
Association between Predicted Effects of TP53 Missense Variants on Protein Conformation and Their Phenotypic Presentation as Li-Fraumeni Syndrome or Hereditary Breast Cancer

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of TP53 Variants
2.2. Analysis of Protein Structure
2.3. Explanatory Variables
2.4. Statistical Analysis
3. Results
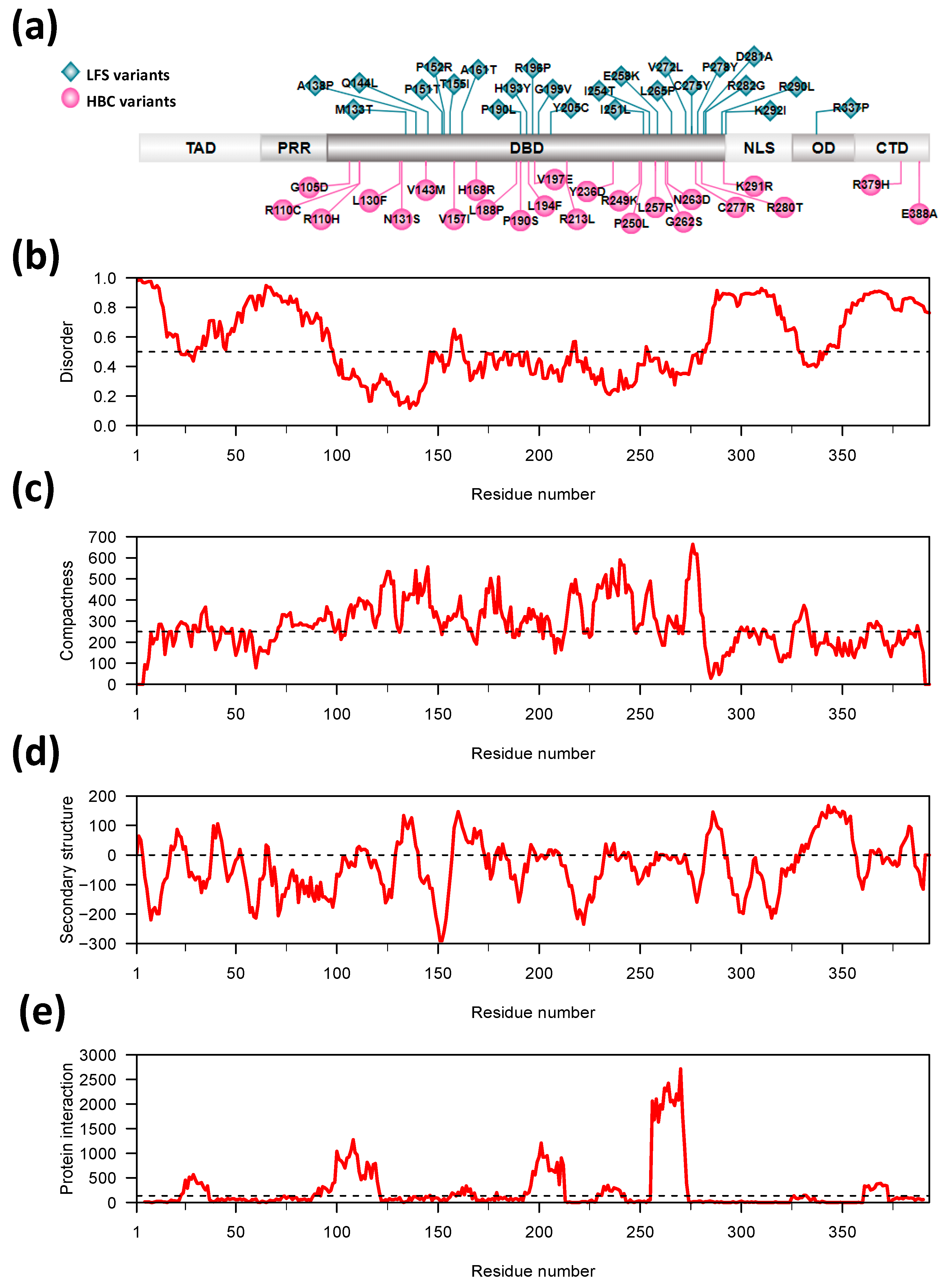
3.1. Characteristics of LFS and HBC Germline TP53 Variants
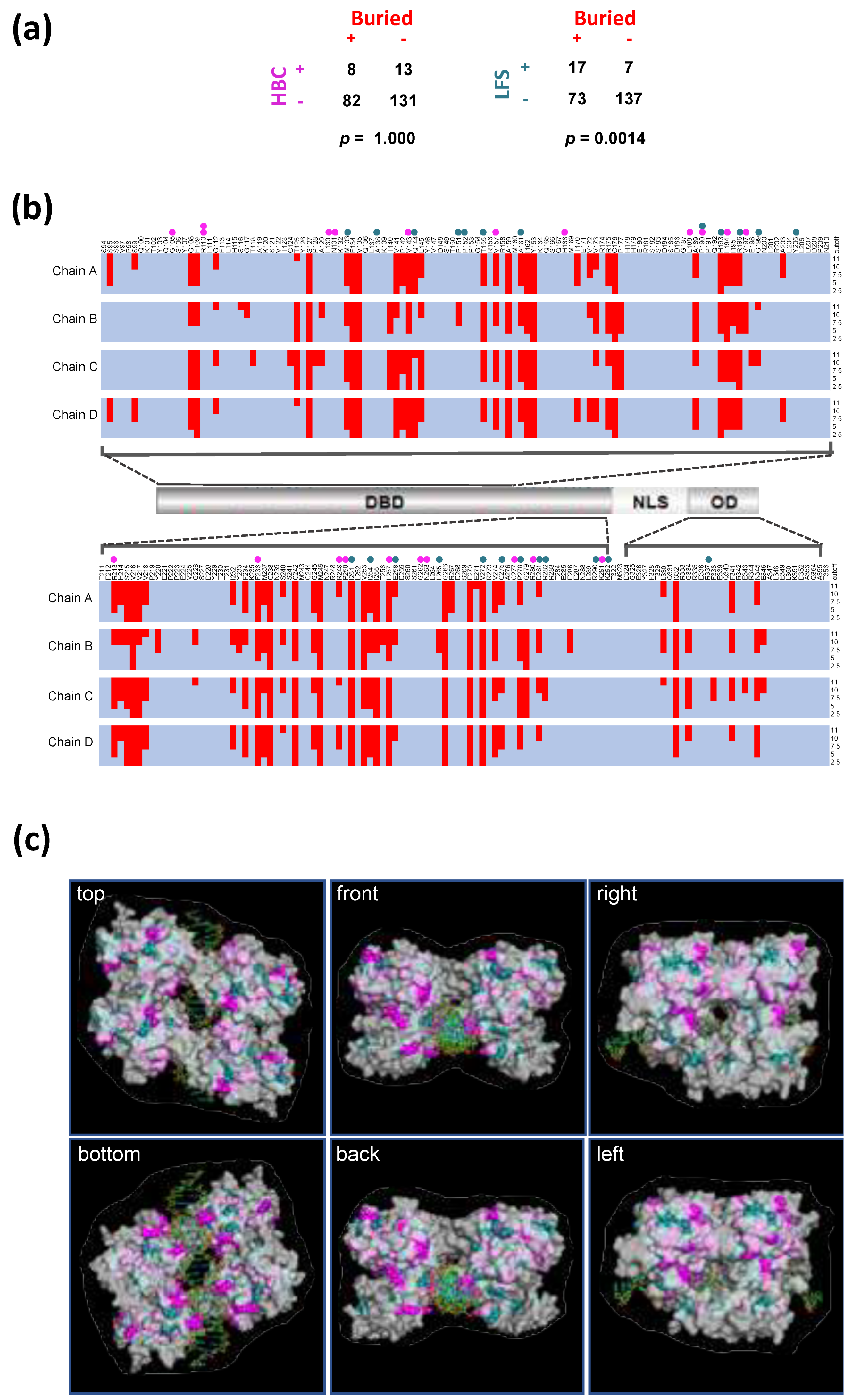
3.2. Location of LFS- and HBC-Residues in Relation to the TP53 Protein Structure
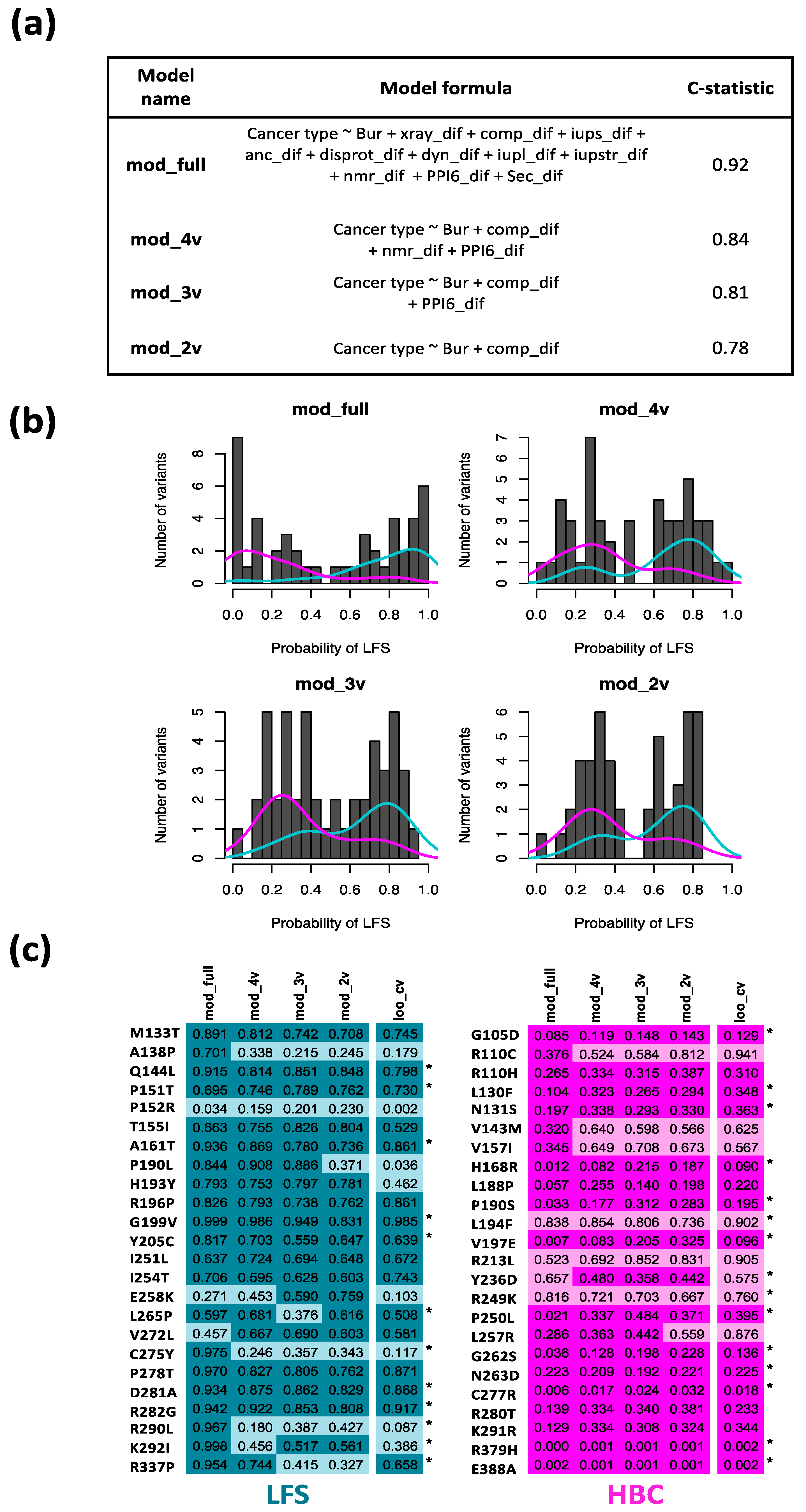
3.3. Association between TP53-Variant-Induced Changes in Protein Conformation Characteristics and Disease Outcome
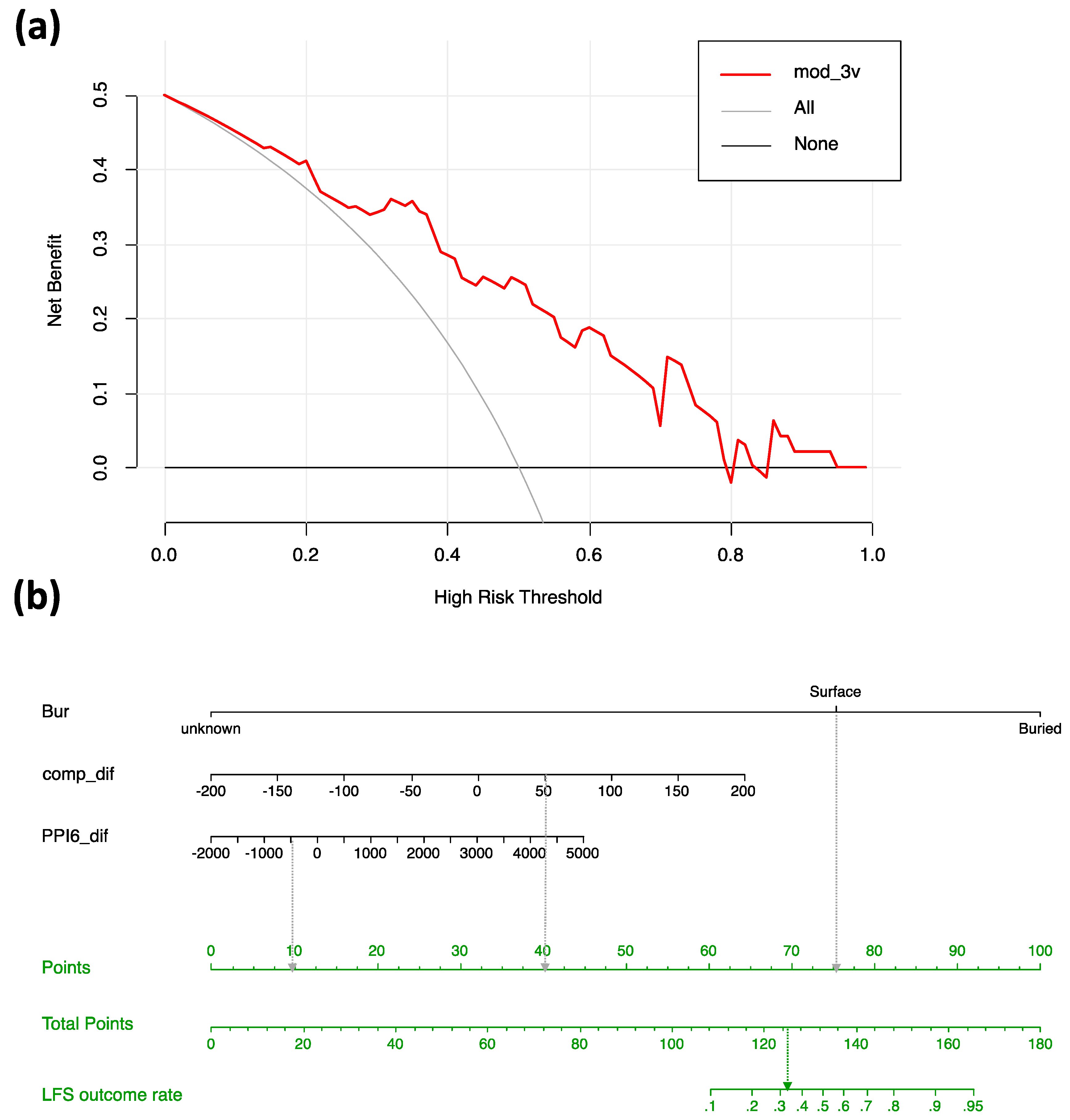
3.4. Potential for the Most Favored Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, F.P. Soft-Tissue Sarcomas, Breast Cancer, and Other Neoplasms. A Familial Syndrome? Ann. Intern. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Malkin, D.; Jolly, K.W.; Barbier, N.; Look, A.T.; Friend, S.H.; Gebhardt, M.C.; Andersen, T.I.; Børresen, A.-L.; Li, F.P.; Garber, J.; et al. Germline Mutations of the p53 Tumor-Suppressor Gene in Children and Young Adults with Second Malignant Neoplasms. N. Engl. J. Med. 1992, 326, 1309–1315. [Google Scholar] [CrossRef]
- Birch, J.M.; Hartley, A.L.; Tricker, K.J.; Prosser, J.; Condie, A.; Kelsey, A.M.; Harris, M.; Jones, P.H.; Binchy, A.; Crowther, D. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994, 54, 1298–1304. [Google Scholar]
- Kleihues, P.; Schäuble, B.; Hausen, A.Z.; Estève, J.; Ohgaki, H. Tumors associated with p53 germline mutations: A synopsis of 91 families. Am. J. Pathol. 1997, 150, 1. [Google Scholar]
- Chompret, A.; Abel, A.; Stoppa-Lyonnet, D.; Brugières, L.; Pagès, S.; Feunteun, J.; Bonaïti-Pellié, C. Sensitivity and predictive value of criteria for p53germline mutation screening. J. Med. Genet. 2001, 38, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.-M.; Kanetsky, A.P.; Amirimani, B.; Colligon, A.T.; Athanasiadis, G.; Shih, A.H.; Gerrero, M.R.; Calzone, K.; Rebbeck, T.R.; Weber, B.L. Germline TP53 mutations in breast cancer families with multiple primary cancers: Is TP53 a modifier of BRCA1? J. Med. Genet. 2003, 40, e34. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical Characteristics of Families With p53 Germline Mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Frebourg, T.; Genturis, T.E.R.N.; Lagercrantz, S.B.; Oliveira, C.; Magenheim, R.; Evans, D.G. Guidelines for the Li–Fraumeni and heritable TP53-related cancer syndromes. Eur. J. Hum. Genet. 2020, 28, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.; Shore, A.; Wasserman, J.; Stephens, D.; Kim, R.H.; Druker, H.; Gallinger, B.; Naumer, A.; Kohlmann, W.; Novokmet, A.; et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol. 2016, 17, 1295–1305. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Petros, A.M.; Gunasekera, A.; Xu, N.; Olejniczak, E.T.; Fesik, S.W. Defining the p53 DNA-binding domain/Bcl-xL-binding interface using NMR. FEBS Lett. 2004, 559, 171–174. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef]
- Wasserman, J.D.; Novokmet, A.; Eichler-Jonsson, C.; Ribeiro, R.C.; Rodriguez-Galindo, C.; Zambetti, G.P.; Malkin, D. Prevalence and Functional Consequence of TP53 Mutations in Pediatric Adrenocortical Carcinoma: A Children’s Oncology Group Study. J. Clin. Oncol. 2015, 33, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.-M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome from TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- Amadou, A.; Achatz, M.I.; Hainaut, P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: Temporal phases of Li–Fraumeni syndrome. Curr. Opin. Oncol. 2018, 30, 23–29. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.; Pavletich, N. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Jurneczko, E.; Cruickshank, F.L.; Porrini, M.; Clarke, D.J.; Campuzano, I.D.G.; Morris, M.; Nikolova, P.V.; Barran, P.E. Probing the Conformational Diversity of Cancer-Associated Mutations in p53 with Ion-Mobility Mass Spectrometry. Angew. Chem. Int. Ed. 2013, 52, 4370–4374. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Olivier, M.; Goldgar, E.D.; Sodha, N.; Ohgaki, H.; Kleihues, P.; Hainaut, P.; Eeles, A.R. Li-Fraumeni and related syndromes: Correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003, 63, 6643–6650. [Google Scholar]
- Li, F.P.; Fraumeni, J.F.; Mulvihill, J.J.; Blattner, A.W.; Dreyfus, M.G.; Tucker, A.M.; Miller, R.W. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988, 48, 5358–5362. [Google Scholar]
- Fortuno, C.; James, P.A.; Spurdle, A.B. Current review ofTP53pathogenic germline variants in breast cancer patients outside Li-Fraumeni syndrome. Hum. Mutat. 2018, 39, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Kharaziha, P.; Ceder, S.; Axell, O.; Krall, M.; Fotouhi, O.; Böhm, S.; Lain, S.; Borg, Å.; Larsson, C.; Wiman, K.G.; et al. Functional characterization of novel germline TP53 variants in Swedish families. Clin. Genet. 2019, 96, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Emamzadah, S.; Tropia, L.; Halazonetis, T.D. Crystal Structure of a Multidomain Human p53 Tetramer Bound to the Natural CDKN1A (p21) p53-Response Element. Mol. Cancer Res. 2011, 9, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, T.J.; Emamzadah, S.; Costantino, L.; Petkova, I.D.; Stavridi, E.S.; Saven, J.G.; Vauthey, E.; Halazonetis, T.D. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 2011, 30, 2167–2176. [Google Scholar] [CrossRef] [Green Version]
- Script Library. PyMOLWiki. Available online: https://pymolwiki.org/index.php/Category:Script_Library (accessed on 29 April 2019).
- Vertrees, J. FindSurfaceResidues—PyMOLWiki. Available online: https://pymolwiki.org/index.php/FindSurfaceResidues (accessed on 26 April 2019).
- Vertrees, J. InterfaceResidues—PyMOLWiki. Available online: https://pymolwiki.org/index.php/InterfaceResidues (accessed on 30 April 2019).
- Torrance, G. Ss–PyMOLWiki. Available online: https://pymolwiki.org/index.php/Ss (accessed on 30 April 2019).
- Konrat, R. The protein meta-structure: A novel concept for chemical and molecular biology. Cell. Mol. Life Sci. 2009, 66, 3625–3639. [Google Scholar] [CrossRef]
- Mayer, C.; Slater, L.; Erat, M.C.; Konrat, R.; Vakonakis, I. Structural Analysis of the Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1) Intracellular Domain Reveals a Conserved Interaction Epitope. J. Biol. Chem. 2012, 287, 7182–7189. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.; Martin, A.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2011, 28, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-Dependent Prediction of Protein Disorder as a Function of Redox State and Protein Binding. Nucleic Acids Res. 2018, 46, W329–W337. Available online: https://academic.oup.com/nar/article/46/W1/W329/5026265 (accessed on 24 September 2020). [CrossRef] [PubMed]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. The DynaMine webserver: Predicting protein dynamics from sequence. Nucleic Acids Res. 2014, 42, W264–W270. [Google Scholar] [CrossRef] [Green Version]
- Vickers, A.J.; Cronin, A.M.; Elkin, E.B.; Gonen, M. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC Med. Inform. Decis. Mak. 2008, 8, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences: Fig. 1. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargallo, P.; Yáñez, Y.; Segura, V.; Juan, A.; Torres, B.; Balaguer, J.; Oltra, S.; Castel, V.; Cañete, A. Li–Fraumeni syndrome heterogeneity. Clin. Transl. Oncol. 2020, 22, 978–988. [Google Scholar] [CrossRef]
- Khoury, M.P.; Bourdon, J.-C. p53 Isoforms: An Intracellular Microprocessor? Genes Cancer 2011, 2, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, S.; De Miranda, N.; Ruano, D.; Barge-Schaapveld, D.; Hes, F.; Tops, C.; Joruiz, S.; Diot, A.; Bourdon, J.; Van Wezel, T. PO-059 Cancer-predisposing variants in alternatively spliced TP53 exons. ESMO Open 2018, 3, A249–A250. [Google Scholar] [CrossRef] [Green Version]
- Eiholzer, R.A.; Mehta, S.; Kazantseva, M.; Drummond, C.J.; McKinney, C.; Young, K.; Slater, D.; Morten, B.C.; Avery-Kiejda, K.A.; Lasham, A.; et al. Intronic TP53 Polymorphisms Are Associated with Increased Δ133TP53 Transcript, Immune Infiltration and Cancer Risk. Cancers 2020, 12, 2472. [Google Scholar] [CrossRef]
- Fortuno, C.; Cipponi, A.; Ballinger, M.L.; Tavtigian, S.V.; Olivier, M.; Ruparel, V.; Haupt, Y.; Haupt, S.; International Sarcoma Kindred Study; Tucker, K.; et al. A quantitative model to predict pathogenicity of missense variants in the TP53 gene. Hum. Mutat. 2019, 40, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Nichols, K.E.; Malkin, D. Genotype Versus Phenotype: The Yin and Yang of Germline TP53 Mutations in Li-Fraumeni Syndrome. J. Clin. Oncol. 2015, 33, 2331–2333. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Characteristics | Variables | Predictor Algorithm [Ref] |
|---|---|---|
| Tertiary structure propensity | ||
| buried/surface | Bur | Pymol-findSurfaceResidues script [28] |
| Intrinsic protein disorder propensity | ||
| disorder (trained on Disprot DB) | disprot_dif | Espritz [35] |
| disorder (trained on NMR structures) | nmr_dif | Espritz [35] |
| disorder (trained on X-ray structures) | xray_dif | Espritz [35] |
| disorder (longer regions) | iupl_dif | IUPred2A [36] |
| disorder (short regions) | iups_dif | IUPred2A [36] |
| Predicted protein backbone flexibility | ||
| protein backbone dynamics | dyn_dif | Dynamine [37] |
| Secondary structure propensity | ||
| alpha-helix/beta-sheet | Sec_dif | Meta-structure [33,34] |
| Compactness propensity | ||
| protein compactness | comp_dif | Meta-structure [33,34] |
| protein globularity | iupstr_dif | IUPred2A [36] |
| Protein interaction propensity | ||
| protein protein interaction | PPI6_dif | Meta-structure-PPI [33,34] |
| protein protein interaction | anc_dif | IUPred2A [36] |
| Intercept and Variable | β | OR (95% CI) | p-Value |
|---|---|---|---|
| Four variables model (mod_4v) | |||
| Intercept | 1.282 | 0.017 * | |
| Bur | |||
| Surface vs. Buried | −2.465 | 0.09 (0.02–0.44) | 0.003 * |
| unknown vs. Buried | −9.897 | 5.03 × 10−5(4.31 × 10−27–5.87× 1017) | 0.703 |
| comp_dif | 0.023 | 3.88 (1.30–11.59) | 0.015 * |
| PPI6_dif | 0.001 | 1.29 (0.99–1.67) | 0.059 |
| nmr_dif | 10.461 | 2.20 (0.79–6.11) | 0.132 |
| Three variables model (mod_3v) | |||
| Intercept | 1.094 | 0.030 * | |
| Bur | |||
| Surface vs. Buried | −2.229 | 0.11 (0.02–0.50) | 0.005 * |
| unknown vs. Buried | −9.03 | 0.00012 (1.18 × 10−26–1.22 × 1018) | 0.727 |
| comp_dif | 0.015 | 2.40 (1.08–5.30) | 0.031 * |
| PPI6_dif | 0.0006 | 1.15 (0.95–1.39) | 0.165 |
| Two variables models (mod_2v) | |||
| Intercept | 0.914 | 0.048 * | |
| Bur | |||
| Surface vs. Buried | −1.886 | 0.15 (0.04–0.62) | 0.009 * |
| unknown vs. Buried | −8.804 | 0.0002 (1.48 × 10−26–1.53 × 1018) | 0.733 |
| comp_dif | 0.014 | 2.30 (1.04–5.09) | 0.039 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Axell, O.; van Leeuwen, T.; Konrat, R.; Kharaziha, P.; Larsson, C.; Wright, A.P.H.; Bajalica-Lagercrantz, S. Association between Predicted Effects of TP53 Missense Variants on Protein Conformation and Their Phenotypic Presentation as Li-Fraumeni Syndrome or Hereditary Breast Cancer. Int. J. Mol. Sci. 2021, 22, 6345. https://doi.org/10.3390/ijms22126345
Liu Y, Axell O, van Leeuwen T, Konrat R, Kharaziha P, Larsson C, Wright APH, Bajalica-Lagercrantz S. Association between Predicted Effects of TP53 Missense Variants on Protein Conformation and Their Phenotypic Presentation as Li-Fraumeni Syndrome or Hereditary Breast Cancer. International Journal of Molecular Sciences. 2021; 22(12):6345. https://doi.org/10.3390/ijms22126345
Chicago/Turabian StyleLiu, Yaxuan, Olga Axell, Tom van Leeuwen, Robert Konrat, Pedram Kharaziha, Catharina Larsson, Anthony P. H. Wright, and Svetlana Bajalica-Lagercrantz. 2021. "Association between Predicted Effects of TP53 Missense Variants on Protein Conformation and Their Phenotypic Presentation as Li-Fraumeni Syndrome or Hereditary Breast Cancer" International Journal of Molecular Sciences 22, no. 12: 6345. https://doi.org/10.3390/ijms22126345