
The Serpin Superfamily and Their Role in the Regulation and Dysfunction of Serine Protease Activity in COPD and Other Chronic Lung Diseases

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Serine Proteases
3. The Serpin Superfamily
3.1. Classification
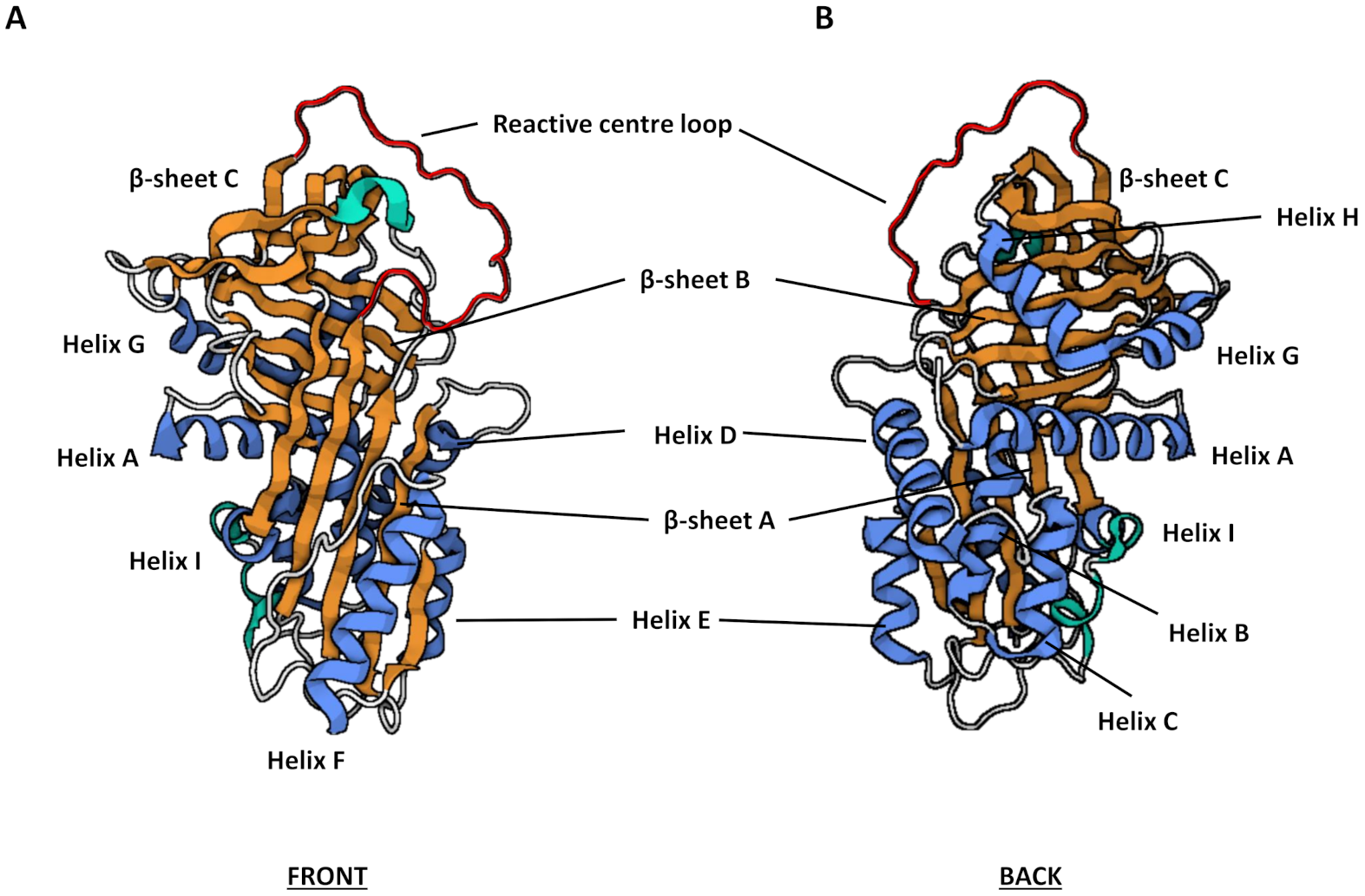
3.2. Structure and Function
3.3. Mechanism of Inhibition
3.4. Regulation of Function
3.5. Physiological Roles
3.6. Pathological Roles
4. Serpins Reported to Have Association with COPD or Other Chronic Airways Conditions
4.1. Clade A Serpins
4.1.1. SERPINA1
4.1.2. SERPINA3
4.1.3. SERPINA5
4.1.4. SERPINA7
4.1.5. SERPINA8
4.1.6. SERPINA12
4.2. Clade B Serpins
4.2.1. SERPINB1
4.2.2. SERPINB2
4.2.3. SERPINB3 and SERPINB4
4.2.4. SERPINB6
4.2.5. SERPINB9
4.2.6. SERPINB10
4.3. Other Serpins
4.3.1. SERPINE1
4.3.2. SERPINE2
4.3.3. SERPINF1
5. Therapeutic Use of Serpins
6. Animal Models of Serpin-Associated Human Disease
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. The Top 10 Causes of Death. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 3 May 2021).
- World Health Organization. Projections of Mortality and Causes of Death, 2016–2060. 2018. Available online: https://www.who.int/healthinfo/global_burden_disease/projections/en/ (accessed on 3 May 2021).
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.-W.; et al. Forecasting life expectancy, years of life lost, and all-Cause and cause-Specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef] [Green Version]
- Hoenderdos, K.; Condliffe, A. The Neutrophil in Chronic Obstructive Pulmonary Disease. Too Little, Too Late or Too Much, Too Soon? Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Paone, G.; Conti, V.; Vestri, A.; Leone, A.; Puglisi, G.; Benassi, F.; Brunetti, G.; Schmid, G.; Cammarella, I.; Terzano, C. Analysis of Sputum Markers in the Evaluation of Lung Inflammation and Functional Impairment in Symptomatic Smokers and COPD Patients. Dis. Mark. 2011, 31, 139493. [Google Scholar] [CrossRef]
- Cavarra, E.; Lucattelli, M.; Gambelli, F.; Bartalesi, B.; Fineschi, S.; Szarka, A.; Giannerini, F.; Martorana, P.A.; Lungarella, G. Human SLPI inactivation after cigarette smoke exposure in a new in vivo model of pulmonary oxidative stress. Am. J. Physiol. Cell. Mol. Physiol. 2001, 281, L412–L417. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Li, Z.; Janciauskiene, S.; Mahadeva, R. Oxidation of Z α1-antitrypsin by cigarette smoke induces polymerization: A novel mechanism of early-onset emphysema. Am. J. Respir. Cell Mol. Biol. 2011, 45, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either Methionine 351 or Methionine 358 in α1-Antitrypsin Causes Loss of Anti-neutrophil Elastase Activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Guay, C.; Laviolette, M.; Tremblay, G.M. Targeting Serine Proteases in Asthma. Curr. Top. Med. Chem. 2006, 6, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Menou, A.; Duitman, J.; Crestani, B. The impaired proteases and anti-proteases balance in Idiopathic Pulmonary Fibrosis. Matrix Biol. 2018, 68–69, 382–403. [Google Scholar] [CrossRef]
- Cochrane, C.G.; Spragg, R.G.; Revak, S.D.; Cohen, A.B.; McGuire, W.W. The presence of neutrophil elastase and evidence of oxidation activity in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1983, 127, 25–27. [Google Scholar]
- Lee, C.T.; Fein, A.M.; Lippmann, M.; Holtzman, H.; Kimbel, P.; Weinbaum, G. Elastolytic Activity in Pulmonary Lavage Fluid from Patients with Adult Respiratory-Distress Syndrome. N. Engl. J. Med. 1981, 304, 192–196. [Google Scholar] [CrossRef]
- Donnelly, S.; MacGregor, I.; Zamani, A.; Gordon, M.W.; Robertson, C.E.; Steedman, D.J.; Little, K.; Haslett, C. Plasma elastase levels and the development of the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 151, 1428–1433. [Google Scholar] [CrossRef]
- Puente, X.S.; Sánchez, L.M.; Overall, C.M.; López-Otín, C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544–558. [Google Scholar] [CrossRef]
- López-Otín, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef]
- Kappelhoff, R.; Puente, X.S.; Wilson, C.H.; Seth, A.; López-Otín, C.; Overall, C.M. Overview of transcriptomic analysis of all human proteases, non-proteolytic homologs and inhibitors: Organ, tissue and ovarian cancer cell line expression profiling of the human protease degradome by the CLIP-CHIP™ DNA microarray. Biochim. Biophys. Acta Molec. Cell Res. 2017, 1864, 2210–2219. [Google Scholar] [CrossRef]
- Page, M.J.; Di Cera, E. Serine peptidases: Classification, structure and function. Cell. Mol. Life Sci. 2008, 65, 1220–1236. [Google Scholar] [CrossRef]
- Di Cera, E. Serine proteases. IUBMB Life 2009, 61, 510–515. [Google Scholar] [CrossRef]
- Adkison, A.M.; Raptis, S.Z.; Kelley, D.G.; Pham, C.T.N. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J. Clin. Investig. 2002, 109, 363–371. [Google Scholar] [CrossRef]
- Khan, M.S.; Singh, P.; Azhar, A.; Naseem, A.; Rashid, Q.; Kabir, M.A.; Jairajpuri, M.A. Serpin Inhibition Mechanism: A Delicate Balance between Native Metastable State and Polymerization. J. Amino Acids 2011, 2011, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Farady, C.J.; Craik, C.S. Mechanisms of Macromolecular Protease Inhibitors. ChemBioChem 2010, 11, 2341–2346. [Google Scholar] [CrossRef] [Green Version]
- Silverman, G.A.; Bird, P.I.; Carrell, R.W.; Church, F.C.; Coughlin, P.B.; Gettins, P.G.W.; Irving, J.A.; Lomas, D.A.; Luke, C.J.; Moyer, R.W.; et al. The Serpins Are an Expanding Superfamily of Structurally Similar but Functionally Diverse Proteins: Evolution, Mechanism Of Inhibition, Novel Functions, and a Revised Nomenclature. J. Biol. Chem. 2001, 276, 33293–33296. [Google Scholar] [CrossRef] [Green Version]
- Heit, C.; Jackson, B.C.; McAndrews, M.; Wright, M.W.; Thompson, D.C.; Silverman, G.A.; Nebert, D.W.; Vasiliou, V. Update of the human and mouse SERPINgene superfamily. Hum. Genom. 2013, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, J.; Pan, G.; Poncz, M.; Wei, J.; Ran, M.; Zhou, Z. Serpin functions in host-pathogen interactions. Peer J. 2018, 6, e4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiyama, T.; Ray, C.A.; Pickup, D.J.; Howard, A.D.; Thornberry, N.A.; Peterson, E.P.; Salvesen, G. Inhibition of interleukin-1 beta converting enzyme by the cowpox virus serpin CrmA. An example of cross-class inhibition. J. Biol. Chem. 1994, 269, 19331–19337. [Google Scholar] [CrossRef]
- Schick, C.; Pemberton, P.A.; Shi, G.-P.; Kamachi, Y.; Çataltepe, S.; Bartuski, A.J.; Gornstein, E.R.; Brömme, D.; Chapman, A.H.A.; Silverman, G.A. Cross-Class Inhibition of the Cysteine Proteinases Cathepsins K, L, and S by the Serpin Squamous Cell Carcinoma Antigen 1: A Kinetic Analysis. Biochemistry 1998, 37, 5258–5266. [Google Scholar] [CrossRef]
- Ksiazek, M.; Mizgalska, D.; Enghild, J.J.; Scavenius, C.; Thogersen, I.B.; Potempa, J. Miropin, a Novel Bacterial Serpin from the Periodontopathogen Tannerella forsythia, Inhibits a Broad Range of Proteases by Using Different Peptide Bonds within the Reactive Center Loop. J. Biol. Chem. 2015, 290, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Goulas, T.; Ksiazek, M.; Garcia-Ferrer, I.; Sochaj-Gregorczyk, A.M.; Waligorska, I.; Wasylewski, M.; Potempa, J.; Gomis-Rüth, F.X. A structure-derived snap-trap mechanism of a multispecific serpin from the dysbiotic human oral microbiome. J. Biol. Chem. 2017, 292, 10883–10898. [Google Scholar] [CrossRef] [Green Version]
- Sochaj-Gregorczyk, A.; Ksiazek, M.; Waligorska, I.; Straczek, A.; Benedyk, M.; Mizgalska, D.; Thøgersen, I.B.; Enghild, J.J.; Potempa, J. Plasmin inhibition by bacterial serpin: Implications in gum disease. FASEB J. 2019, 34, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Gettins, P.G.W. Serpin Structure, Mechanism, and Function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef]
- Sanrattana, W.; Maas, C.; de Maat, S. SERPINs—From Trap to Treatment. Front. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Bock, S.C.; Skriver, K.; Nielsen, E.; Thoegersen, H.C.; Wiman, B.; Donaldson, V.H.; Eddy, R.L.; Marrinan, J.; Radziejewska, E. Human, C.hivin.1 inhibitor: Primary structure, cDNA cloning, and chromosomal localization. Biochemistry 1986, 25, 4292–4301. [Google Scholar] [CrossRef]
- Law, R.H.P.; Zhang, Q.; McGowan, S.; Buckle, A.M.; Silverman, G.A.; Wong, W.; Rosado, C.J.; Langendorf, C.G.; Pike, R.N.; Bird, P.I.; et al. An overview of the serpin superfamily. Genome Biol. 2006, 7, 216. [Google Scholar] [CrossRef] [Green Version]
- Gatto, M.; Iaccarino, L.; Ghirardello, A.; Bassi, N.; Pontisso, P.; Punzi, L.; Shoenfeld, Y.; Doria, A. Serpins, Immunity and Autoimmunity: Old Molecules, New Functions. Clin. Rev. Allergy Immunol. 2013, 45, 267–280. [Google Scholar] [CrossRef]
- Dementiev, A.; Dobó, J.; Gettins, P.G.W. Active Site Distortion Is Sufficient for Proteinase Inhibition by Serpins: Structure of The Covalent Complex Of α1-Proteinase Inhibitor With Porcine Pancreatic Elastase. J. Biol. Chem. 2006, 281, 3452–3457. [Google Scholar] [CrossRef] [Green Version]
- Hirsh, J.; Anand, S.S.; Halperin, J.L.; Fuster, V. Mechanism of Action and Pharmacology of Unfractionated Heparin. Arter. Thromb. Vasc. Biol. 2001, 21, 1094–1096. [Google Scholar] [CrossRef] [Green Version]
- Maimone, M.M.; Tollefsen, D.M. Structure of a dermatan sulfate hexasaccharide that binds to heparin cofactor II with high affinity. J. Biol. Chem. 1990, 265, 18263–18271. [Google Scholar] [CrossRef]
- Pratt, C.W.; Church, F.C. Heparin binding to protein C inhibitor. J. Biol. Chem. 1992, 267, 8789–8794. [Google Scholar] [CrossRef]
- Mkaouar, H.; Akermi, N.; Kriaa, A.; Abraham, A.-L.; Jablaoui, A.; Soussou, S.; Mokdad-Gargouri, R.; Maguin, E.; Rhimi, M. Serine protease inhibitors and human wellbeing interplay: New insights for old friends. Peer J. 2019, 7, e7224. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.; Smith, C.L.; Goping, I.S.; Underhill, D.; Harley, M.J.; Reventos, J.; Musto, N.A.; Gunsalus, G.L.; Bardin, C.W. Primary structure of human corticosteroid binding globulin, deduced from hepatic and pulmonary cDNAs, exhibits homology with serine protease inhibitors. Proc. Natl. Acad. Sci. USA 1987, 84, 5153–5157. [Google Scholar] [CrossRef] [Green Version]
- Flink, I.L.; Bailey, T.J.; Gustafson, T.A.; Markham, B.E.; Morkin, E. Complete amino acid sequence of human thyroxine-binding globulin deduced from cloned DNA: Close homology to the serine antiproteases. Proc. Natl. Acad. Sci. USA 1986, 83, 7708–7712. [Google Scholar] [CrossRef] [Green Version]
- Nagata, K. Hsp47: A collagen-specific molecular chaperone. Trends Biochem. Sci. 1996, 21, 22–26. [Google Scholar] [CrossRef]
- Zou, Z.; Anisowicz, A.; Hendrix, M.J.; Thor, A.; Neveu, M.; Sheng, S.; Rafidi, K.; Seftor, E.; Sager, R. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science 1994, 263, 526–529. [Google Scholar] [CrossRef]
- Pike, R.N.; Bottomley, S.P.; Irving, J.A.; Bird, P.I.; Whisstock, J.C. Serpins: Finely Balanced Conformational Traps. IUBMB Life 2002, 54, 1–7. [Google Scholar] [CrossRef]
- Lomas, R.; Belorgey, D.; Mallya, M.; Miranda, E.; Kinghorn, K.; Sharp, L.; Phillips, R.; Page, R.; Robertson, A.; Crowther, D. Molecular mousetraps and the serpinopathies. Biochem. Soc. Trans. 2005, 33, 321–330. [Google Scholar] [CrossRef]
- Lomas, D.A.; Li-Evans, D.; Finch, J.T.; Carrell, R.W. The mechanism of Z α1-antitrypsin accumulation in the liver. Nat. Cell Biol. 1992, 357, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Bruce, D.; Perry, D.J.; Borg, J.Y.; Carrell, R.W.; Wardell, M.R. Thromboembolic disease due to thermolabile conformational changes of antithrombin Rouen-VI (187 Asn--Asp). J. Clin. Investig. 1994, 94, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Aulak, K.S.; Pemberton, P.A.; Rosen, F.S.; Carrell, R.W.; Lachmann, P.J.; Harrison, R.A. Dysfunctional C1-inhibitor(At), isolated from a type II hereditary-angio-oedema plasma, contains a P1 ’reactive centre’ (Arg444----His) mutation. Biochem. J. 1988, 253, 615–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.L.; Shrimpton, A.E.; Holohan, P.D.; Bradshaw, C.; Feiglin, D.; Collins, G.H.; Sonderegger, P.; Kinter, J.; Becker, L.M.; Lacbawan, F.; et al. Familial dementia caused by polymerization of mutant neuroserpin. Nat. Cell Biol. 1999, 401, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Luisetti, M.; Seersholm, N. α1-Antitrypsin deficiency: Epidemiology of α1-antitrypsin deficiency. Thorax 2004, 59, 164–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fregonese, L.; Stolk, J. Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. Orphanet J. Rare Dis. 2008, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Cichy, J.; Potempa, J.; Travis, J. Biosynthesis of α1-Proteinase Inhibitor by Human Lung-derived Epithelial Cells. J. Biol. Chem. 1997, 272, 8250–8255. [Google Scholar] [CrossRef] [Green Version]
- Beatty, K.; Bieth, J.; Travis, J. Kinetics of association of serine proteinases with native and oxidized alpha-1-proteinase inhibitor and alpha-1-antichymotrypsin. J. Biol. Chem. 1980, 255, 3931–3934. [Google Scholar] [CrossRef]
- Carrell, R.W. alpha 1-Antitrypsin: Molecular pathology, leukocytes, and tissue damage. J. Clin. Investig. 1986, 78, 1427–1431. [Google Scholar] [CrossRef]
- Janciauskiene, S.; Zelvyte, I.; Jansson, L.; Stevens, T. Divergent effects of α1-antitrypsin on neutrophil activation, in vitro. Biochem. Biophys. Res. Commun. 2004, 315, 288–296. [Google Scholar] [CrossRef]
- Jonigk, D.; Al-Omari, M.; Maegel, L.; Müller, M.; Izykowski, N.; Hong, J.; Hong, K.; Soo-Hyun, K.; Dorsch, M.; Mahadeva, R.; et al. Anti-inflammatory and immunomodulatory properties of α1-antitrypsin without inhibition of elastase. Proc. Natl. Acad. Sci. USA 2013, 110, 15007–15012. [Google Scholar] [CrossRef] [Green Version]
- Petrache, I.; Fijalkowska, I.; Medler, T.R.; Skirball, J.; Cruz, P.; Zhen, L.; Petrache, H.I.; Flotte, T.R.; Tuder, R.M. alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am. J. Pathol 2006, 169, 1155–1166. [Google Scholar] [CrossRef] [Green Version]
- Sarabhai, T.; Peter, C.; Bär, A.-K.; Windolf, J.; Relja, B.; Wesselborg, S.; Wahlers, T.; Paunel-Görgülü, A. Serum α-1 Antitrypsin (AAT) antagonizes intrinsic apoptosis induction in neutrophils from patients with systemic inflammatory response syndrome. PLoS ONE 2017, 12, e0177450. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, N.; Tumpara, S.; Wrenger, S.; Ercetin, E.; Hamacher, J.; Welte, T.; Janciauskiene, S. Alpha1-antitrypsin protects lung cancer cells from staurosporine-induced apoptosis: The role of bacterial lipopolysaccharide. Sci. Rep. 2020, 10, 9563. [Google Scholar] [CrossRef]
- Laurell, C.-B.; Eriksson, S. The Electrophoretic α1-Globulin Pattern of Serum in α1-Antitrypsin Deficiency. COPD J. Chronic Obstr. Pulm. Dis. 2013, 10, 3–8. [Google Scholar] [CrossRef]
- Stoller, J.K. Clinical Features and Natural History of Severe α1-Antitrypsin Deficiency. Chest 1997, 111, 123S–128S. [Google Scholar] [CrossRef]
- Bartels, C.L.; Marchetti, A.L.; Highsmith, W.E.; Tsongalis, G.J. Real time PCR detection of the PI*Z and PI*S mutations associated with alpha-1 antitrypsin deficiency. Am. J. Transl. Res. 2009, 1, 406–411. [Google Scholar]
- Stolk, J.; Seersholm, N.; Kalsheker, N. Alpha1-antitrypsin deficiency: Current perspective on research, diagnosis, and management. Int. J. Chronic Obstr. Pulm. Dis. 2006, 1, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häggblom, J.; Kettunen, K.; Karjalainen, J.; Heliövaara, M.; Jousilahti, P.; Saarelainen, S. Prevalence of PI*Z and PI*S alleles of alpha-1-antitrypsin deficiency in Finland. Eur. Clin. Respir. J. 2015, 2, 28829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooptu, B.; Lomas, D.A. Polymers and inflammation: Disease mechanisms of the serpinopathies. J. Exp. Med. 2008, 205, 1529–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, L.; Burdon, J.G.; Brenton, S.; Knight, K.R.; Janus, E.D. Kinetic characterisation of ALPHA-1-Antitrypsin F as an inhibitor of human neutrophil elastase. Pathology 1996, 28, 242–247. [Google Scholar] [CrossRef]
- Mahadeva, R.; Atkinson, C.; Li, Z.; Stewart, S.; Janciauskiene, S.; Kelley, D.G.; Parmar, J.; Pitman, R.; Shapiro, S.D.; Lomas, D.A. Polymers of Z α1-Antitrypsin Co-Localize with Neutrophils in Emphysematous Alveoli and Are Chemotactic in Vivo. Am. J. Pathol. 2005, 166, 377–386. [Google Scholar] [CrossRef]
- Askew, D.J.; A Silverman, G. Intracellular and extracellular serpins modulate lung disease. J. Perinatol. 2008, 28, S127–S135. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.; Travis, J. The oxidative inactivation of human alpha-1-proteinase inhibitor. Further evidence for methionine at the reactive center. J. Biol. Chem. 1979, 254, 4022–4026. [Google Scholar]
- Janoff, A.; Carp, H.; Lee, D.; Drew, R. Cigarette smoke inhalation decreases alpha 1-antitrypsin activity in rat lung. Science 1979, 206, 1313–1314. [Google Scholar] [CrossRef]
- Hwang, S.-R.; Steineckert, B.; Kohn, A.; Palkovits, M.; Hook, V.Y.H. Molecular Studies Define the Primary Structure of α1-Antichymotrypsin (ACT) Protease Inhibitor in Alzheimer’s Disease Brains: Comparison of Actin in Hippocampus and Liver. J. Biol. Chem. 1999, 274, 1821–1827. [Google Scholar] [CrossRef] [Green Version]
- Poller, W.; Faber, J.-P.; Weidinger, S.; Tief, K.; Scholz, S.; Fischer, M.; Olek, K.; Kirchgesser, M.; Heidtmann, H.-H. A Leucine-to-Proline Substitution Causes a Defective α1-Antichymotrypsin Allele Associated with Familial Obstructive Lung Disease. Genoms 1993, 17, 740–743. [Google Scholar] [CrossRef]
- Travis, J.; Bowen, J.; Baugh, R. Human α-1-antichymotrypsin: Interaction with chymotrypsin-like proteinases. Biochemistry 1978, 17, 5651–5656. [Google Scholar] [CrossRef]
- Christensson, A.; Laurell, C.-B.; Lilja, H. Enzymatic activity of prostate-specific antigen and its reactions with extracellular serine proteinase inhibitors. JBIC J. Biol. Inorg. Chem. 1990, 194, 755–763. [Google Scholar] [CrossRef]
- Frenette, G.; Deperthes, D.; Tremblay, R.R.; Lazure, C.; Dubé, J.Y. Purification of enzymatically active kallikrein hK2 from human seminal plasma. Biochim. Biophys. Acta Gen. Subj. 1997, 1334, 109–115. [Google Scholar] [CrossRef]
- Håkansson, H.O.; Ohlsson, K. Interactions in vitro and in vivo between human and porcine cationic pancreatic elastase and plasma protease inhibitors. Biol. Chem. Hoppe Seyler 1988, 369, 309–315. [Google Scholar] [CrossRef]
- Aronsen, K.F.; Ekelund, G.; Kindmark, C.O.; Laurell, C.B. Sequential changes of plasma proteins after surgical trauma. Scand. J. Clin. Lab. Investig. Suppl. 1972, 124, 127–136. [Google Scholar] [CrossRef]
- Kalsheker, N.; Morgan, K.; Chappell, S. Proteinase inhibitors. Antichymotrypsin. In Encyclopedia of Respiratory Medicine; Elsevier BV: Amsterdam, The Netherlands, 2006; pp. 507–511. [Google Scholar]
- Eriksson, S.; Lindmark, B.; Lilja, H. Familial alpha 1-antichymotrypsin deficiency. Acta Med. Scand. 1986, 220, 447–453. [Google Scholar] [CrossRef]
- Faber, J.-P.; Poller, W.; Olek, K.; Baumann, U.; Carlson, J.; Lindmark, B.; Eriksson, S. The molecular basis of α1-antichymotrypsin deficiency in a heterozygote with liver and lung disease. J. Hepatol. 1993, 18, 313–321. [Google Scholar] [CrossRef]
- Gooptu, B.; Hazes, B.; Chang, W.-S.W.; Dafforn, T.R.; Carrell, R.W.; Read, R.; Lomas, D.A. Inactive conformation of the serpin alpha 1-antichymotrypsin indicates two-stage insertion of the reactive loop: Implications for inhibitory function and conformational disease. Proc. Natl. Acad. Sci. USA 2000, 97, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Mahadeva, R.; Sharples, L.; Ross-Russell, R.I.; Webb, A.K.; Bilton, D.; Lomas, D.A. Association of α1-antichymotrypsin deficiency with milder lung disease in patients with cystic fibrosis. Thorax 2001, 56, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Sandford, A.J.; Chagani, T.; Weir, T.D.; Paré, P.D. Alpha 1-antichymotrypsin mutations in patients with chronic obstructive pulmonary disease. Dis. Mark. 1998, 13, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, N.; Cooperman, B.S.; Wang, Z.-M.; Liu, X.-Z.; Rubin, H. Identification of Lysines within α1-Antichymotrypsin Important for DNA Binding. An unusual combination of dna-binding elements. J. Biol. Chem. 1995, 270, 14548–14555. [Google Scholar] [CrossRef] [Green Version]
- Laurell, M.; Christensson, A.; Abrahamsson, P.A.; Stenflo, J.; Lilja, H. Protein C inhibitor in human body fluids. Seminal plasma is rich in inhibitor antigen deriving from cells throughout the male reproductive system. J. Clin. Investig. 1992, 89, 1094–1101. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Nishioka, J.; Kusumoto, H.; Hashimoto, S. Mechanism of inhibition of activated protein C by protein C inhibitor. J. Biochem. 1984, 95, 187–195. [Google Scholar] [CrossRef]
- Rezaie, A.R.; Cooper, S.T.; Church, F.C.; Esmon, C.T. Protein C Inhibitor Is a Potent Inhibitor of the Thrombin-Thrombomodulin Complex. J. Biol. Chem. 1995, 270, 25336–25339. [Google Scholar] [CrossRef] [Green Version]
- Stief, T.W.; Radtke, K.-P.; Heimburger, N. Inhibition of Urokinase by Protein C-Inhibitor (PCI). Evidence for Identity of PCI and Plasminogen Activator Inhibitor. Biol. Chem. Hoppe-Seyler 1987, 368, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Nishioka, J.; Nakagawa, N.; Kamada, H.; Gabazza, E.C.; Kobayashi, T.; Hattori, A.; Suzuki, K. Protein C inhibitor directly and potently inhibits activated hepatocyte growth factor activator. J. Thromb. Haemost. 2007, 5, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, T.A.; Wahlmüller, F.C.; Furtmüller, M.; Geiger, M. Interaction of Protein C Inhibitor with the Type II Transmembrane Serine Protease Enteropeptidase. PLoS ONE 2012, 7, e39262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Adams, T.E.; Kjellberg, M.; Stenflo, J.; Huntington, J.A. Structure of Native Protein C Inhibitor Provides Insight into Its Multiple Functions. J. Biol. Chem. 2007, 282, 13759–13768. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Nishioka, J.; Kamada, H.; Asanuma, K.; Kondo, H.; Gabazza, E.C.; Ido, M.; Suzuki, K. Characterization of a novel human protein C inhibitor (PCI) gene transgenic mouse useful for studying the role of PCI in physiological and pathological conditions. J. Thromb. Haemost. 2004, 2, 949–961. [Google Scholar] [CrossRef]
- Nishii, Y.; Gabazza, E.; Fujimoto, H.; Nakahara, H.; Takagi, T.; Bruno, N.; D’Alessandro-Gabazza, C.N.; Maruyama, J.; Maruyama, K.; Hayashi, T.; et al. Protective role of protein C inhibitor in monocrotaline-induced pulmonary hypertension. J. Thromb. Haemost. 2006, 4, 2331–2339. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Church, F.C. Is protein C inhibitor antithrombotic and protective in pulmonary hypertension? J. Thromb. Haemost. 2006, 4, 2327–2330. [Google Scholar] [CrossRef]
- Fujimoto, H.; Gabazza, E.C.; Hataji, O.; Yuda, H.; D’Alessandro-Gabazza, C.N.; Nakano, M.; Franco, O.E.; Hayashi, T.; Suzuki, K.; Adachi, Y.; et al. Thrombin-activatable Fibrinolysis Inhibitor and Protein C Inhibitor in Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1687–1694. [Google Scholar] [CrossRef]
- Goodman, H.M. Thyroid Gland. In Basic Medical Endocrinology; Elsevier BV: Amsterdam, The Netherlands, 2009; pp. 43–59. [Google Scholar]
- Carolan, B.J.; Hughes, G.; Morrow, J.; Hersh, C.P.; O’Neal, W.K.; Rennard, S.; Pillai, S.G.; Belloni, P.; Cockayne, D.A.; Comellas, A.P.; et al. The association of plasma biomarkers with computed tomography-assessed emphysema phenotypes. Respir. Res. 2014, 15, 127. [Google Scholar] [CrossRef] [Green Version]
- Diao, W.; Shen, N.; Du, Y.; Sun, X.; Liu, B.; Xu, M.; He, B. Identification of thyroxine-binding globulin as a candidate plasma marker of chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1549–1564. [Google Scholar] [CrossRef] [Green Version]
- Gimenez-Roqueplo, A.-P.; Célérier, J.; Lucarelli, G.; Corvol, P.; Jeunemaitre, X. Role of N-Glycosylation in Human Angiotensinogen. J. Biol. Chem. 1998, 273, 21232–21238. [Google Scholar] [CrossRef] [Green Version]
- Turgut, S.; Akın, F.; Akcılar, R.; Ayada, C.; Turgut, G.; Akin, F. Angiotensin converting enzyme I/D, angiotensinogen M235T and AT1-R A/C1166 gene polymorphisms in patients with acromegaly. Mol. Biol. Rep. 2010, 38, 569–576. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, L.; Wang, H.-W.; Wang, X.-Y.; Li, X.-Q.; Zhang, L.-L. The M235T polymorphism in the angiotensinogen gene and heart failure: A meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. 2014, 15, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Ayada, C.; Toru, Ümran; Genç, O.; Şahin, S.; Turgut, S.; Turgut, G. Angiotensinogen gene M235T and angiotensin II-type 1 receptor gene A/C1166 polymorphisms in chronic obtructive pulmonary disease. Int. J. Clin. Exp. Med. 2015, 8, 4521–4526. [Google Scholar]
- Kyrou, I.; Mattu, H.S.; Chatha, K.; Randeva, H.S. Fat Hormones, Adipokines. In Endocrinology of the Heart in Health and Disease; Schisler, J.C., Lang, C.H., Willis, M.S., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 167–205. [Google Scholar]
- Wang, C. Obesity, Inflammation, and Lung Injury (OILI): The Good. Mediat. Inflamm. 2014, 2014, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.-K.; Han, T.-K.; Kang, H.-S. Combined effects of body mass index and cardio/respiratory fitness on serum vaspin concentrations in Korean young men. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 108, 347–353. [Google Scholar] [CrossRef]
- Jackson, V.E.; Ntalla, I.; Sayers, I.; Morris, R.; Whincup, P.; Casas, J.-P.; Amuzu, A.; Choi, M.; Dale, C.; Kumari, M.; et al. Exome-wide analysis of rare coding variation identifies novel associations with COPD and airflow limitation inMOCS3,IFIT3andSERPINA. Thorax 2016, 71, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Sivaprasad, U.; Askew, D.J.; Ericksen, M.B.; Gibson, A.M.; Stier, M.T.; Brandt, E.; Bass, S.A.; Daines, M.O.; Chakir, J.; Stringer, K.F.; et al. A nonredundant role for mouse Serpinb3a in the induction of mucus production in asthma. J. Allergy Clin. Immunol. 2011, 127, 254–261.e6. [Google Scholar] [CrossRef] [Green Version]
- Cooley, J.; Takayama, T.K.; Shapiro, S.D.; Schechter, N.M.; Remold-O’Donnell, E. The Serpin MNEI Inhibits Elastase-like and Chymotrypsin-like Serine Proteases through Efficient Reactions at Two Active Sites. Biochemistry 2001, 40, 15762–15770. [Google Scholar] [CrossRef] [PubMed]
- Benarafa, C.; Priebe, G.P.; Remold-O’Donnell, E. The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomonas aeruginosa infection. J. Exp. Med. 2007, 204, 1901–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooley, J.; Sontag, M.K.; Accurso, F.J.; Remoldodonnell, E. SerpinB1 in cystic fibrosis airway fluids: Quantity, molecular form and mechanism of elastase inhibition. Eur. Respir. J. 2010, 37, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Medcalf, R.L.; Stasinopoulos, S.J. The undecided serpin. FEBS J. 2005, 272, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Kotani, I.; Sato, A.; Hayakawa, H.; Urano, T.; Takada, Y.; Takada, A. Increased procoagulant and antifibrinolytic activities in the lungs with idiopathic pulmonary fibrosis. Thromb. Res. 1995, 77, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, P.G.; Boushey, H.A.; Dolganov, G.M.; Barker, C.S.; Yang, Y.H.; Donnelly, S.; Ellwanger, A.; Sidhu, S.S.; Dao-Pick, T.P.; Pantoja, C.; et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc. Natl. Acad. Sci. USA 2007, 104, 15858–15863. [Google Scholar] [CrossRef] [Green Version]
- Peters, M.C.; Mekonnen, Z.; Yuan, S.; Bhakta, N.R.; Woodruff, P.G.; Fahy, J.V. Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J. Allergy Clin. Immunol. 2014, 133, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, P.G.; Modrek, B.; Choy, D.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Arron, J.R.; Koth, L.L.; Fahy, J.V. T-helper Type 2–driven Inflammation Defines Major Subphenotypes of Asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395. [Google Scholar] [CrossRef]
- Barnes, R.C.; Worrall, D. Identification of a novel human serpin gene; cloning sequencing and expression of leupin. FEBS Lett. 1995, 373, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Schneider, S.S.; Schick, C.; Fish, K.E.; Miller, E.; Pena, J.C.; Treter, S.D.; Hui, S.M.; Silverman, G.A. A serine proteinase inhibitor locus at 18q21.3 contains a tandem duplication of the human squamous cell carcinoma antigen gene. Proc. Natl. Acad. Sci. USA 1995, 92, 3147–3151. [Google Scholar] [CrossRef] [Green Version]
- Cataltepe, S.; Gornstein, E.R.; Schick, C.; Kamachi, Y.; Chatson, K.; Fries, J.; Silverman, G.A.; Upton, M.P. Co-expression of the Squamous Cell Carcinoma Antigens 1 and 2 in Normal Adult Human Tissues and Squamous Cell Carcinomas. J. Histochem. Cytochem. 2000, 48, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Sheshadri, N.; Zong, W.-X. SERPINB3 and B4: From biochemistry to biology. Semin. Cell Dev. Biol. 2017, 62, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Schick, C.; Kamachi, Y.; Bartuski, A.J.; Çataltepe, S.; Schechter, N.M.; Pemberton, P.A.; Silverman, G.A. Squamous Cell Carcinoma Antigen 2 Is a Novel Serpin That Inhibits the Chymotrypsin-like Proteinases Cathepsin G and Mast Cell Chymase. J. Biol. Chem. 1997, 272, 1849–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, F.L.; Hirst, C.E.; Sun, J.; Bird, C.H.; Bottomley, S.P.; I Bird, P. The intracellular serpin proteinase inhibitor 6 is expressed in monocytes and granulocytes and is a potent inhibitor of the azurophilic granule protease, cathepsin G. Blood 1999, 93, 2089–2097. [Google Scholar] [CrossRef]
- Ray, R.; Choi, M.; Zhang, Z.; Silverman, G.A.; Askew, D.; Mukherjee, A.B. Uteroglobin Suppresses SCCA Gene Expression Associated with Allergic Asthma. J. Biol. Chem. 2005, 280, 9761–9764. [Google Scholar] [CrossRef] [Green Version]
- Yuyama, N.; Davies, N.E.; Akaiwa, M.; Matsui, K.; Hamasaki, Y.; Suminami, Y.; Yoshida, N.L.; Maeda, M.; Pandit, A.; Lordan, J.L.; et al. Analysis of novel disease-related genes in bronchial asthma. Cytokine 2002, 19, 287–296. [Google Scholar] [CrossRef]
- Molina, R.; Filella, X.; Torres, M.D.; Ballesta, A.M.; Mengual, P.; Cases, A.; Balaque, A. SCC antigen measured in malignant and nonmalignant diseases. Clin. Chem. 1990, 36, 251–254. [Google Scholar] [CrossRef]
- Kato, H. Squamous Cell Carcinoma Antigen. In Serological Cancer Markers; Springer: Berlin/Heidelberg, Germany, 1992; Volume 11, pp. 437–451. [Google Scholar]
- Franciosi, L.; Postma, D.S.; Berge, M.V.D.; Govorukhina, N.; Horvatovich, P.L.; Fusetti, F.; Poolman, B.; Lodewijk, M.E.; Timens, W.; Bischoff, R.; et al. Susceptibility to COPD: Differential Proteomic Profiling after Acute Smoking. PLoS ONE 2014, 9, e102037. [Google Scholar] [CrossRef]
- Coughlin, P.; Sun, J.; Cerruti, L.; Salem, H.H.; Bird, P. Cloning and molecular characterization of a human intracellular serine proteinase inhibitor. Proc. Natl. Acad. Sci. USA 1993, 90, 9417–9421. [Google Scholar] [CrossRef] [Green Version]
- Scott, F.L.; Sun, J.; Whisstock, J.C.; Kato, K.; Bird, P.I. SerpinB6 is an inhibitor of kallikrein-8 in keratinocytes. J. Biochem. 2007, 142, 435–442. [Google Scholar] [CrossRef]
- Strik, M.C.M.; Wolbink, A.; Wouters, D.; Bladergroen, B.A.; Verlaan, A.R.; van Houdt, I.S.; Hijlkema, S.; Hack, C.E.; Kummer, J.A. Intracellular serpin SERPINB6 (PI6) is abundantly expressed by human mast cells and forms complexes with β-tryptase monomers. Blood 2004, 103, 2710–2717. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; Schuurhuis, D.H.; Rea, D.; van Tongeren, J.; de Jong, J.; Bres, S.A.; Laban, S.; Toes, R.E.; Toebes, M.; Schumacher, T.N.; et al. Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte-induced apoptosis: Differential modulation by T helper type 1 and type 2 cells. J. Exp. Med. 2001, 194, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.; Opferman, J.T.; Shah, R.; Liu, N.; Froelich, C.J.; Ashton-Rickardt, P.G. A role for the granzyme B inhibitor serine protease inhibitor 6 in CD8+ memory cell homeostasis. J. Immunol. 2004, 173, 3801–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Bird, C.H.; Sutton, V.; McDonald, L.; Coughlin, P.B.; De Jong, T.A.; Trapani, J.A.; Bird, P.I. A Cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier a is present in cytotoxic lymphocytes. J. Biol. Chem. 1996, 271, 27802–27809. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Trapani, J.A. Granzymes: Exogenous proteinases that induce target cell apoptosis. Immunol. Today 1995, 16, 202–206. [Google Scholar] [CrossRef]
- Eriksson Ström, J.; Pourazar, J.; Linder, R.; Blomberg, A.; Lindberg, A.; Bucht, A.; Behndig, A.F. Cytotoxic lymphocytes in COPD airways: Increased NK cells associated with disease, iNKT and NKT-like cells with current smoking. Respir. Res. 2018, 19, 244. [Google Scholar] [CrossRef] [Green Version]
- Freeman, C.M.; Han, M.K.; Martinez, F.J.; Murray, S.; Liu, L.X.; Chensue, S.W.; Polak, T.J.; Sonstein, J.; Todt, J.C.; Ames, T.M.; et al. Cytotoxic potential of lung CD8+ T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J. Immunol. 2010, 184, 6504–6513. [Google Scholar] [CrossRef] [Green Version]
- Hodge, S.; Hodge, G.; Nairn, J.; Holmes, M.; Reynolds, P.N. Increased airway granzyme b and perforin in current and ex-smoking COPD subjects. COPD 2006, 3, 179–187. [Google Scholar] [CrossRef]
- Riewald, M.; Schleef, R.R. Molecular Cloning of Bomapin (Protease Inhibitor 10), a Novel Human Serpin That Is Expressed Specifically in the Bone Marrow. J. Biol. Chem. 1995, 270, 26754–26757. [Google Scholar] [CrossRef] [Green Version]
- Przygodzka, P.; Ramstedt, B.; Tengel, T.; Larsson, G.; Wilczynska, M. Bomapin is a redox-sensitive nuclear serpin that affects responsiveness of myeloid progenitor cells to growth environment. BMC Cell Biol. 2010, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Mo, Y.; Zhang, K.; Feng, Y.; Yi, L.; Liang, Y.; Wu, W.; Zhao, J.-P.; Zhang, Z.; Xu, Y.; Hu, Q.; et al. Epithelial SERPINB10, a novel marker of airway eosinophilia in asthma, contributes to allergic airway inflammation. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L245–L254. [Google Scholar] [CrossRef]
- Chuang, T.L.; Schleef, R.R. Identification of a Nuclear Targeting Domain in the Insertion between Helices C and D in Protease Inhibitor. J. Biol. Chem. 1999, 274, 11194–11198. [Google Scholar] [CrossRef] [Green Version]
- Zhen, G.; Park, S.W.; Nguyenvu, L.T.; Rodriguez, M.W.; Barbeau, R.; Paquet, A.C.; Erle, D.J. IL-13 and Epidermal Growth Factor Receptor Have Critical but Distinct Roles in Epithelial Cell Mucin Production. Am. J. Respir. Cell Mol. Biol. 2007, 36, 244–253. [Google Scholar] [CrossRef]
- Rezaie, A.R. Vitronectin Functions as a Cofactor for Rapid Inhibition of Activated Protein C by Plasminogen Activator Inhibitor. J. Biol. Chem. 2001, 276, 15567–15570. [Google Scholar] [CrossRef] [Green Version]
- van Meijer, M.; Smilde, A.; Tans, G.; Nesheim, M.E.; Pannekoek, H.; Horrevoets, A.J. The suicide substrate reaction between plasminogen activator inhibitor 1 and thrombin is regulated by the cofactors vitronectin and heparin. Blood 1997, 90, 1874–1882. [Google Scholar] [CrossRef] [Green Version]
- Chapman, H.A.; Yang, X.L.; Sailor, L.Z.; Sugarbaker, D.J. Developmental expression of plasminogen activator inhibitor type 1 by human alveolar macrophages. Possible role in lung injury. J. Immunol. 1990, 145, 3398–3405. [Google Scholar]
- Choi, G.; Schultz, M.J.; Van Till, J.O.; Bresser, P.; van der Zee, J.; Boermeester, M.A.; Levi, M.; Marcel, M.; Van Der Poll, T. Disturbed alveolar fibrin turnover during pneumonia is restricted to the site of infection. Eur. Respir. J. 2004, 24, 786–789. [Google Scholar] [CrossRef]
- El-Solh, A.A.; Okada, M.; Pietrantoni, C.; Aquilina, A.; Berbary, E. Procoagulant and fibrinolytic activity in ventilator-associated pneumonia: Impact of inadequate antimicrobial therapy. Intensiv. Care Med. 2004, 30, 1914–1920. [Google Scholar] [CrossRef]
- Nie, W.; Li, B.; Xiu, Q.-Y. The −675 4G/5G Polymorphism in Plasminogen Activator Inhibitor-1 Gene Is Associated with Risk of Asthma: A Meta-Analysis. PLoS ONE 2012, 7, e34385. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.H.; Hall, I.; Wheatley, A.; Dewar, J.; Abraha, D.; Del Mundo, J.; Lee, H.; Oh, C.K. Possible role of the 4G/5G polymorphism of the plasminogen activator inhibitor 1 gene in the development of asthma. J. Allergy Clin. Immunol. 2001, 108, 212–214. [Google Scholar] [CrossRef]
- Cho, S.; Kang, J.; Lyttle, C.; Harris, K.; Daley, B.; Grammer, L.; Avila, P.; Kumar, R.; Schleimer, R. Association of elevated plasminogen activator inhibitor 1 levels with diminished lung function in patients with asthma. Ann. Allergy Asthma Immunol. 2011, 106, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Tapson, V.F. The Role of Smoking in Coagulation and Thromboembolism in Chronic Obstructive Pulmonary Disease. Proc. Am. Thorac. Soc. 2005, 2, 71–77. [Google Scholar] [CrossRef]
- Xiao, W.; Hsu, Y.-P.; Ishizaka, A.; Kirikae, T.; Moss, R.B. Sputum Cathelicidin, Urokinase Plasminogen Activation System Components, and Cytokines Discriminate Cystic Fibrosis, COPD, and Asthma Inflammation. Chest 2005, 128, 2316–2326. [Google Scholar] [CrossRef] [Green Version]
- Christ, G.; Graf, S.; Huber-Beckmann, R.; Zorn, G.; Lang, I.; Kneussi, M.; Binder, B.R.; Huber, K. Impairment of the plasmin activation system in primary pulmonary hypertension: Evidence for gender differences. Thromb. Haemost. 2001, 86, 557–562. [Google Scholar]
- Dimova, E.Y.; Samoylenko, A.; Kietzmann, T. Oxidative Stress and Hypoxia: Implications for Plasminogen Activator Inhibitor-1 Expression. Antiox. Redox Signal. 2004, 6, 777–791. [Google Scholar] [CrossRef]
- Wu, Y.P.; Wei, R.; Liu, Z.H.; Chen, B.; Lisman, T.; Ren, D.L.; Han, J.J.; Xia, Z.L.; Zhang, F.S.; Xu, W.B.; et al. Analysis of thrombotic factors in severe acute respiratory syndrome (SARS) patients. Thromb. Haemost. 2006, 96, 100–101. [Google Scholar] [CrossRef] [Green Version]
- de Lang, A.; Baas, T.; Teal, T.; Leijten, L.M.; Rain, B.; Osterhaus, A.D.; Haagmans, B.L.; Katze, M.G. Functional Genomics Highlights Differential Induction of Antiviral Pathways in the Lungs of SARS-CoV–Infected Macaques. PLoS Pathog. 2007, 3, e112. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.-Y. Pulmonary Pathology of Early-Phase 2019 Novel Coronavirus (COVID-19) Pneumonia in Two Patients with Lung Cancer. J. Thorac. Oncol. 2020, 15, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Eitzman, D.T.; McCoy, R.D.; Zheng, X.; Fay, W.P.; Shen, T.; Ginsburg, D.; Simon, R.H. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J. Clin. Investig. 1996, 97, 232–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, N.; Degen, J.L.; Sisson, T.H.; Liu, H.; Moore, B.B.; Pandrangi, R.G.; Simon, R.H.; Drew, A.F. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J. Clin. Investig. 2000, 106, 1341–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang-Tsai, S.; Sisson, T.H.; Hattori, N.; Tsai, C.G.; Subbotina, N.M.; Hanson, K.E.; Simon, R.H. Reduction in Fibrotic Tissue Formation in Mice Genetically Deficient in Plasminogen Activator Inhibitor. Am. J. Pathol. 2003, 163, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Eren, M.; Vaughan, U.E.; Schleimer, R.P.; Cho, S.H. A Plasminogen Activator Inhibitor-1 Inhibitor Reduces Airway Remodeling in a Murine Model of Chronic Asthma. Am. J. Respir. Cell Mol. Biol. 2012, 46, 842–846. [Google Scholar] [CrossRef]
- Baker, J.B.; Low, D.A.; Simmer, R.L.; Cunningham, D.D. Protease-nexin: A cellular component that links thrombin and plasminogen activator and mediates their binding to cells. Cell 1980, 21, 37–45. [Google Scholar] [CrossRef]
- Scott, R.W.; Bergman, B.L.; Bajpai, A.; Hersh, R.T.; Rodriguez, H.; Jones, B.N.; Barreda, C.; Watts, S.; Baker, J.B. Protease nexin. Properties and a modified purification procedure. J. Biol. Chem. 1985, 260, 7029–7034. [Google Scholar] [CrossRef]
- Houenou, L.J.; Turner, P.L.; Li, L.; Oppenheim, R.W.; Festoff, B.W. A serine protease inhibitor, protease nexin I, rescues motoneurons from naturally occurring and axotomy-induced cell death. Proc. Natl. Acad. Sci. USA 1995, 92, 895–899. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, P.; Ho-Tin-Noé, B.; Vranckx, R.; Bouton, M.-C.; Meilhac, O.; Lijnen, H.R.; Guillin, M.-C.; Michel, J.-B.; Anglés-Cano, E. Protease Nexin-1 Inhibits Plasminogen Activation-induced Apoptosis of Adherent Cells. J. Biol. Chem. 2004, 279, 10346–10356. [Google Scholar] [CrossRef] [Green Version]
- DeMeo, D.L.; Mariani, T.J.; Lange, C.; Srisuma, S.; Litonjua, A.A.; Celedón, J.C.; Lake, S.L.; Reilly, J.J.; Chapman, H.A.; Mecham, B.H.; et al. The SERPINE2 Gene Is Associated with Chronic Obstructive Pulmonary Disease. Am. J. Hum. Genet. 2006, 78, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Chappell, S.; Daly, L.; Morgan, K.; Baranes, T.G.; Roca, J.; Rabinovich, R.; Millar, A.; Donnelly, S.C.; Keatings, V.; MacNee, W.; et al. The SERPINE2 Gene and Chronic Obstructive Pulmonary Disease. Am. J. Hum. Genet. 2006, 79, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Warren, L.; Aponte, J.; Gulsvik, A.; Bakke, P.; Anderson, W.H.; Lomas, D.A.; Silverman, E.K.; Pillai, S.G. The International COPD Genetics Network (ICGN) Investigators TheSERPINE2Gene Is Associated with Chronic Obstructive Pulmonary Disease in Two Large Populations. Am. J. Respir. Crit. Care Med. 2007, 176, 167–173. [Google Scholar] [CrossRef]
- Cha, S.I.; Kang, H.-G.; Choi, J.E.; Kim, M.J.; Park, J.; Lee, W.K.; Kim, C.H.; Jung, T.H.; Park, J.Y. SERPINE2 Polymorphisms and Chronic Obstructive Pulmonary Disease. J. Korean Med. Sci. 2009, 24, 1119–1125. [Google Scholar] [CrossRef]
- Cosgrove, G.P.; Brown, K.K.; Schiemann, W.P.; Serls, A.E.; Parr, J.E.; Geraci, M.W.; Schwarz, M.I.; Cool, C.D.; Worthen, G.S. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: A role in aberrant angiogenesis. Am. J. Respir. Crit. Care Med. 2004, 170, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.; Millar, A. Vascular remodelling in the pathogenesis of idiopathic pulmonary fibrosis. QJM Int. J. Med. 2014, 107, 515–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wewers, M.D.; Casolaro, M.A.; Sellers, S.E.; Swayze, S.C.; McPhaul, K.M.; Wittes, J.T.; Crystal, R.G. Replacement Therapy for Alpha1-Antitrypsin Deficiency Associated with Emphysema. N. Engl. J. Med. 1987, 316, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.L.; Downey, D.; Bilton, D.; Keogan, M.T.; Edgar, J.; Elborn, J.S. on behalf of the Recombinant AAT CF Study Team Safety and Efficacy of Recombinant Alpha1-Antitrypsin Therapy in Cystic Fibrosis. Pediatr. Pulmonol. 2006, 41, 177–183. [Google Scholar] [CrossRef]
- McElvaney, N.; Hubbard, R.; Birrer, P.; Crystal, R.; Chernick, M.; Frank, M.; Caplan, D. Aerosol α1 -antitrypsin treatment for cystic fibrosis. Lancet 1991, 337, 392–394. [Google Scholar] [CrossRef]
- Gaggar, A.; Chen, J.; Chmiel, J.F.; Dorkin, H.L.; Flume, P.A.; Griffin, R.; Nichols, D.; Donaldson, S.H. Inhaled alpha 1 -proteinase inhibitor therapy in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Gallus, A. Replacement Therapy in Antithrombin III Deficiency. Transfus. Med. Rev. 1989, 3, 253–263. [Google Scholar] [CrossRef]
- Hada, K.; Isshiki, K.; Matsuda, S.; Yuasa, K.; Tsuji, A. Engineering of α1-antitrypsin variants with improved specificity for the proprotein convertase furin using site-directed random mutagenesis. Prot. Eng. Des. Sel. 2012, 26, 123–131. [Google Scholar] [CrossRef]
- Byrne, S.M.; Aucher, A.; Alyahya, S.; Elder, M.; Olson, S.T.; Davis, D.M.; Ashton-Rickardt, P.G. Cathepsin B controls the persistence of memory CD8+ T lymphocytes. J. Immunol. 2012, 189, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Byrne, S.M.; Rainville, N.; Su, S.; Jachimowicz, E.; Aucher, A.; Davis, D.M.; Ashton-Rickardt, P.G.; Wojchowski, D.M. Brief report: Serpin Spi2A as a novel modulator of hematopoietic progenitor cell formation. Stem Cells 2014, 32, 2550–2556. [Google Scholar] [CrossRef] [Green Version]
- Safdar, H.; Cheung, K.L.; Salvatori, D.; Versteeg, H.H.; Laghmani, E.H.; Wagenaar, G.T.M.; Reitsma, P.H.; van Vlijmen, B.J.M. Acute and severe coagulopathy in adult mice following silencing of hepatic antithrombin and protein C production. Blood 2013, 121, 4413–4416. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.; Pham, C.T.N.; Benarafa, C. SerpinB1 is critical for neutrophil survival through cell-autonomous inhibition of cathepsin G. Blood 2013, 121, 3900–3907. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhang, G.; Lu, Z.; Geurts, A.M.; Usa, K.; Jacob, H.J.; Cowley, A.W.; Wang, N.; Liang, M. Antithrombin III/SerpinC1 insufficiency exacerbates renal ischemia/reperfusion injury. Kidney Int. 2015, 88, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Zechmeister-Machhart, M.; Hufnagl, P.; Uhrin, P.; Xu, J.; Geiger, M.; Binder, B.R. Molecular cloning and tissue distribution of mouse protein C inhibitor (PCI). Immunopharmacology 1996, 32, 96–98. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelly-Robinson, G.A.; Reihill, J.A.; Lundy, F.T.; McGarvey, L.P.; Lockhart, J.C.; Litherland, G.J.; Thornbury, K.D.; Martin, S.L. The Serpin Superfamily and Their Role in the Regulation and Dysfunction of Serine Protease Activity in COPD and Other Chronic Lung Diseases. Int. J. Mol. Sci. 2021, 22, 6351. https://doi.org/10.3390/ijms22126351
Kelly-Robinson GA, Reihill JA, Lundy FT, McGarvey LP, Lockhart JC, Litherland GJ, Thornbury KD, Martin SL. The Serpin Superfamily and Their Role in the Regulation and Dysfunction of Serine Protease Activity in COPD and Other Chronic Lung Diseases. International Journal of Molecular Sciences. 2021; 22(12):6351. https://doi.org/10.3390/ijms22126351
Chicago/Turabian StyleKelly-Robinson, Gillian A., James A. Reihill, Fionnuala T. Lundy, Lorcan P. McGarvey, John C. Lockhart, Gary J. Litherland, Keith D. Thornbury, and S. Lorraine Martin. 2021. "The Serpin Superfamily and Their Role in the Regulation and Dysfunction of Serine Protease Activity in COPD and Other Chronic Lung Diseases" International Journal of Molecular Sciences 22, no. 12: 6351. https://doi.org/10.3390/ijms22126351
APA StyleKelly-Robinson, G. A., Reihill, J. A., Lundy, F. T., McGarvey, L. P., Lockhart, J. C., Litherland, G. J., Thornbury, K. D., & Martin, S. L. (2021). The Serpin Superfamily and Their Role in the Regulation and Dysfunction of Serine Protease Activity in COPD and Other Chronic Lung Diseases. International Journal of Molecular Sciences, 22(12), 6351. https://doi.org/10.3390/ijms22126351