; Sequeiros, J. Improvement in the molecular diagnosis of Machado-Joseph disease. Arch. Neurol. 2001, 58, 1821–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watchon, M.; Yuan, K.C.; Mackovski, N.; Svahn, A.J.; Cole, N.J.; Goldsbury, C.; Rinkwitz, S.; Becker, T.S.; Nicholson, G.A.; Laird, A.S. Calpain inhibition is protective in machado–joseph disease zebrafish due to induction of autophagy. J. Neurosci. 2017, 37, 7782–7794. [Google Scholar] [CrossRef] [Green Version]
- Hübener, J.; Vauti, F.; Funke, C.; Wolburg, H.; Ye, Y.; Schmidt, T.; Wolburg-Buchholz, K.; Schmitt, I.; Gardyan, A.; Drießen, S. N-terminal ataxin-3 causes neurological symptoms with inclusions, endoplasmic reticulum stress and ribosomal dislocation. Brain 2011, 134, 1925–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, V.F.C.; Goti, D.; Pearce, D.; Gonzalez, G.A.; Gao, H.; de Leon, M.B.; Jenkins, N.A.; Copeland, N.G.; Ross, C.A.; Brown, D.R. A mutant ataxin-3 fragment results from processing at a site N-terminal to amino acid 190 in brain of Machado–Joseph disease-like transgenic mice. Neurobiol. Dis. 2007, 27, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, R.; Galarraga, J.; Orozco, G.; Nodarse, A.; Auburger, G. Spinocerebellar ataxia 2 (SCA2): Morphometric analyses in 11 autopsies. Acta Neuropathol. 1999, 97, 306–310. [Google Scholar] [CrossRef]
- Wang, P.-S.; Liu, R.-S.; Yang, B.-H.; Soong, B.-W. Regional patterns of cerebral glucose metabolism in spinocerebellar ataxia type 2, 3 and 6. J. Neurol. 2007, 254, 838–845. [Google Scholar] [CrossRef]
- Rodrigues, A.-J.; Coppola, G.; Santos, C.; Costa, M.d.C.; Ailion, M.; Sequeiros, J.; Geschwind, D.H.; Mariel, P. Functional genomics and biochemical characterization of the C. elegans orthologue of the Machado-Joseph disease protein ataxin-3. FASEB J. 2007, 21, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Asano, T.; Kuma, A.; Mizushima, N. Cycloheximide inhibits starvation-induced autophagy through mTORC1 activation. Biochem. Biophys. Res. Commun. 2014, 445, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, G.; Chen, S.; Gou, Y.; Dong, Z.; Zhang, X. Homocysteine aggravates cortical neural cell injury through neuronal autophagy overactivation following rat cerebral ischemia-reperfusion. Int. J. Mol. Sci. 2016, 17, 1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Song, Y.; Li, Y.; Du, Y.; Zhang, X.; Fu, J. The role of autophagy in the correlation between neuron damage and cognitive impairment in rat chronic cerebral hypoperfusion. Mol. Neurobiol. 2018, 55, 776–791. [Google Scholar] [CrossRef]
- Pattison, J.S.; Osinska, H.; Robbins, J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ. Res. 2011, 109, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.; Liu, L.; Zhu, W.; Li, D.; Yang, L.; Duan, J.; Cai, Z.; Nie, Y.; Zhang, Y.; Gong, Q. Iron oxide nanoparticles promote macrophage autophagy and inflammatory response through activation of toll-like Receptor-4 signaling. Biomaterials 2019, 203, 23–30. [Google Scholar] [CrossRef]
- Yang, H.-H.; Xu, Y.-X.; Chen, J.-Y.; Harn, H.-J.; Chiou, T.-W. n-Butylidenephthalide inhibits the phenotypic switch of VSMCs through activation of AMPK and prevents stenosis in an arteriovenous fistula rat model. Int. J. Mol. Sci. 2020, 21, 7403. [Google Scholar] [CrossRef] [PubMed]
- Marcelo, A.; Brito, F.; Carmo-Silva, S.; Matos, C.A.; Alves-Cruzeiro, J.; Vasconcelos-Ferreira, A.; Koppenol, R.; Mendonça, L.; de Almeida, L.P.; Nóbrega, C. Cordycepin activates autophagy through AMPK phosphorylation to reduce abnormalities in Machado–Joseph disease models. Hum. Mol. Genet. 2019, 28, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.-Y.; Chuang, H.-M.; Huang, M.-H.; Lin, S.-Z.; Chiou, T.-W.; Harn, H.-J. n-Butylidenephthalide regulated tumor stem cell genes EZH2/AXL and reduced its migration and invasion in glioblastoma. Int. J. Mol. Sci. 2017, 18, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Setter, S.M.; Iltz, J.L.; Thams, J.; Campbell, R.K. Metformin hydrochloride in the treatment of type 2 diabetes mellitus: A clinical review with a focus on dual therapy. Clin. Ther. 2003, 25, 2991–3026. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell 2018, 71, 606–620. [Google Scholar] [CrossRef] [Green Version]
- Bisulli, F.; Muccioli, L.; d’Orsi, G.; Canafoglia, L.; Freri, E.; Licchetta, L.; Mostacci, B.; Riguzzi, P.; Pondrelli, F.; Avolio, C. Treatment with metformin in twelve patients with Lafora disease. Orphanet. J. Rare Dis. 2019, 14, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Huttunen, K.M.; Sikora, J. Metformin and its sulphonamide derivative simultaneously potentiateanti-cholinesterase activity of donepezil and inhibit beta-amyloid aggregation. J. Enzym. Inhib. Med. Ch. 2018, 33, 1309–1322. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, X.; Zhao, M.; Wei, Z.; Li, X.; Zhang, X.; Liu, Z.; Gong, Y.; Shao, C. Berberine induces senescence of human glioblastoma cells by downregulating the EGFR–MEK–ERK signaling pathway. Mol. Cancer Ther. 2015, 14, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Liu, S.; Ma, Q.; Xiao, D.; Chen, L. Berberine enhances the AMPK activation and autophagy and mitigates high glucose-induced apoptosis of mouse podocytes. Eur. J. Pharmacol. 2017, 794, 106–114. [Google Scholar] [CrossRef]
- Wang, J.; Jin, D. Berberine alleviates amyloid beta-induced injury in Alzheimer′s disease by miR-107/ZNF217. RSC Adv. 2019, 9, 25232–25239. [Google Scholar] [CrossRef] [Green Version]
- Loening, A.M.; Gambhir, S.S. AMIDE: A free software tool for multimodality medical image analysis. Mol. Imaging 2003, 2, 131–137. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-H.; Lin, S.-Y.; Liu, J.-W.; Lin, S.-Z.; Harn, H.-J.; Chiou, T.-W. n-Butylidenephthalide Modulates Autophagy to Ameliorate Neuropathological Progress of Spinocerebellar Ataxia Type 3 through mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 6339. https://doi.org/10.3390/ijms22126339
Lee J-H, Lin S-Y, Liu J-W, Lin S-Z, Harn H-J, Chiou T-W. n-Butylidenephthalide Modulates Autophagy to Ameliorate Neuropathological Progress of Spinocerebellar Ataxia Type 3 through mTOR Pathway. International Journal of Molecular Sciences. 2021; 22(12):6339. https://doi.org/10.3390/ijms22126339
Chicago/Turabian StyleLee, Jui-Hao, Si-Yin Lin, Jen-Wei Liu, Shinn-Zong Lin, Horng-Jyh Harn, and Tzyy-Wen Chiou. 2021. "n-Butylidenephthalide Modulates Autophagy to Ameliorate Neuropathological Progress of Spinocerebellar Ataxia Type 3 through mTOR Pathway" International Journal of Molecular Sciences 22, no. 12: 6339. https://doi.org/10.3390/ijms22126339