
Let’s Talk about Placental Sex, Baby: Understanding Mechanisms That Drive Female- and Male-Specific Fetal Growth and Developmental Outcomes

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. From Blastocyst to Neonate: Ontogeny of Sex-Specific Intrauterine Growth
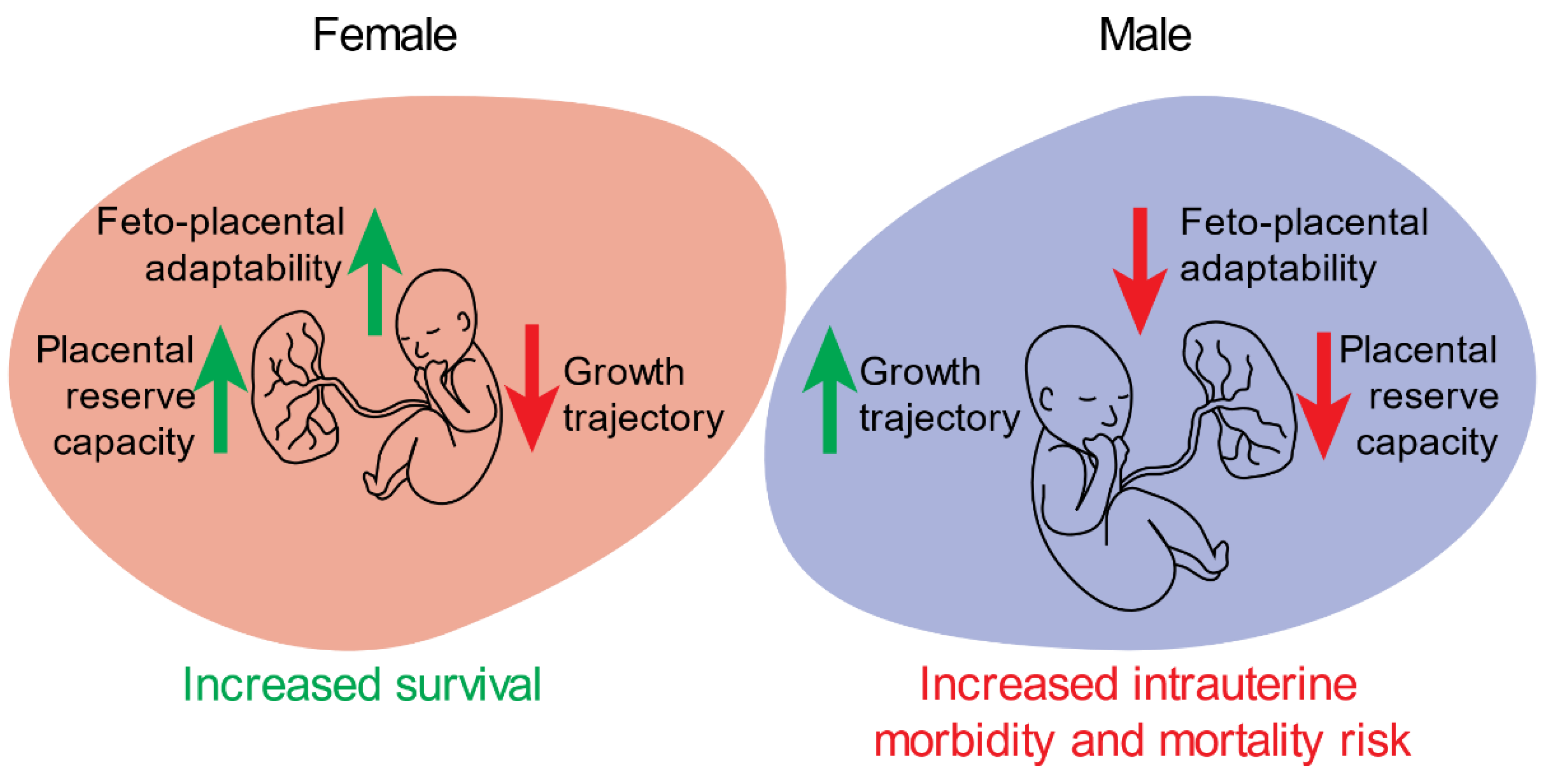
3. Sex-Specific Differences in Placental Function and Structure Contribute to Adverse Intrauterine and Neonatal Outcomes
4. Changes to the Feto-Placental Hormonal Milieu Contribute to Sex-Specific Intrauterine Growth Outcomes
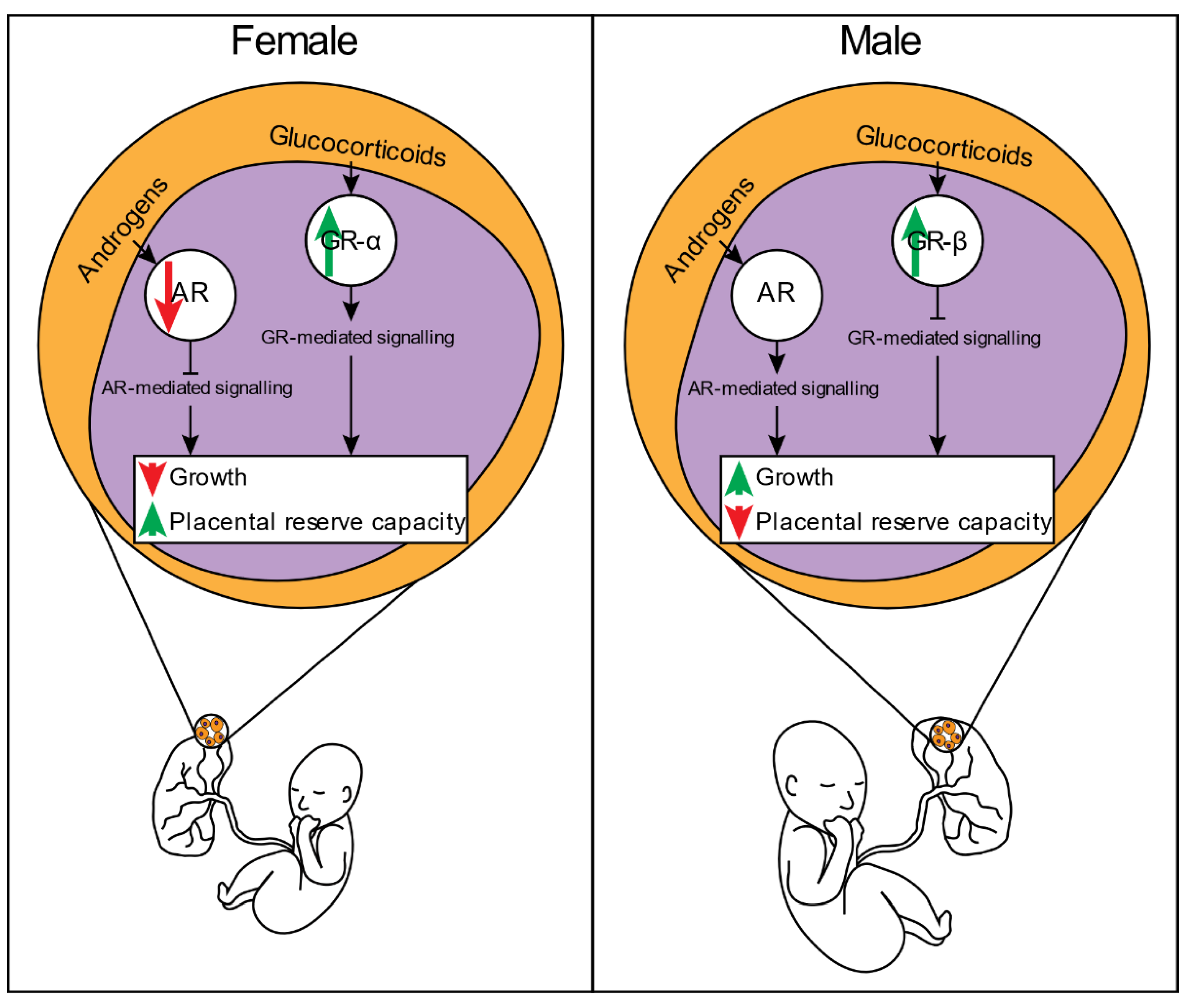
4.1. Placental Glucocorticoid Signalling and Its Impact on Fetal Growth and Developmental Outcomes
4.2. Placental Androgen Signalling: A Potential Regulator of Sex-Specific Intrauterine Growth and Developmental Outcomes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pedersen, J.F. Ultrasound evidence of sexual difference in fetal size in first trimester. Br. Med. J. 1980, 281, 1253. [Google Scholar] [CrossRef] [Green Version]
- Mittwoch, U. Blastocysts prepare for the race to be male. Hum. Reprod. 1993, 8, 1550–1555. [Google Scholar] [CrossRef]
- Ray, P.F.; Conaghan, J.; Winston, R.M.; Handyside, A.H. Increased number of cells and metabolic activity in male human preimplantation embryos following in vitro fertilization. J. Reprod. Fertil. 1995, 104, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, C.L.D.; Gardosi, J.; Baldwin, C.; Francis, A.; Dekker, G.A.; van Geijn, H.P. Fetal weight gain in a serially scanned high-risk population. Ultrasound Obstet. Gynecol. 1998, 11, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Álvarez, P.; Rizos, D.; Rath, D.; Lonergan, P.; Gutierrez-Adan, A. Epigenetic differences between male and female bovine blastocysts produced in vitro. Physiol. Genom. 2008, 32, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Clifton, V.L. Review: Sex and the human placenta: Mediating differential strategies of fetal growth and survival. Placenta 2010, 31, S33–S39. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.A.; Moritz, K.M.; Walker, D.W.; Dickinson, H. Sexually dimorphic placental development throughout gestation in the spiny mouse (Acomys cahirinus). Placenta 2013, 34, 119–126. [Google Scholar] [CrossRef]
- Brown, Z.A.; Schalekamp-Timmermans, S.; Tiemeier, H.W.; Hofman, A.; Jaddoe, V.W.V.; Steegers, E.A.P. Fetal sex specific differences in human placentation: A prospective cohort study. Placenta 2014, 35, 359–364. [Google Scholar] [CrossRef]
- Rosenfeld, C.S. Sex-Specific Placental Responses in Fetal Development. Endocrinology 2015, 156, 3422–3434. [Google Scholar] [CrossRef] [Green Version]
- Broere-Brown, Z.A.; Baan, E.; Schalekamp-Timmermans, S.; Verburg, B.O.; Jaddoe, V.W.V.; Steegers, E.A.P. Sex-specific differences in fetal and infant growth patterns: A prospective population-based cohort study. Biol. Sex Differ. 2016, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Kalisch-Smith, J.I.; Simmons, D.G.; Pantaleon, M.; Moritz, K.M. Sex differences in rat placental development: From pre-implantation to late gestation. Biol. Sex Differ. 2017, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Galjaard, S.; Ameye, L.; Lees, C.C.; Pexsters, A.; Bourne, T.; Timmerman, D.; Devlieger, R. Sex differences in fetal growth and immediate birth outcomes in a low-risk Caucasian population. Biol. Sex Differ. 2019, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Saoi, M.; Kennedy, K.M.; Gohir, W.; Sloboda, D.M.; Britz-McKibbin, P. Placental Metabolomics for Assessment of Sex-specific Differences in Fetal Development During Normal Gestation. Sci. Rep. 2020, 10, 9399. [Google Scholar] [CrossRef]
- Clarke, J.; Price, R., XVII. Observations on some causes of the excess of the mortality of males above that of females. By Joseph Clarke, M.D. Physician to the Lying-in Hospital at Dublin. Communicated by the Rev. Richard Price, D.D.F.R.S. in a letter to Charles Blagden, M.D. Sec. R. S. Philos. Trans. R. Soc. Lond. 1786, 76, 349–362. [Google Scholar] [CrossRef]
- Eriksson, J.G.; Kajantie, E.; Osmond, C.; Thornburg, K.; Barker, D.J. Boys live dangerously in the womb. Am. J. Hum. Biol. 2010, 22, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Murphy, V.E.; Gibson, P.; Talbot, P.I.; Clifton, V.L. Severe Asthma Exacerbations During Pregnancy. Obstet. Gynecol. 2005, 106, 9. [Google Scholar] [CrossRef] [PubMed]
- Murphy, V.E.; Clifton, V.L.; Gibson, P.G. Asthma exacerbations during pregnancy: Incidence and association with adverse pregnancy outcomes. Thorax 2006, 61, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, V.E.; Gibson, P.G.; Giles, W.B.; Zakar, T.; Smith, R.; Bisits, A.M.; Kessell, C.G.; Clifton, V.L. Maternal asthma is associated with reduced female fetal growth. Am. J. Respir. Crit. Care Med. 2003, 168, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.J.; Clifton, V.L.; Wright, I.M. Neonates born to mothers with preeclampsia exhibit sex-specific alterations in microvascular function. Pediatr. Res. 2009, 65, 292–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trudell, A.S.; Cahill, A.G.; Tuuli, M.G.; Macones, G.A.; Odibo, A.O. Stillbirth and the small fetus: Use of a sex-specific versus a non-sex-specific growth standard. J. Perinatol. 2015, 35, 566–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broere-Brown, Z.A.; Adank, M.C.; Benschop, L.; Tielemans, M.; Muka, T.; Gonçalves, R.; Bramer, W.M.; Schoufour, J.D.; Voortman, T.; Steegers, E.A.P.; et al. Fetal sex and maternal pregnancy outcomes: A systematic review and meta-analysis. Biol. Sex Differ. 2020, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Otte, A.P.; Allis, C.D.; Reinberg, D.; Heard, E. Epigenetic Dynamics of Imprinted X Inactivation During Early Mouse Development. Science 2004, 303, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Alvarez, P.; Rizos, D.; Rath, D.; Lonergan, P.; Gutierrez-Adan, A. Sex determines the expression level of one third of the actively expressed genes in bovine blastocysts. Proc. Natl. Acad. Sci. USA 2010, 107, 3394–3399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffin, G.J.; Rieger, D.; Betteridge, K.J.; Yadav, B.R.; King, W.A. Glucose and glutamine metabolism in pre-attachment cattle embryos in relation to sex and stage of development. J. Reprod. Fertil. 1991, 93, 125–132. [Google Scholar] [CrossRef]
- Gonzalez, T.L.; Sun, T.; Koeppel, A.F.; Lee, B.; Wang, E.T.; Farber, C.R.; Rich, S.S.; Sundheimer, L.W.; Buttle, R.A.; Chen, Y.I.; et al. Sex differences in the late first trimester human placenta transcriptome. Biol. Sex Differ. 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Gonzalez, T.L.; Deng, N.; DiPentino, R.; Clark, E.L.; Lee, B.; Tang, J.; Wang, Y.; Stripp, B.R.; Yao, C.; et al. Sexually Dimorphic Crosstalk at the Maternal-Fetal Interface. J. Clin. Endocrinol. Metab. 2020, 105, e4831–e4847. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.F.; Mantoni, M. Difference in fetal size in the first trimester. Br. Med. J. 1985, 291, 1278. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, B.A.; Moritz, K.M.; Roberts, C.T.; Walker, D.W.; Dickinson, H. The Placental Response to Excess Maternal Glucocorticoid Exposure Differs Between the Male and Female Conceptus in Spiny Mice1. Biol. Reprod. 2011, 85, 1040–1047. [Google Scholar] [CrossRef]
- Woods, L.; Perez-Garcia, V.; Hemberger, M. Regulation of Placental Development and Its Impact on Fetal Growth—New Insights From Mouse Models. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Tunster, S.J.; Creeth, H.D.J.; John, R.M. The imprinted Phlda2 gene modulates a major endocrine compartment of the placenta to regulate placental demands for maternal resources. Dev. Biol. 2016, 409, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Cuffe, J.S.M.; Walton, S.L.; Singh, R.R.; Spiers, J.G.; Bielefeldt-Ohmann, H.; Wilkinson, L.; Little, M.H.; Moritz, K.M. Mid- to late term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways and nutrient transporters in a sex-specific manner. J. Physiol. 2014, 592, 3127–3141. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Zhang, X.; Sieli, P.T.; Falduto, M.T.; Torres, K.E.; Rosenfeld, C.S. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc. Natl. Acad. Sci. USA 2010, 107, 5557–5562. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-J.; Huang, L.-T.; Tsai, C.-C.; Sheen, J.-M.; Tiao, M.-M.; Yu, H.-R.; Lin, I.C.; Tain, Y.-L. Maternal high-fat diet sex-specifically alters placental morphology and transcriptome in rats: Assessment by next-generation sequencing. Placenta 2019, 78, 44–53. [Google Scholar] [CrossRef]
- Adu-Gyamfi, E.A.; Czika, A.; Gorleku, P.N.; Ullah, A.; Panhwar, Z.; Ruan, L.L.; Ding, Y.B.; Wang, Y.X. The Involvement of Cell Adhesion Molecules, Tight Junctions, and Gap Junctions in Human Placentation. Reprod. Sci. 2021, 28, 305–320. [Google Scholar] [CrossRef]
- Buckberry, S.; Bianco-Miotto, T.; Bent, S.J.; Dekker, G.A.; Roberts, C.T. Integrative transcriptome meta-analysis reveals widespread sex-biased gene expression at the human fetal-maternal interface. Mol. Hum. Reprod. 2014, 20, 810–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, K.S.; Gilchrist, R.L.; Koo, Y.B.; Ji, I.; Ji, T.H. Gene, interaction, signal generation, signal divergence and signal transduction of the LH/CG receptor. Int. J. Gynaecol. Obstet. 1998, 60 (Suppl. S1), S9–S20. [Google Scholar] [CrossRef]
- Depoix, C.; Tee, M.K.; Taylor, R.N. Molecular regulation of human placental growth factor (PlGF) gene expression in placental villi and trophoblast cells is mediated via the protein kinase a pathway. Reprod. Sci. 2011, 18, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Herr, F.; Baal, N.; Reisinger, K.; Lorenz, A.; McKinnon, T.; Preissner, K.T.; Zygmunt, M. hCG in the Regulation of Placental Angiogenesis. Results of an In Vitro Study. Placenta 2007, 28, S85–S93. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Smeester, L.; Bommarito, P.A.; Grace, M.R.; Boggess, K.; Kuban, K.; Karagas, M.R.; Marsit, C.J.; O’Shea, T.M.; Fry, R.C. Sexual epigenetic dimorphism in the human placenta: Implications for susceptibility during the prenatal period. Epigenomics 2017, 9, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Johnson, M.D.; Dopierala, J.; Gaccioli, F.; Sovio, U.; Constância, M.; Smith, G.C.S.; Charnock-Jones, D.S. Genome-wide oxidative bisulfite sequencing identifies sex-specific methylation differences in the human placenta. Epigenetics 2018, 13, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.E.; Muench, K.L.; Robinson, B.G.; Wang, A.; Palmer, T.D.; Winn, V.D. Examining Sex Differences in the Human Placental Transcriptome During the First Fetal Androgen Peak. Reprod. Sci. 2021, 28, 801–818. [Google Scholar] [CrossRef] [PubMed]
- Meakin, A.S.; Saif, Z.; Seedat, N.; Clifton, V.L. The impact of maternal asthma during pregnancy on fetal growth and development: A review. Expert Rev. Respir. Med. 2020, 14, 1207–1216. [Google Scholar] [CrossRef]
- Paranavitana, L.; Walker, M.; Chandran, A.R.; Milligan, N.; Shinar, S.; Whitehead, C.L.; Hobson, S.R.; Serghides, L.; Parks, W.T.; Baschat, A.A.; et al. Sex differences in uterine artery Doppler during gestation in pregnancies complicated by placental dysfunction. Biol. Sex Differ. 2021, 12, 19. [Google Scholar] [CrossRef]
- Broere-Brown, Z.A.; Schalekamp-Timmermans, S.; Hofman, A.; Jaddoe, V.; Steegers, E. Fetal sex dependency of maternal vascular adaptation to pregnancy: A prospective population-based cohort study. BJOG Int. J. Obstet. Gynaecol. 2016, 123, 1087–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andraweera, P.H.; Dekker, G.A.; Roberts, C.T. The vascular endothelial growth factor family in adverse pregnancy outcomes. Hum. Reprod. Update 2012, 18, 436–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osei-Kumah, A.; Smith, R.; Jurisica, I.; Caniggia, I.; Clifton, V.L. Sex-specific differences in placental global gene expression in pregnancies complicated by asthma. Placenta 2011, 32, 570–578. [Google Scholar] [CrossRef]
- Mayhew, T.M.; Jenkins, H.; Todd, B.; Clifton, V.L. Maternal asthma and placental morphometry: Effects of severity, treatment and fetal sex. Placenta 2008, 29, 366–373. [Google Scholar] [CrossRef]
- Ahmed, A.; Dunk, C.; Ahmad, S.; Khaliq, A. Regulation of placental vascular endothelial growth factor (VEGF) and placenta growth factor (PIGF) and soluble Flt-1 by oxygen—A review. Placenta 2000, 21, S16–S24. [Google Scholar] [CrossRef]
- Khaliq, A.; Dunk, C.; Jiang, J.; Shams, M.; Li, X.F.; Acevedo, C.; Weich, H.; Whittle, M.; Ahmed, A. Hypoxia down-regulates placenta growth factor, whereas fetal growth restriction up-regulates placenta growth factor expression: Molecular evidence for “placental hyperoxia” in intrauterine growth restriction. Lab. Investig. J. Tech. Methods Pathol. 1999, 79, 151–170. [Google Scholar]
- Cao, Y.; Chen, H.; Zhou, L.; Chiang, M.K.; Anand-Apte, B.; Weatherbee, J.A.; Wang, Y.; Fang, F.; Flanagan, J.G.; Tsang, M.L. Heterodimers of placenta growth factor/vascular endothelial growth factor. Endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. J. Biol. Chem. 1996, 271, 3154–3162. [Google Scholar] [CrossRef] [Green Version]
- Procianoy, R.S.; Hentges, C.R.; Silveira, R.C. Vascular Endothelial Growth Factor/Placental Growth Factor Heterodimer Levels in Preterm Infants with Bronchopulmonary Dysplasia. Am. J. Perinatol. 2016, 33, 480–485. [Google Scholar] [CrossRef]
- Meakin, A.S.; Saif, Z.; Tuck, A.R.; Clifton, V.L. Human placental androgen receptor variants: Potential regulators of male fetal growth. Placenta 2019, 80, 18–26. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Maloyan, A.; Myatt, L. Evidence of sexual dimorphism in the placental function with severe preeclampsia. Placenta 2013, 34, 1183–1189. [Google Scholar] [CrossRef] [Green Version]
- Reddy, T.E.; Pauli, F.; Sprouse, R.O.; Neff, N.F.; Newberry, K.M.; Garabedian, M.J.; Myers, R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009, 19, 2163–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- Guichard, A.; Humbert, P.; Tissot, M.; Muret, P.; Courderot-Masuyer, C.; Viennet, C. Effects of topical corticosteroids on cell proliferation, cell cycle progression and apoptosis: In vitro comparison on HaCaT. Int. J. Pharm. 2015, 479, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.D. Mechanisms of glucocorticoid inhibition of growth. Kidney Int. 1978, 14, 330–333. [Google Scholar] [CrossRef] [Green Version]
- Lu, J. The anti-proliferation mechanism of glucocorticoid mediated by glucocorticoid receptor-regulating gene expression. Pathophysiology 2009, 16, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Mandl, M.; Ghaffari-Tabrizi, N.; Haas, J.; Nohammer, G.; Desoye, G. Differential glucocorticoid effects on proliferation and invasion of human trophoblast cell lines. Reproduction 2006, 132, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.P.; Sandman, C.A. The Timing of Prenatal Exposure to Maternal Cortisol and Psychosocial Stress is Associated with Human Infant Cognitive Development. Child. Dev. 2010, 81, 131–148. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.J.; Waddell, B.J. P-glycoprotein restricts access of cortisol and dexamethasone to the glucocorticoid receptor in placental BeWo cells. Endocrinology 2006, 147, 5147–5152. [Google Scholar] [CrossRef] [Green Version]
- Saif, Z.; Hodyl, N.A.; Stark, M.J.; Fuller, P.J.; Cole, T.; Lu, N.; Clifton, V.L. Expression of eight glucocorticoid receptor isoforms in the human preterm placenta vary with fetal sex and birthweight. Placenta 2015, 36, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifton, V.L.; Cuffe, J.; Moritz, K.M.; Cole, T.J.; Fuller, P.J.; Lu, N.Z.; Kumar, S.; Chong, S.; Saif, Z. Review: The role of multiple placental glucocorticoid receptor isoforms in adapting to the maternal environment and regulating fetal growth. Placenta 2017, 54, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Nixon, M.; Mackenzie, S.D.; Taylor, A.I.; Homer, N.Z.; Livingstone, D.E.; Mouras, R.; Morgan, R.A.; Mole, D.J.; Stimson, R.H.; Reynolds, R.M.; et al. ABCC1 confers tissue-specific sensitivity to cortisol versus corticosterone: A rationale for safer glucocorticoid replacement therapy. Sci. Transl. Med. 2016, 8, 352ra109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharm. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Wieczorek, A.; Perani, C.V.; Nixon, M.; Constancia, M.; Sandovici, I.; Zazara, D.E.; Leone, G.; Zhang, M.-Z.; Arck, P.C.; Solano, M.E. Sex-specific regulation of stress-induced fetal glucocorticoid surge by the mouse placenta. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E109–E120. [Google Scholar] [CrossRef] [PubMed]
- Hodyl, N.A.; Stark, M.J.; Butler, M.; Clifton, V.L. Placental P-glycoprotein is unaffected by timing of antenatal glucocorticoid therapy but reduced in SGA preterm infants. Placenta 2013, 34, 325–330. [Google Scholar] [CrossRef]
- Wyrwoll, C.S.; Seckl, J.R.; Holmes, M.C. Altered Placental Function of 11β-Hydroxysteroid Dehydrogenase 2 Knockout Mice. Endocrinology 2009, 150, 1287–1293. [Google Scholar] [CrossRef] [Green Version]
- Cuffe, J.S.M.; O’Sullivan, L.; Simmons, D.G.; Anderson, S.T.; Moritz, K.M. Maternal Corticosterone Exposure in the Mouse Has Sex-Specific Effects on Placental Growth and mRNA Expression. Endocrinology 2012, 153, 5500–5511. [Google Scholar] [CrossRef] [Green Version]
- Mericq, V.; Medina, P.; Kakarieka, E.; Márquez, L.; Johnson, M.C.; Iñiguez, G. Differences in expression and activity of 11β-hydroxysteroid dehydrogenase type 1 and 2 in human placentas of term pregnancies according to birth weight and gender. Eur. J. Endocrinol. 2009, 161, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Stark, M.J.; Wright, I.M.; Clifton, V.L. Sex-specific alterations in placental 11beta-hydroxysteroid dehydrogenase 2 activity and early postnatal clinical course following antenatal betamethasone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R510–R514. [Google Scholar] [CrossRef]
- Cole, T.J.; Blendy, J.A.; Monaghan, A.P.; Krieglstein, K.; Schmid, W.; Aquzzi, A.; Fantuzzi, G.; Hummler, E.; Unsicker, K.; Schütz, G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rog-Zielinska, E.A.; Thomson, A.; Kenyon, C.J.; Brownstein, D.G.; Moran, C.M.; Szumska, D.; Michailidou, Z.; Richardson, J.; Owen, E.; Watt, A.; et al. Glucocorticoid receptor is required for foetal heart maturation. Hum. Mol. Genet. 2013, 22, 3269–3282. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, D.P.; Mark, P.J.; Waddell, B.J. Glucocorticoids Prevent the Normal Increase in Placental Vascular Endothelial Growth Factor Expression and Placental Vascularity during Late Pregnancy in the Rat. Endocrinology 2006, 147, 5568–5574. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Fang, M.; Zhuang, S.; Qiao, Y.; Huang, W.; Gong, Q.; Xu, D.; Zhang, Y.; Wang, H. Prenatal dexamethasone exposure exerts sex-specific effect on placental oxygen and nutrient transport ascribed to the differential expression of IGF2. Ann. Transl. Med. 2020, 8, 233. [Google Scholar] [CrossRef]
- Cuffe, J.S.M.; Dickinson, H.; Simmons, D.G.; Moritz, K.M. Sex specific changes in placental growth and MAPK following short term maternal dexamethasone exposure in the mouse. Placenta 2011, 32, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Cuffe, J.S.M.; Saif, Z.; Perkins, A.V.; Moritz, K.M.; Clifton, V.L. Dexamethasone and sex regulate placental glucocorticoid receptor isoforms in mice. J. Endocrinol. 2017, 234, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Ain, R.; Canham, L.N.; Soares, M.J. Dexamethasone-induced intrauterine growth restriction impacts the placental prolactin family, insulin-like growth factor-II and the Akt signaling pathway. J. Endocrinol. 2005, 185, 253–263. [Google Scholar] [CrossRef]
- Fowden, A.L. The insulin-like growth factors and feto-placental growth. Placenta 2003, 24, 803–812. [Google Scholar] [CrossRef]
- Ozmen, A.; Unek, G.; Kipmen-Korgun, D.; Mendilcioglu, I.; Sanhal, C.; Sakıncı, M.; Korgun, E.T. Glucocorticoid effects on angiogenesis are associated with mTOR pathway activity. Biotech. Histochem. 2016, 91, 296–306. [Google Scholar] [CrossRef]
- Morrison, J.L. Sheep models of intrauterine growth restriction: Fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008, 35, 730–743. [Google Scholar] [CrossRef]
- Hahn, T.; Barth, S.; Graf, R.; Engelmann, M.; Beslagic, D.; Reul, J.M.H.M.; Holsboer, F.; Dohr, G.; Desoye, G. Placental Glucose Transporter Expression Is Regulated by Glucocorticoids1. J. Clin. Endocrinol. Metab. 1999, 84, 1445–1452. [Google Scholar] [CrossRef] [Green Version]
- Menconi, M.; Gonnella, P.; Petkova, V.; Lecker, S.; Hasselgren, P.O. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J. Cell. Biochem. 2008, 105, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Yu, I.T.; Lee, S.-H.; Lee, Y.-S.; Son, H. Differential effects of corticosterone and dexamethasone on hippocampal neurogenesis in vitro. Biochem. Biophys. Res. Commun. 2004, 317, 484–490. [Google Scholar] [CrossRef]
- Ishiguro, H.; Kawahara, T.; Zheng, Y.; Kashiwagi, E.; Li, Y.; Miyamoto, H. Differential regulation of bladder cancer growth by various glucocorticoids: Corticosterone and prednisone inhibit cell invasion without promoting cell proliferation or reducing cisplatin cytotoxicity. Cancer Chemother. Pharmacol. 2014, 74, 249–255. [Google Scholar] [CrossRef]
- Hutter, S.; Hepp, P.; Hofmann, S.; Kuhn, C.; Messner, J.; Andergassen, U.; Mayr, D.; Emilia Solano, M.; Obermeier, V.; Mahner, S.; et al. Glucocorticoid receptors α and β are modulated sex specifically in human placentas of intrauterine growth restriction (IUGR). Arch. Gynecol. Obstet. 2019, 300, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Saif, Z.; Hodyl, N.A.; Hobbs, E.; Tuck, A.R.; Butler, M.S.; Osei-Kumah, A.; Clifton, V.L. The human placenta expresses multiple glucocorticoid receptor isoforms that are altered by fetal sex, growth restriction and maternal asthma. Placenta 2014, 35, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Clifton, V.L.; McDonald, M.; Morrison, J.L.; Holman, S.L.; Lock, M.C.; Saif, Z.; Meakin, A.; Wooldridge, A.L.; Gatford, K.L.; Wallace, M.J.; et al. Placental glucocorticoid receptor isoforms in a sheep model of maternal allergic asthma. Placenta 2019, 83, 33–36. [Google Scholar] [CrossRef]
- Leung, D.Y.M.; Hamid, Q.; Vottero, A.; Szefler, S.J.; Surs, W.; Minshall, E.; CHrousos, G.P.; Klemm, D.J. Association of Glucocorticoid Insensitivity with Increased Expression of Glucocorticoid Receptor β. J. Exp. Med. 1997, 186, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Jewell, C.M.; Yudt, M.R.; Bofetiado, D.M.; Cidlowski, J.A. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificty and mechanisms of action. J. Biol. Chem. 1999, 274, 10. [Google Scholar] [CrossRef] [Green Version]
- Yudt, M.R.; Jewell, C.M.; Bienstock, R.J.; Cidlowski, J.A. Molecular Origins for the Dominant Negative Function of Human Glucocorticoid Receptor Beta. Mol. Cell. Biol. 2003, 23, 4319–4330. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D.; Peck, B.; Shek, E.; Stroup, S.; Hinson, J.; Arthur, S.; Marino, J.S. Overexpression of Glucocorticoid Receptor beta Enhances Myogenesis and Reduces Catabolic Gene Expression. Int. J. Mol. Sci. 2016, 17, 232. [Google Scholar] [CrossRef] [Green Version]
- Meakin, A.S.; Clifton, V.L. Review: Understanding the role of androgens and placental AR variants: Insight into steroid-dependent fetal-placental growth and development. Placenta 2019. [Google Scholar] [CrossRef]
- Makieva, S.; Saunders, P.T.; Norman, J.E. Androgens in pregnancy: Roles in parturition. Hum. Reprod. Update 2014, 20, 542–559. [Google Scholar] [CrossRef] [Green Version]
- Maccoby, E.E.; Doering, C.H.; Jacklin, C.N.; Kraemer, H. Concentrations of sex hormones in umbilical-cord blood: Their relation to sex and birth order of infants. Child. Dev. 1979, 50, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Gitau, R.; Adams, D.; Fisk, N.M.; Glover, V. Fetal plasma testosterone correlates positively with cortisol. Arch. Dis. Child. Fetal. Neonatal Ed. 2005, 90, F166–F169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shaughnessy, P.J.; Antignac, J.P.; Le Bizec, B.; Morvan, M.L.; Svechnikov, K.; Söder, O.; Savchuk, I.; Monteiro, A.; Soffientini, U.; Johnston, Z.C.; et al. Alternative (backdoor) androgen production and masculinization in the human fetus. PLoS Biol. 2019, 17, e3000002. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, E.D.; Robb, V.A.; Pepe, G.J. Regulation of placental vascular endothelial growth/permeability factor expression and angiogenesis by estrogen during early baboon pregnancy. J. Clin. Endocrinol. Metab. 2004, 89, 5803–5809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robb, V.A.; Pepe, G.J.; Albrecht, E.D. Acute temporal regulation of placental vascular endothelial growth/permeability factor expression in baboons by estrogen. Biol. Reprod. 2004, 71, 1694–1698. [Google Scholar] [CrossRef]
- Das, A.; Mantena, S.R.; Kannan, A.; Evans, D.B.; Bagchi, M.K.; Bagchi, I.C. De novo synthesis of estrogen in pregnant uterus is critical for stromal decidualization and angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12542–12547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, J.J.; Nicholson, C.; Sweeney, M.; Charnock, J.C.; Robson, S.C.; Westwood, M.; Taggart, M.J. Human uterine and placental arteries exhibit tissue-specific acute responses to 17β-estradiol and estrogen-receptor-specific agonists. Mol. Hum. Reprod. 2014, 20, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Escobar, J.C.; Patel, S.S.; Beshay, V.E.; Suzuki, T.; Carr, B.R. The human placenta expresses CYP17 and generates androgens de novo. J. Clin. Endocrinol. Metab. 2011, 96, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.T.; Hirst, J.J.; Stark, M.; Wright, I.M.; Palliser, H.K.; Hodyl, N.; Clifton, V.L. Changes in human placental 5alpha-reductase isoenzyme expression with advancing gestation: Effects of fetal sex and glucocorticoid exposure. Reprod. Fertil. Dev. 2009, 21, 599–607. [Google Scholar] [CrossRef]
- Lek, N.; Miles, H.; Bunch, T.; Pilfold-Wilkie, V.; Tadokoro-Cuccaro, R.; Davies, J.; Ong, K.K.; Hughes, I.A. Low frequency of androgen receptor gene mutations in 46 XY DSD, and fetal growth restriction. Arch. Dis. Child. 2014, 99, 358–361. [Google Scholar] [CrossRef] [PubMed]
- de Zegher, F.; Francois, I.; Boehmer, A.L.; Saggese, G.; Muller, J.; Hiort, O.; Sultan, C.; Clayton, P.; Brauner, R.; Cacciari, E.; et al. Androgens and fetal growth. Horm. Res. 1998, 50, 243–244. [Google Scholar] [CrossRef]
- Zhou, X. Roles of androgen receptor in male and female reproduction: Lessons from global and cell-specific androgen receptor knockout (ARKO) mice. J. Androl. 2010, 31, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Cicognani, A.; Alessandroni, R.; Pasini, A.; Pirazzoli, P.; Cassio, A.; Barbieri, E.; Cacciari, E. Low birth weight for gestational age and subsequent male gonadal function. J. Pediatr. 2002, 141, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, S.M.; Jacobsen, G.; Romundstad, P. Maternal testosterone levels during pregnancy are associated with offspring size at birth. Eur. J. Endocrinol. 2006, 155, 365–370. [Google Scholar] [CrossRef]
- Voegtline, K.M.; Costigan, K.A.; Kivlighan, K.T.; Henderson, J.L.; DiPietro, J.A. Sex-specific associations of maternal prenatal testosterone levels with birth weight and weight gain in infancy. J. Dev. Orig. Health Dis. 2013, 4, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Salamalekis, E.; Bakas, P.; Vitoratos, N.; Eleptheriadis, M.; Creatsas, G. Androgen levels in the third trimester of pregnancy in patients with preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2006, 126, 16–19. [Google Scholar] [CrossRef]
- Acromite, M.T.; Mantzoros, C.S.; Leach, R.E.; Hurwitz, J.; Dorey, L.G. Androgens in preeclampsia. Am. J. Obstet. Gynecol. 1999, 180, 60–63. [Google Scholar] [CrossRef]
- Sathishkumar, K.; Balakrishnan, M.; Chinnathambi, V.; Chauhan, M.; Hankins, G.D.; Yallampalli, C. Fetal sex-related dysregulation in testosterone production and their receptor expression in the human placenta with preeclampsia. J. Perinatol. 2012, 32, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Maliqueo, M.; Lara, H.E.; Sanchez, F.; Echiburu, B.; Crisosto, N.; Sir-Petermann, T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 166, 151–155. [Google Scholar] [CrossRef]
- Shozu, M.; Akasofu, K.; Harada, T.; Kubota, Y. A new cause of female pseudohermaphroditism: Placental aromatase deficiency. J. Clin. Endocrinol. Metab. 1991, 72, 560–566. [Google Scholar] [CrossRef]
- Stalberg, K.; Gonzales, R.; Lang-Muritano, M.; Gobet, R. Aromatase Deficiency in Fetal Virilization. J. Pediatr. Urol. 2010, 6, S85. [Google Scholar] [CrossRef]
- Jones, M.E.E.; Boon, W.C.; McInnes, K.; Maffei, L.; Carani, C.; Simpson, E.R. Recognizing rare disorders: Aromatase deficiency. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 414. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Abbott, D.H.; Padmanabhan, V. Polycystic ovary syndrome and its developmental origins. Rev. Endocr. Metab. Disord. 2007, 8, 127–141. [Google Scholar] [CrossRef]
- Padmanabhan, V.; Veiga-Lopez, A. Animal models of the polycystic ovary syndrome phenotype. Steroids 2013, 78, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, A.K.; Hoang, V.; Padmanabhan, V.; Gilbreath, E.; Mietelka, K.A. Prenatal programming: Adverse cardiac programming by gestational testosterone excess. Sci. Rep. 2016, 6, 28335. [Google Scholar] [CrossRef] [Green Version]
- Beckett, E.M.; Astapova, O.; Steckler, T.L.; Veiga-Lopez, A.; Padmanabhan, V. Developmental programing: Impact of testosterone on placental differentiation. Reproduction 2014, 148, 199–209. [Google Scholar] [CrossRef]
- Cleys, E.R.; Halleran, J.L.; Enriquez, V.A.; da Silveira, J.C.; West, R.C.; Winger, Q.A.; Anthony, R.V.; Bruemmer, J.E.; Clay, C.M.; Bouma, G.J. Androgen receptor and histone lysine demethylases in ovine placenta. PLoS ONE 2015, 10, e0117472. [Google Scholar] [CrossRef] [PubMed]
- Blesson, C.S.; Chinnathambi, V.; Hankins, G.D.; Yallampalli, C.; Sathishkumar, K. Prenatal testosterone exposure induces hypertension in adult females via androgen receptor-dependent protein kinase Cdelta-mediated mechanism. Hypertension 2015, 65, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, K.; Mishra, J.S.; Chinnathambi, V.; Vincent, K.L.; Patrikeev, I.; Motamedi, M.; Saade, G.R.; Hankins, G.D.; Sathishkumar, K. Elevated testosterone reduces uterine blood flow, spiral artery elongation and placental oxygenation in pregnant rats. Hypertension 2016, 67, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Sathishkumar, K.; Elkins, R.; Chinnathambi, V.; Gao, H.; Hankins, G.D.; Yallampalli, C. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod. Biol. Endocrinol. 2011, 9, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, A.S.; Puttabyatappa, M.; Ciarelli, J.N.; Zeng, L.; Smith, Y.R.; Lieberman, R.; Pennathur, S.; Padmanabhan, V. Prenatal Testosterone Excess Disrupts Placental Function in a Sheep Model of Polycystic Ovary Syndrome. Endocrinology 2019, 160, 2663–2672. [Google Scholar] [CrossRef]
- Izumi, K.; Fang, L.Y.; Mizokami, A.; Namiki, M.; Li, L.; Lin, W.J.; Chang, C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 2013, 5, 1383–1401. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hwang, T.H.; Oseth, L.A.; Hauge, A.; Vessella, R.L.; Schmechel, S.C.; Hirsch, B.; Beckman, K.B.; Silverstein, K.A.; Dehm, S.M. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene 2012, 31, 4759–4767. [Google Scholar] [CrossRef] [Green Version]
- Nyquist, M.D.; Li, Y.; Hwang, T.H.; Manlove, L.S.; Vessella, R.L.; Silverstein, K.A.; Voytas, D.F.; Dehm, S.M. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17492–17497. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Luo, J. Decoding the androgen receptor splice variants. Transl. Androl. Urol. 2013, 2, 178–186. [Google Scholar] [CrossRef]
- Hu, D.G.; Hickey, T.E.; Irvine, C.; Wijayakumara, D.D.; Lu, L.; Tilley, W.D.; Selth, L.A.; Mackenzie, P.I. Identification of androgen receptor splice variant transcripts in breast cancer cell lines and human tissues. Horm. Cancer 2014, 5, 61–71. [Google Scholar] [CrossRef]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J. 2004, 272, 74–84. [Google Scholar] [CrossRef]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stope, M.B.; Bradl, J.; Peters, S.; Streitborger, A.; Weiss, M.; Zimmermann, U.; Walther, R.; Lillig, C.H.; Burchardt, M. Shortened isoforms of the androgen receptor are regulated by the cytoprotective heat-shock protein HSPB1 and the tumor-suppressive microRNA miR-1 in prostate cancer cells. Anticancer. Res. 2013, 33, 4921–4926. [Google Scholar]
- Horie, K.; Takakura, K.; Imai, K.; Liao, S.; Mori, T. Immunohistochemical localization of androgen receptor in the human endometrium, decidua, placenta and pathological conditions of the endometrium. Hum. Reprod. 1992, 7, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.; Lan, K.; Tsai, C.; Ou, C.; Cheng, B.; Tsai, M.; Kang, H.; Tung, Y.; Wong, Y.; Huang, K. Expression of Androgen Receptor in Human Placentas From Normal and Preeclamptic Pregnancies. Taiwan J. Obstet. Gynecol. 2009, 48, 5. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meakin, A.S.; Cuffe, J.S.M.; Darby, J.R.T.; Morrison, J.L.; Clifton, V.L. Let’s Talk about Placental Sex, Baby: Understanding Mechanisms That Drive Female- and Male-Specific Fetal Growth and Developmental Outcomes. Int. J. Mol. Sci. 2021, 22, 6386. https://doi.org/10.3390/ijms22126386
Meakin AS, Cuffe JSM, Darby JRT, Morrison JL, Clifton VL. Let’s Talk about Placental Sex, Baby: Understanding Mechanisms That Drive Female- and Male-Specific Fetal Growth and Developmental Outcomes. International Journal of Molecular Sciences. 2021; 22(12):6386. https://doi.org/10.3390/ijms22126386
Chicago/Turabian StyleMeakin, Ashley S., James S. M. Cuffe, Jack R. T. Darby, Janna L. Morrison, and Vicki L. Clifton. 2021. "Let’s Talk about Placental Sex, Baby: Understanding Mechanisms That Drive Female- and Male-Specific Fetal Growth and Developmental Outcomes" International Journal of Molecular Sciences 22, no. 12: 6386. https://doi.org/10.3390/ijms22126386