
HIF-1α Dependent Upregulation of ZIP8, ZIP14, and TRPA1 Modify Intracellular Zn2+ Accumulation in Inflammatory Synoviocytes


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
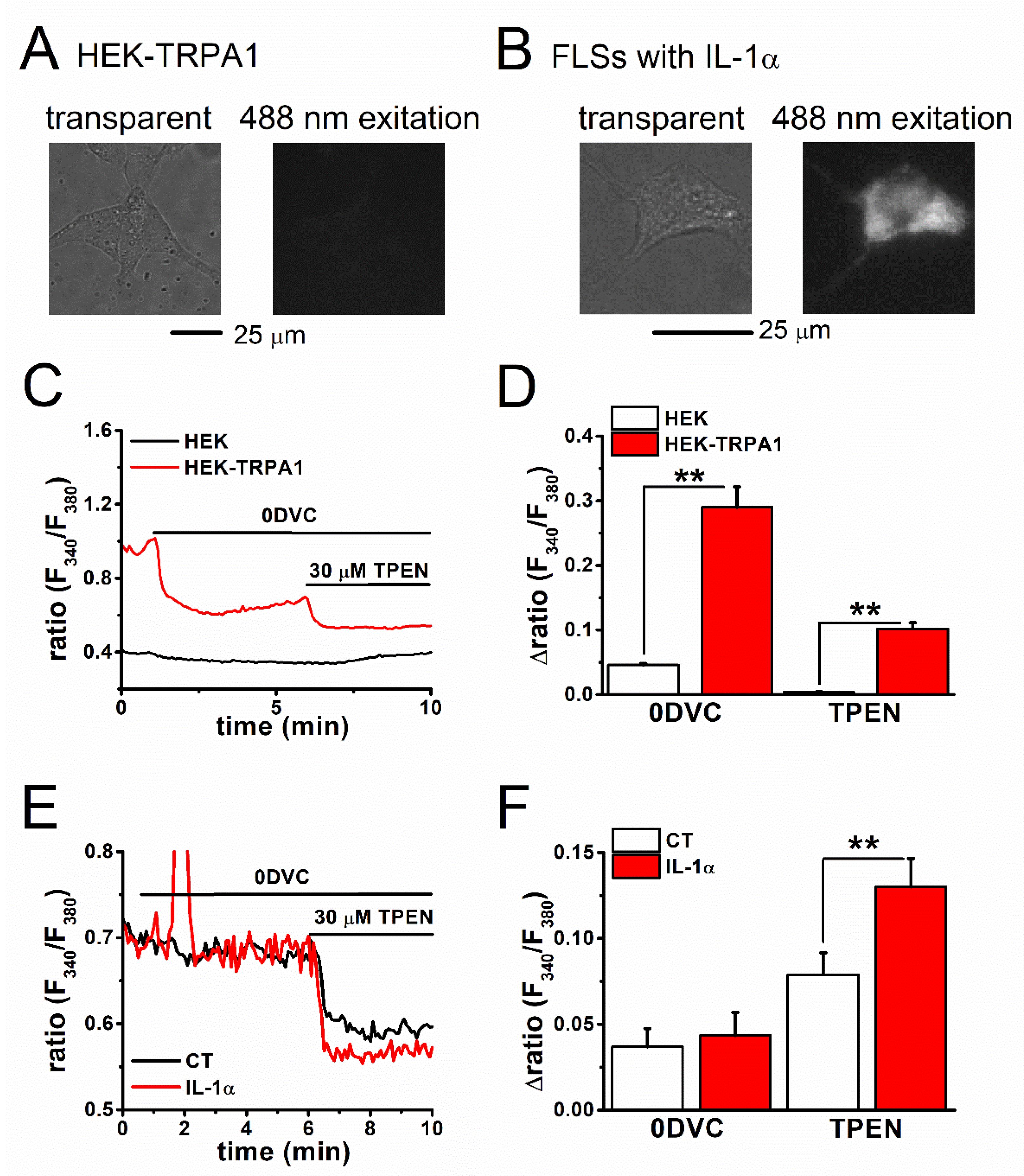
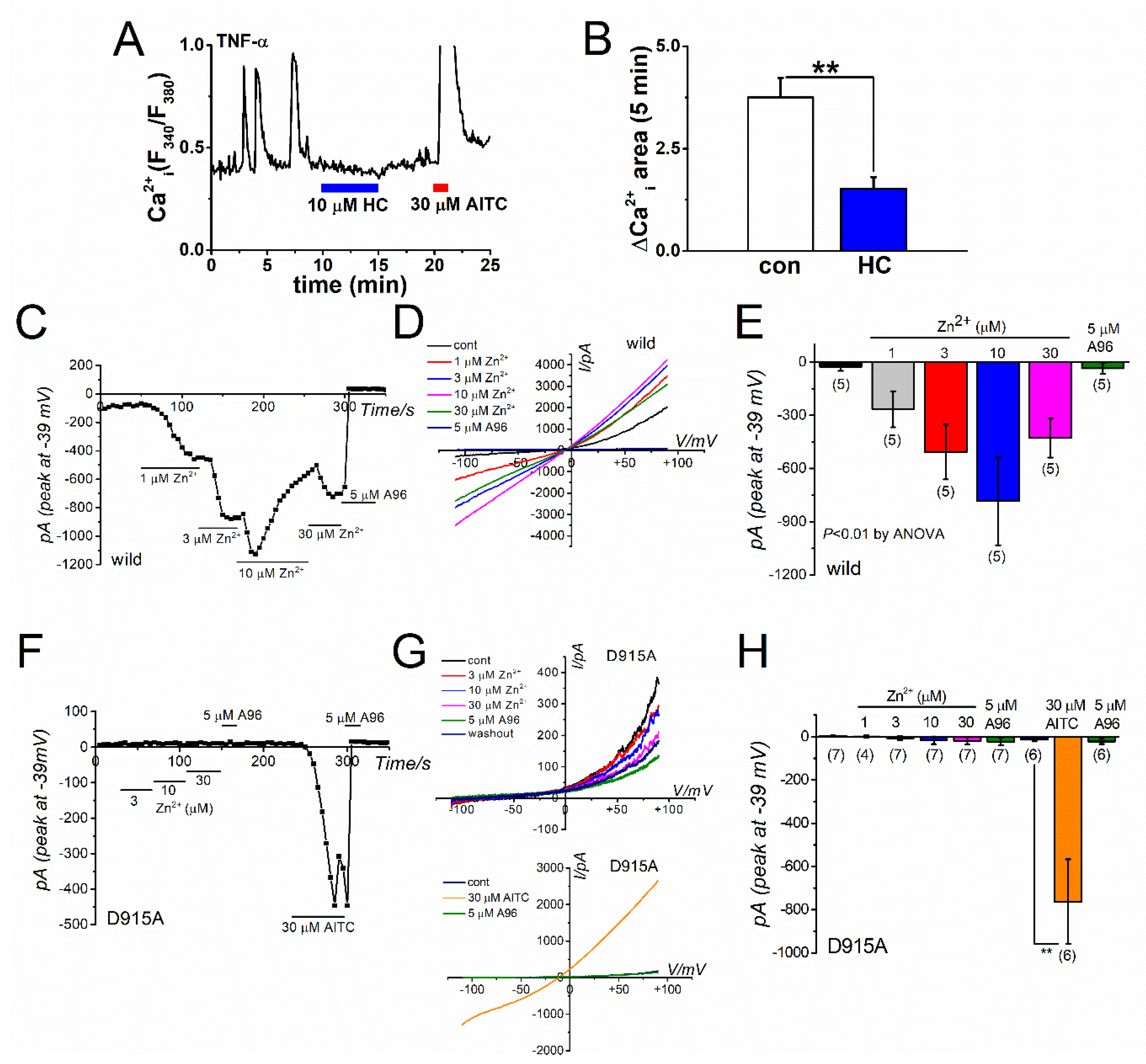
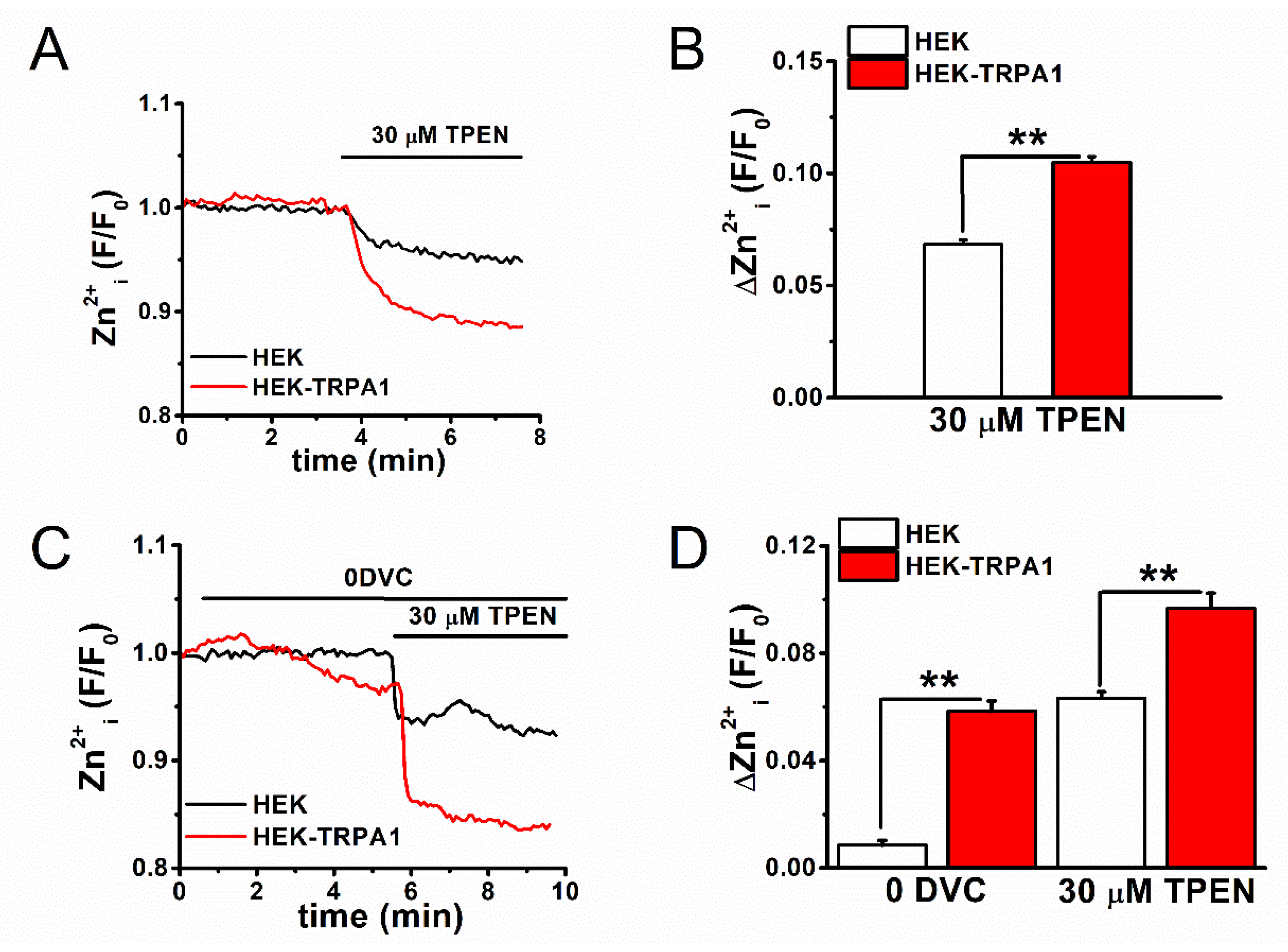
2.1. Zn2+-Dependent TRPA1 Activation and [Zn2+]i Components in TRPA1-Expressing Cells
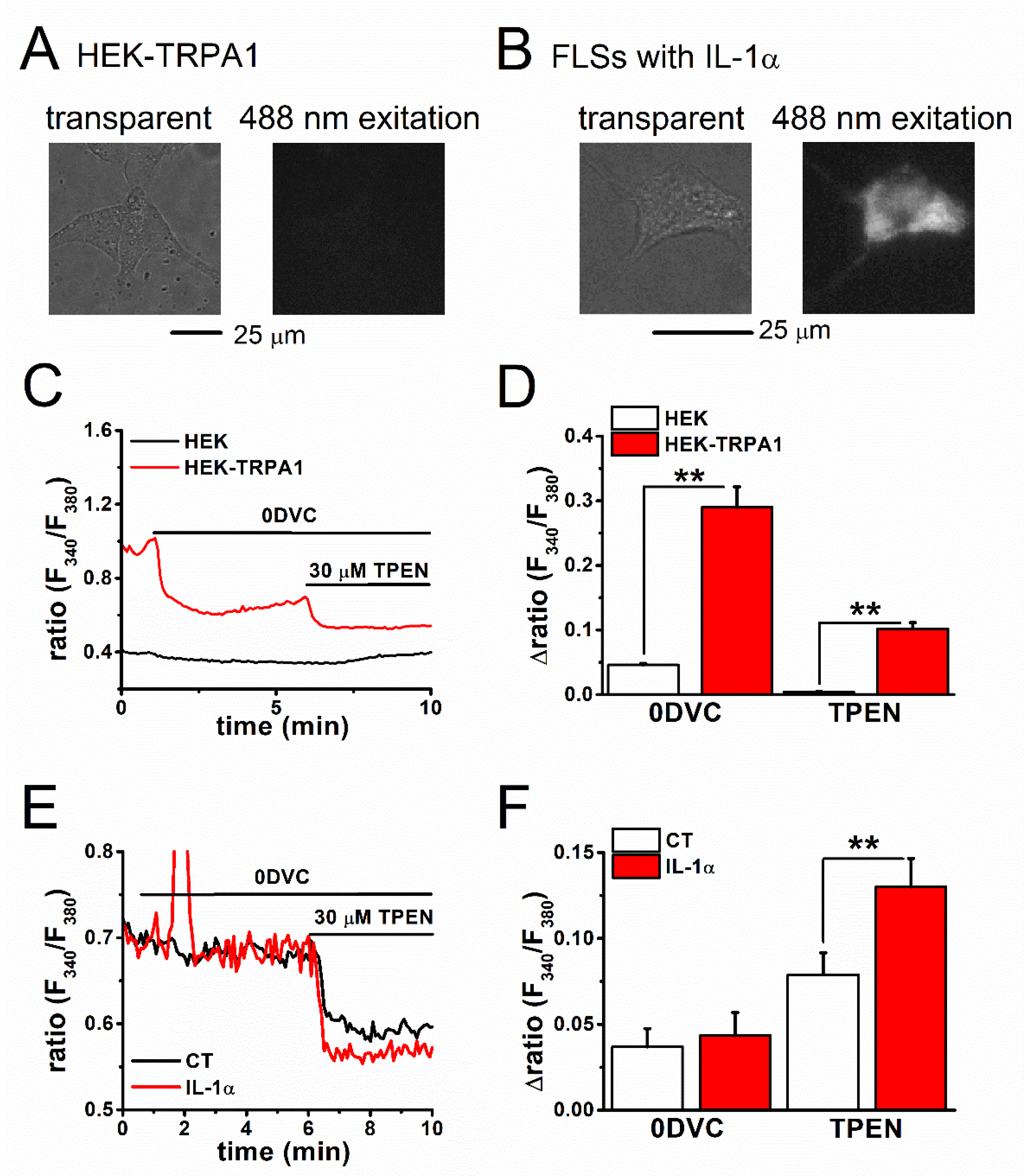
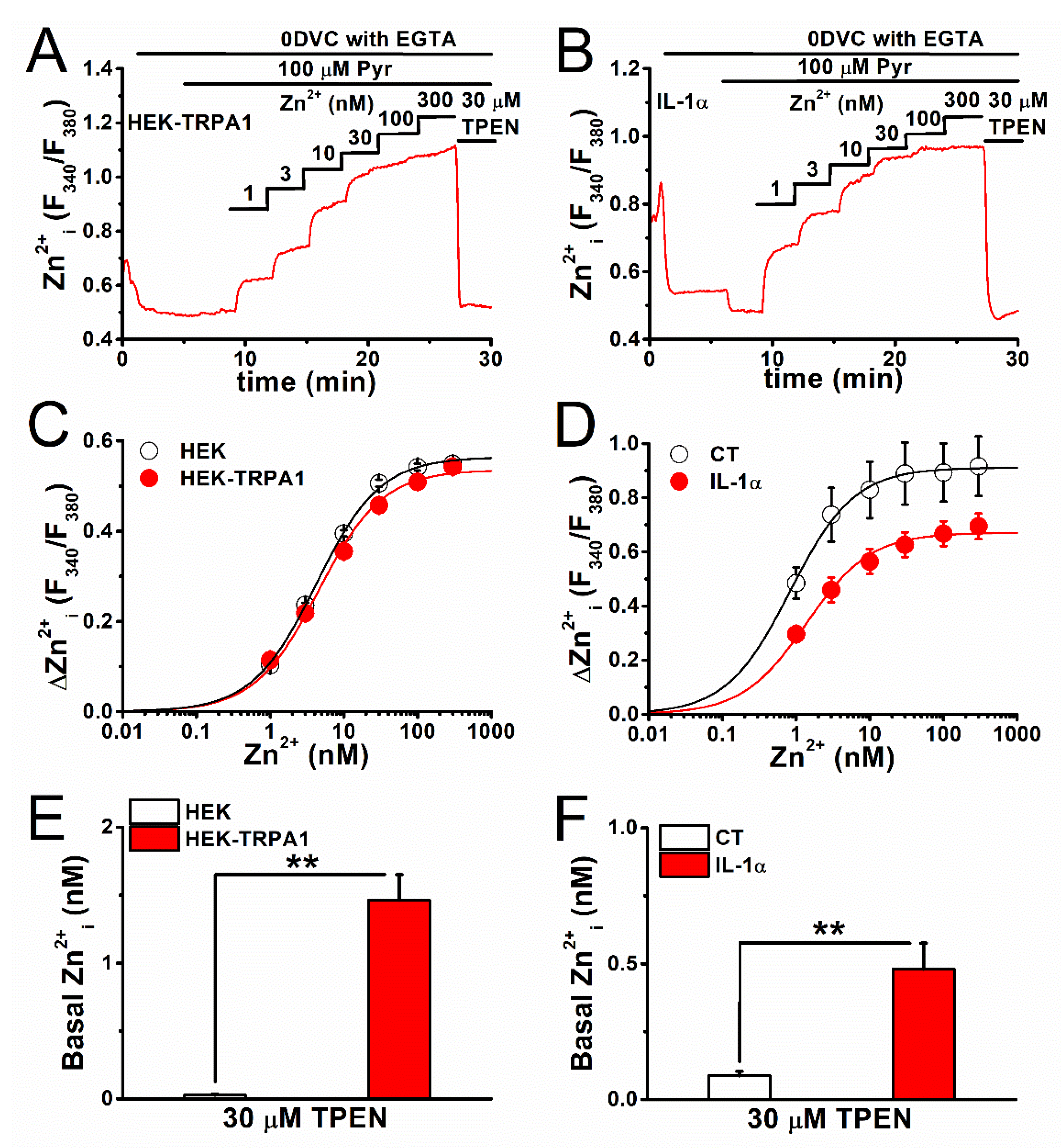
2.2. Estimation of Basal [Zn2+]i Concentration in TRPA1-Expressing Cells
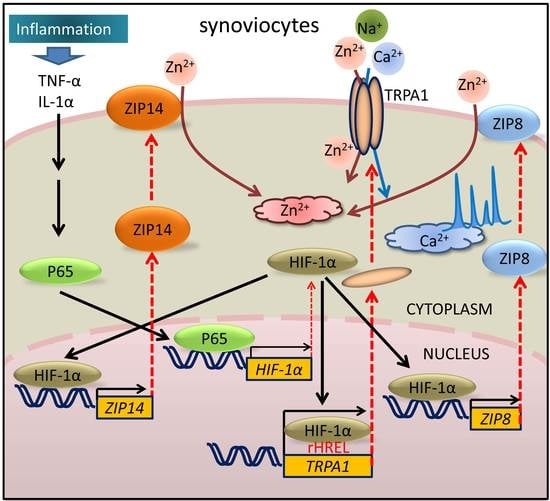
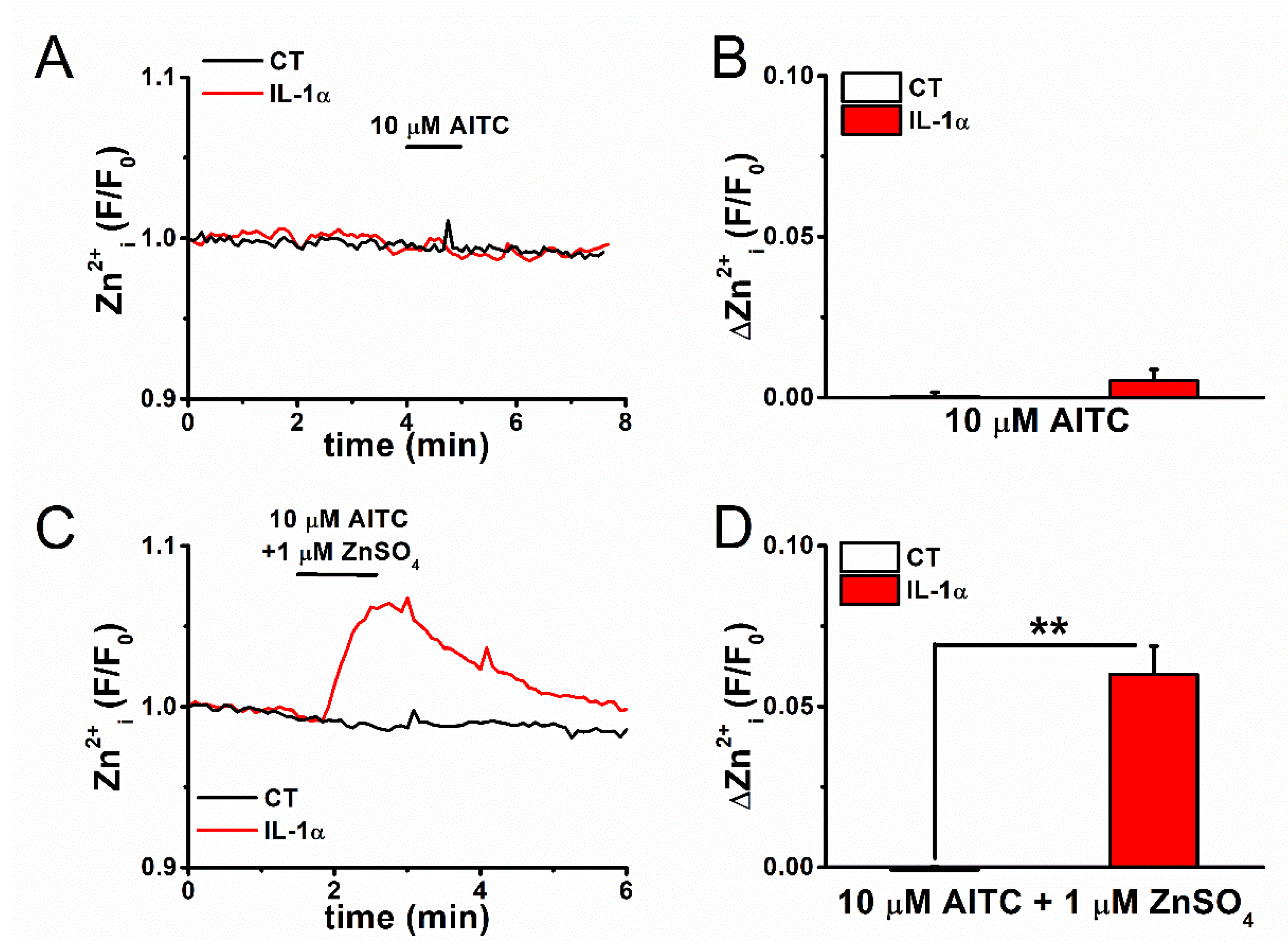
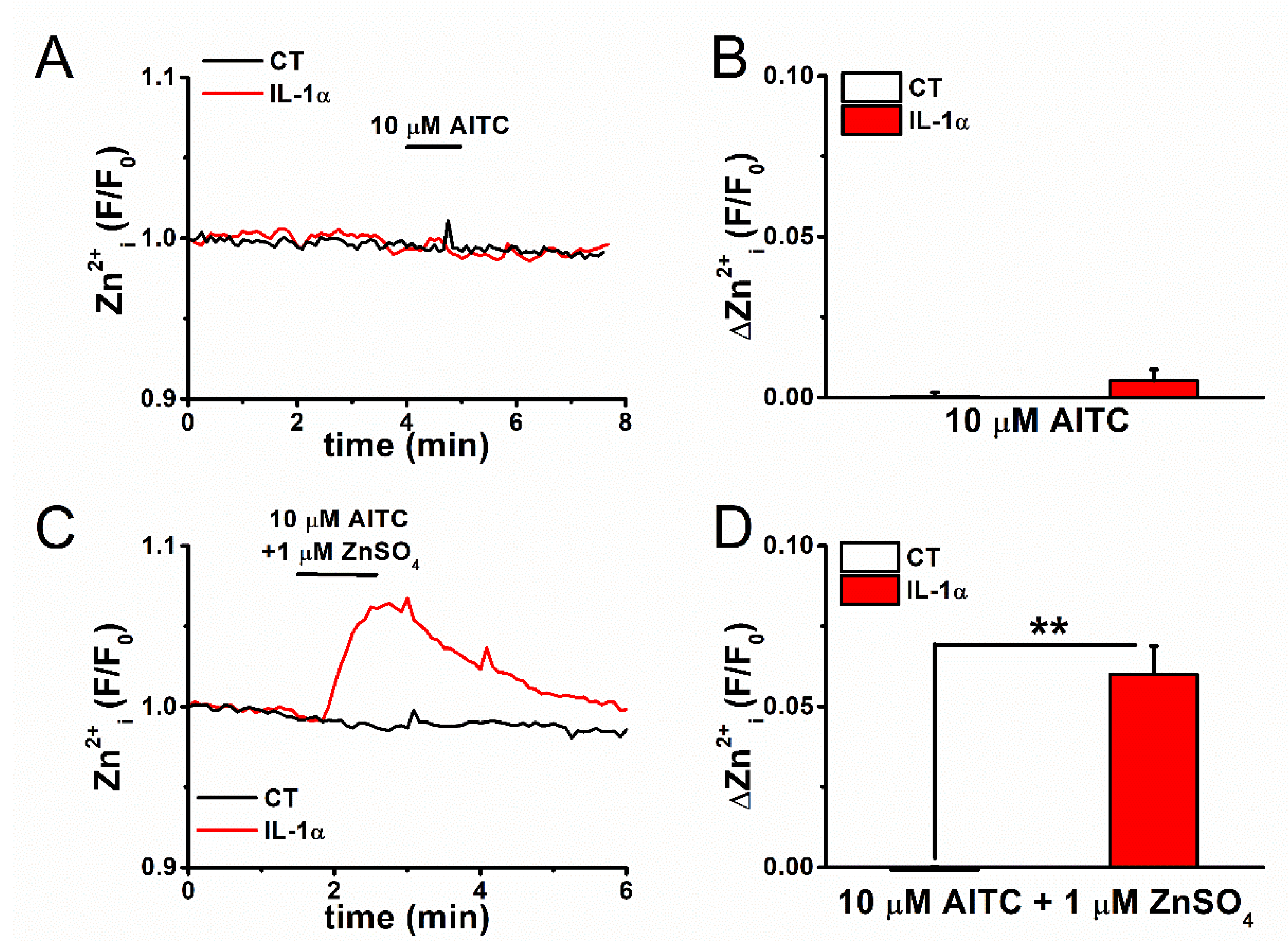
2.3. Mechanism of Elevation of Basal [Zn2+]i Concentration in Inflammatory FLSs
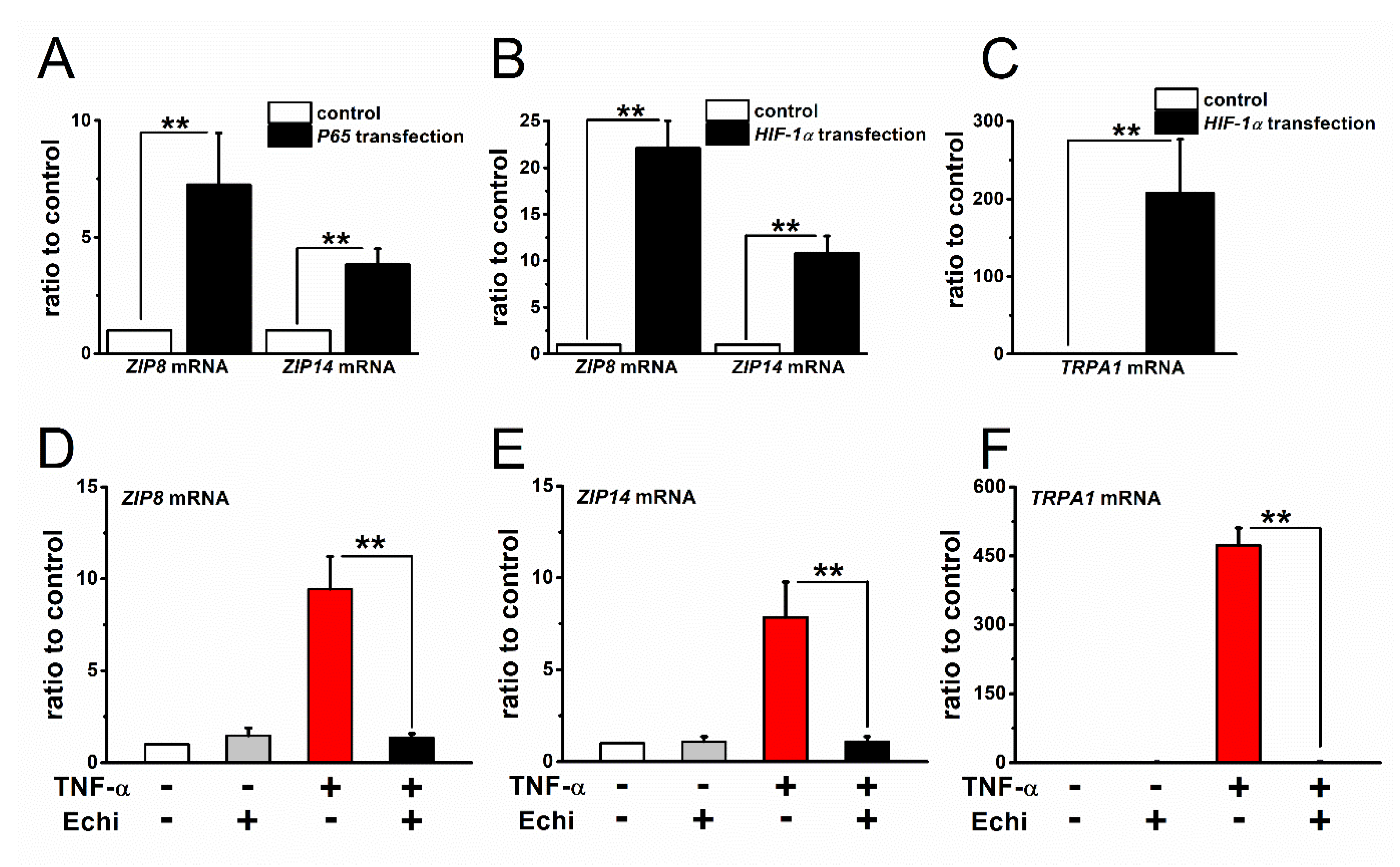
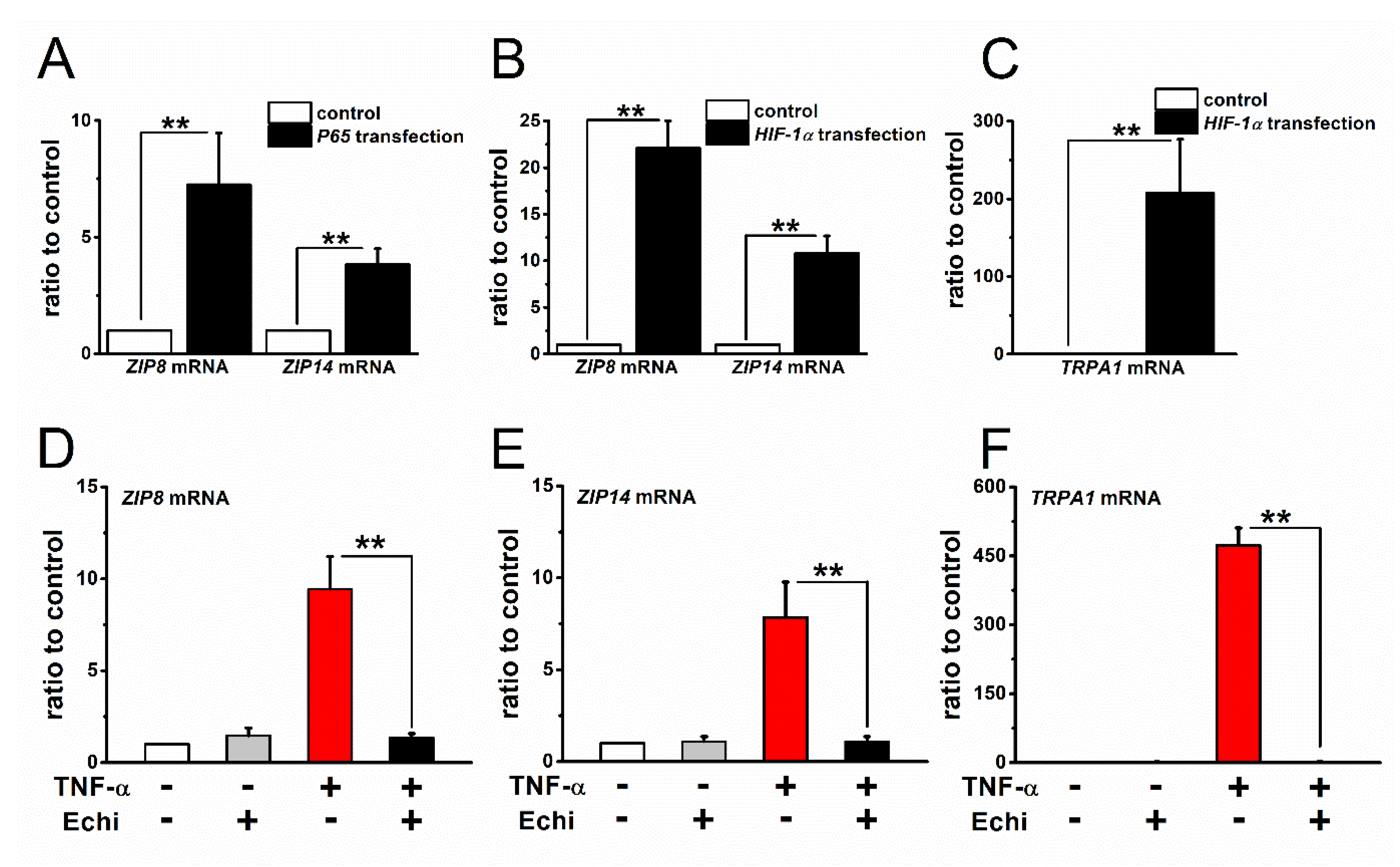
2.4. Expression and Inflammation-Mediated Transcriptional Regulation of Zn2+ Transporters in FLSs
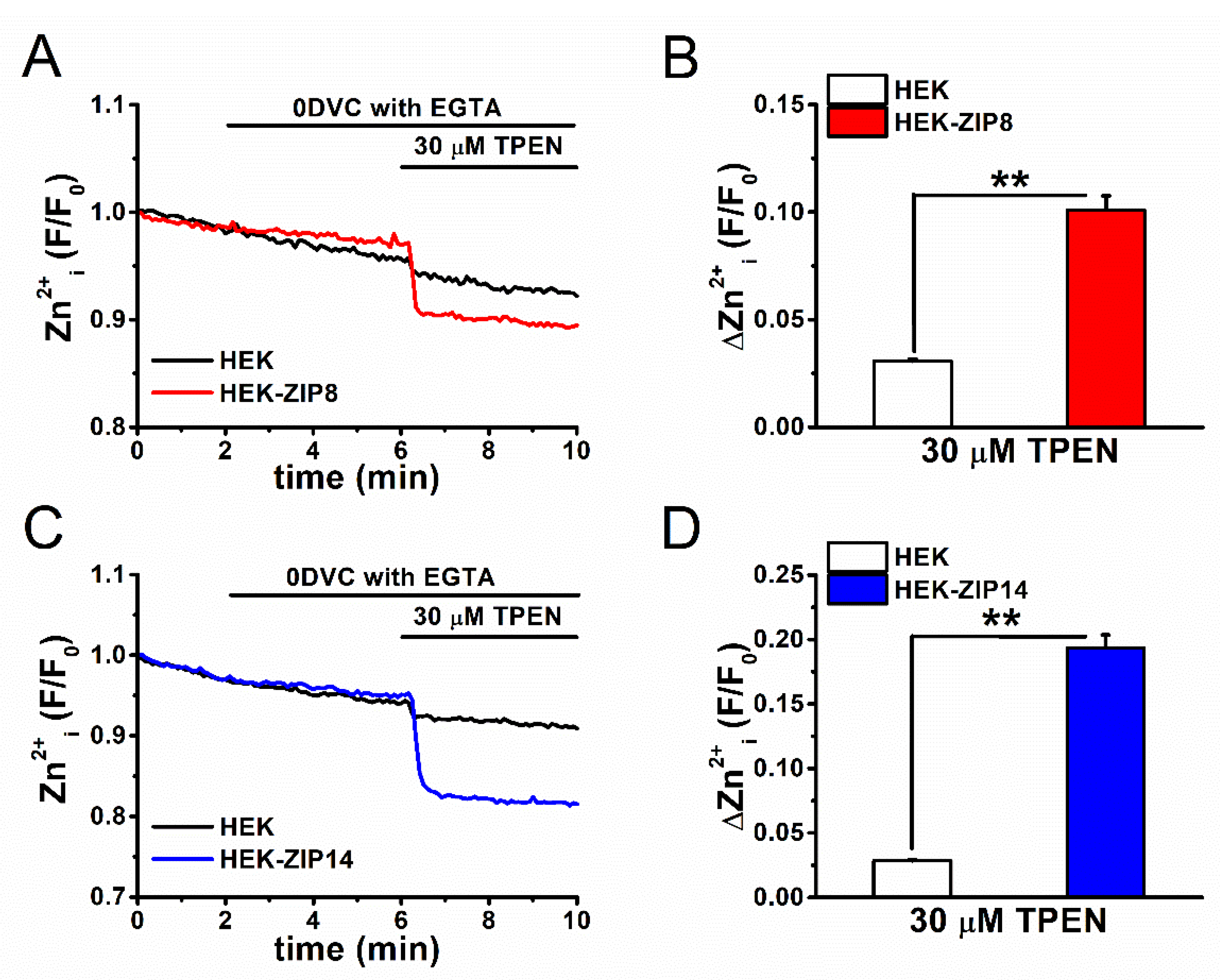
2.5. Basal [Zn2+]i Components in ZIP8- and ZIP14-Expressing Cells
3. Discussion
4. Experimental Procedures
4.1. Reagents
4.2. Cell Culture
4.3. Quantitative PCR
4.4. Recombinant Expression of Wild and Mutant TRPA1, P65, HIF-1α, ZIP8, and ZIP14 in HEK Cells and FLSs
4.5. Patch Clamp Electrophysiology
4.6. Measurement of Ca2+ Fluorescence Ratio and Auto-Fluorescence Images of Cells
4.7. Fluorescence-Based Measurement of Zn2+
4.8. Estimation of Zn2+ Concentration in SBS
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| [Zn2+]i | intracellular free zinc |
| HIF-1α | hypoxia-induced factor 1α |
| TRPA1 | transient receptor potential ankyrin 1 |
| FLSs | human fibroblast-like synoviocytes |
| TNF-α | tumor necrosis factor-α |
| IL-1α | interleukin-1α |
| NF-κB | nuclear factor-κB |
| [Zn2+]e | extracellular free Zn2+ |
References
- Rink, L.; Haase, H. Zinc homeostasis and immunity. Trends Immunol. 2007, 28, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.H.; Ruffin, R.E.; Murgia, C.; Li, L.; Krilis, S.A.; Zalewski, P.D. Labile zinc and zinc transporter ZnT4 in mast cell granules: Role in regulation of caspase activation and NF-kappaB translocation. J. Immunol. 2004, 172, 7750–7760. [Google Scholar] [CrossRef] [Green Version]
- Kabu, K.; Yamasaki, S.; Kamimura, D.; Ito, Y.; Hasegawa, A.; Sato, E.; Kitamura, H.; Nishida, K.; Hirano, T. Zinc is required for Fc epsilon RI-mediated mast cell activation. J. Immunol. 2006, 177, 1296–1305. [Google Scholar] [CrossRef] [Green Version]
- Haase, H.; Ober-Blöbaum, J.L.; Engelhardt, G.; Hebel, S.; Heit, A.; Heine, H.; Rink, L. Zinc Signals Are Essential for Lipopolysaccharide-Induced Signal Transduction in Monocytes. J. Immunol. 2008, 181, 6491–6502. [Google Scholar] [CrossRef] [Green Version]
- Besecker, B.; Bao, S.; Bohacova, B.; Papp, A.; Sadee, W.; Knoell, D.L. The human zinc transporter SLC39A8 (Zip8) is critical in zinc-mediated cytoprotection in lung epithelia. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L1127–L1136. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Koh, J.; Choi, D. Brief exposure to zinc is toxic to cortical neurons. Neurosci. Lett. 1986, 71, 351–355. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Bush, A.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef]
- Liu, M.J.; Bao, S.; Galvez-Peralta, M.; Pyle, C.J.; Rudawsky, A.C.; Pavlovicz, R.E.; Killilea, D.W.; Li, C.; Nebert, D.W.; Wewers, M.D.; et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-kappaB. Cell Rep. 2013, 3, 386–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyle, C.J.; Akhter, S.; Bao, S.; Dodd, C.E.; Schlesinger, L.S.; Knoell, D.L. Zinc Modulates Endotoxin-Induced Human Macrophage Inflammation through ZIP8 Induction and C/EBPbeta Inhibition. PLoS ONE 2017, 12, e0169531. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-H.; Aydemir, T.B.; Kim, J.; Cousins, R.J. Hepatic ZIP14-mediated zinc transport is required for adaptation to endoplasmic reticulum stress. Proc. Natl. Acad. Sci. USA 2017, 114, E5805–E5814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellomo, E.A.; Meur, G.; Rutter, G.A. Glucose regulates free cytosolic Zn2+ concentration, Slc39 (ZiP), and metallothionein gene expression in primary pancreatic islet beta-cells. J. Biol. Chem. 2011, 286, 25778–25789. [Google Scholar] [CrossRef] [Green Version]
- Liuzzi, J.P.; Lichten, L.A.; Rivera, S.; Blanchard, R.K.; Aydemir, T.B.; Knutson, M.D.; Ganz, T.; Cousins, R.J. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc. Natl. Acad. Sci. USA 2005, 102, 6843–6848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordt, S.-E.; Bautista, D.M.; Chuang, H.-H.; McKemy, D.D.; Zygmunt, P.M.; Högestätt, E.D.; Meng, I.D.; Julius, D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 2004, 427, 260–265. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, L.J.; Dubin, A.E.; Evans, M.J.; Marr, F.; Schultz, P.G.; Cravatt, B.F.; Patapoutian, A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Story, G.M.; Peier, A.M.; Reeve, A.J.; Eid, S.R.; Mosbacher, J.; Hricik, T.R.; Earley, T.J.; Hergarden, A.C.; Andersson, D.A.; Hwang, S.W.; et al. ANKTM1, a TRP-like Channel Expressed in Nociceptive Neurons, Is Activated by Cold Temperatures. Cell 2003, 112, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Hatano, N.; Muraki, Y.; Itoh, Y.; Kimura, S.; Hayashi, H.; Onozaki, K.; Ohi, Y.; Haji, A.; Muraki, K. The NADPH oxidase inhibitor diphenyleneiodonium activates the human TRPA1 nociceptor. Am. J. Physiol. Physiol. 2014, 307, C384–C394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraki, K.; Sekine, T.; Ando, Y.; Suzuki, H.; Hatano, N.; Hirata, T.; Muraki, Y. An environmental pollutant, 9,10-phenanthrenequinone, activates human TRPA1 via critical cysteines 621 and 665. Pharmacol. Res. Perspect. 2017, 5, e00342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Camino, D.; Murphy, S.; Heiry, M.; Barrett, L.B.; Earley, T.J.; Cook, C.A.; Petrus, M.J.; Zhao, M.; D’Amours, M.; Deering, N.; et al. TRPA1 Contributes to Cold Hypersensitivity. J. Neurosci. 2010, 30, 15165–15174. [Google Scholar] [CrossRef]
- Fernandes, E.S.; Russell, F.A.; Spina, D.; McDougall, J.J.; Graepel, R.; Gentry, C.; Staniland, A.A.; Mountford, D.M.; Keeble, J.E.; Malcangio, M.; et al. A distinct role for transient receptor potential ankyrin 1, in addition to transient receptor potential vanilloid 1, in tumor necrosis factor alpha-induced inflammatory hyperalgesia and Freund’s complete adjuvant-induced monarthritis. Arthritis Rheum. 2011, 63, 819–829. [Google Scholar] [CrossRef]
- Karashima, Y.; Talavera, K.; Everaerts, W.; Janssens, A.; Kwan, K.Y.; Vennekens, R.; Nilius, B.; Voets, T. TRPA1 acts as a cold sensor in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 1273–1278. [Google Scholar] [CrossRef] [Green Version]
- Kwan, K.Y.; Allchorne, A.J.; Vollrath, M.A.; Christensen, A.P.; Zhang, D.-S.; Woolf, C.J.; Corey, D.P. TRPA1 Contributes to Cold, Mechanical, and Chemical Nociception but Is Not Essential for Hair-Cell Transduction. Neuron 2006, 50, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Obata, K.; Katsura, H.; Mizushima, T.; Yamanaka, H.; Kobayashi, K.; Dai, Y.; Fukuoka, T.; Tokunaga, A.; Tominaga, M.; Noguchi, K. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J. Clin. Investig. 2005, 115, 2393–2401. [Google Scholar] [CrossRef] [Green Version]
- Bautista, D.M.; Jordt, S.-E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 Mediates the Inflammatory Actions of Environmental Irritants and Proalgesic Agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [Green Version]
- Mäki-Opas, I.; Hämäläinen, M.; Moilanen, L.J.; Haavikko, R.; Ahonen, T.J.; Alakurtti, S.; Moreira, V.M.; Muraki, K.; Yli-Kauhaluoma, J.; Moilanen, E. Pyrazine-Fused Triterpenoids Block the TRPA1 Ion Channel in Vitro and Inhibit TRPA1-Mediated Acute Inflammation in Vivo. ACS Chem. Neurosci. 2019, 10, 2848–2857. [Google Scholar] [CrossRef]
- Moilanen, L.J.; Hamalainen, M.; Lehtimaki, L.; Nieminen, R.M.; Muraki, K.; Moilanen, E. Pinosylvin Inhibits TRPA1-Induced Calcium Influx In Vitro and TRPA1-Mediated Acute Paw Inflammation In Vivo. Bas. Clin. Pharmacol. Toxicol. 2016, 118, 238–242. [Google Scholar] [CrossRef]
- Hatano, N.; Itoh, Y.; Suzuki, H.; Muraki, Y.; Hayashi, H.; Onozaki, K.; Wood, I.C.; Beech, D.; Muraki, K. Hypoxia-inducible Factor-1α (HIF1α) Switches on Transient Receptor Potential Ankyrin Repeat 1 (TRPA1) Gene Expression via a Hypoxia Response Element-like Motif to Modulate Cytokine Release. J. Biol. Chem. 2012, 287, 31962–31972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, D.A.; Gentry, C.; Moss, S.; Bevan, S. Clioquinol and pyrithione activate TRPA1 by increasing intracellular Zn2+. Proc. Natl. Acad. Sci. USA 2009, 106, 8374–8379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinkenborg, J.L.; Nicolson, T.J.; Bellomo, E.; Koay, M.S.; Rutter, G.A.; Merkx, M. Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat. Methods 2009, 6, 737–740. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Bandell, M.; Petrus, M.J.; Zhu, M.X.; Patapoutian, A. Zinc activates damage-sensing TRPA1 ion channels. Nat. Chem. Biol. 2009, 5, 183–190. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Chang, R.B.; Waters, H.N.; McKemy, D.D.; Liman, E.R. The Nociceptor Ion Channel TRPA1 Is Potentiated and Inactivated by Permeating Calcium Ions. J. Biol. Chem. 2008, 283, 32691–32703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurborg, S.; Yurgionas, B.; Jira, J.A.; Caspani, O.; Heppenstall, P.A. Direct activation of the ion channel TRPA1 by Ca2+. Nat. Neurosci. 2007, 10, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Yazar, M.; Sarban, S.; Kocyigit, A.; Isikan, U.E. Synovial Fluid and Plasma Selenium, Copper, Zinc, and Iron Concentrations in Patients with Rheumatoid Arthritis and Osteoarthritis. Biol. Trace Element Res. 2005, 106, 123–132. [Google Scholar] [CrossRef]
- Gu, Q.; Lin, R.-L. Heavy metals zinc, cadmium, and copper stimulate pulmonary sensory neurons via direct activation of TRPA1. J. Appl. Physiol. 2010, 108, 891–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krężel, A.; Maret, W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. JBIC J. Biol. Inorg. Chem. 2006, 11, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Colvin, R.A.; Bush, A.; Volitakis, I.; Fontaine, C.P.; Thomas, D.; Kikuchi, K.; Holmes, W.R. Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am. J. Physiol. Physiol. 2008, 294, C726–C742. [Google Scholar] [CrossRef]
- Lichten, L.A.; Liuzzi, J.P.; Cousins, R.J. Interleukin-1beta contributes via nitric oxide to the upregulation and functional activity of the zinc transporter Zip14 (Slc39a14) in murine hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G860–G867. [Google Scholar] [CrossRef] [Green Version]
- Frede, S.; Stockmann, C.; Freitag, P.; Fandrey, J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J. 2006, 396, 517–527. [Google Scholar] [CrossRef]
- Muz, B.; Khan, M.N.; Kiriakidis, S.; Paleolog, E.M. The role of hypoxia and HIF-dependent signalling events in rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, 201. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Schmid, T.; Brune, B. Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. Mol. Biol. Cell 2003, 14, 2216–2225. [Google Scholar] [CrossRef]
- Colvin, R.A.; Holmes, W.R.; Fontaine, C.P.; Maret, W. Cytosolic zinc buffering and muffling: Their role in intracellular zinc homeostasis. Metallomics 2010, 2, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Muraki, K.; Ohnishi, K.; Takezawa, A.; Suzuki, H.; Hatano, N.; Muraki, Y.; Hamzah, N.; Foster, R.; Waldmann, H.; Nussbaumer, P.; et al. Na+ entry through heteromeric TRPC4/C1 channels mediates (-)Englerin A-induced cytotoxicity in synovial sarcoma cells. Sci. Rep. 2017, 7, 16988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatano, N.; Matsubara, M.; Suzuki, H.; Muraki, Y.; Muraki, K. HIF-1α Dependent Upregulation of ZIP8, ZIP14, and TRPA1 Modify Intracellular Zn2+ Accumulation in Inflammatory Synoviocytes. Int. J. Mol. Sci. 2021, 22, 6349. https://doi.org/10.3390/ijms22126349
Hatano N, Matsubara M, Suzuki H, Muraki Y, Muraki K. HIF-1α Dependent Upregulation of ZIP8, ZIP14, and TRPA1 Modify Intracellular Zn2+ Accumulation in Inflammatory Synoviocytes. International Journal of Molecular Sciences. 2021; 22(12):6349. https://doi.org/10.3390/ijms22126349
Chicago/Turabian StyleHatano, Noriyuki, Masaki Matsubara, Hiroka Suzuki, Yukiko Muraki, and Katsuhiko Muraki. 2021. "HIF-1α Dependent Upregulation of ZIP8, ZIP14, and TRPA1 Modify Intracellular Zn2+ Accumulation in Inflammatory Synoviocytes" International Journal of Molecular Sciences 22, no. 12: 6349. https://doi.org/10.3390/ijms22126349
APA StyleHatano, N., Matsubara, M., Suzuki, H., Muraki, Y., & Muraki, K. (2021). HIF-1α Dependent Upregulation of ZIP8, ZIP14, and TRPA1 Modify Intracellular Zn2+ Accumulation in Inflammatory Synoviocytes. International Journal of Molecular Sciences, 22(12), 6349. https://doi.org/10.3390/ijms22126349