
The Role of Sphingolipids in Cancer Immunotherapy



Abstract
1. Introduction
2. The Multifaceted Functions of Sphingolipids in the Interplay between Cancer Cells and Immune System
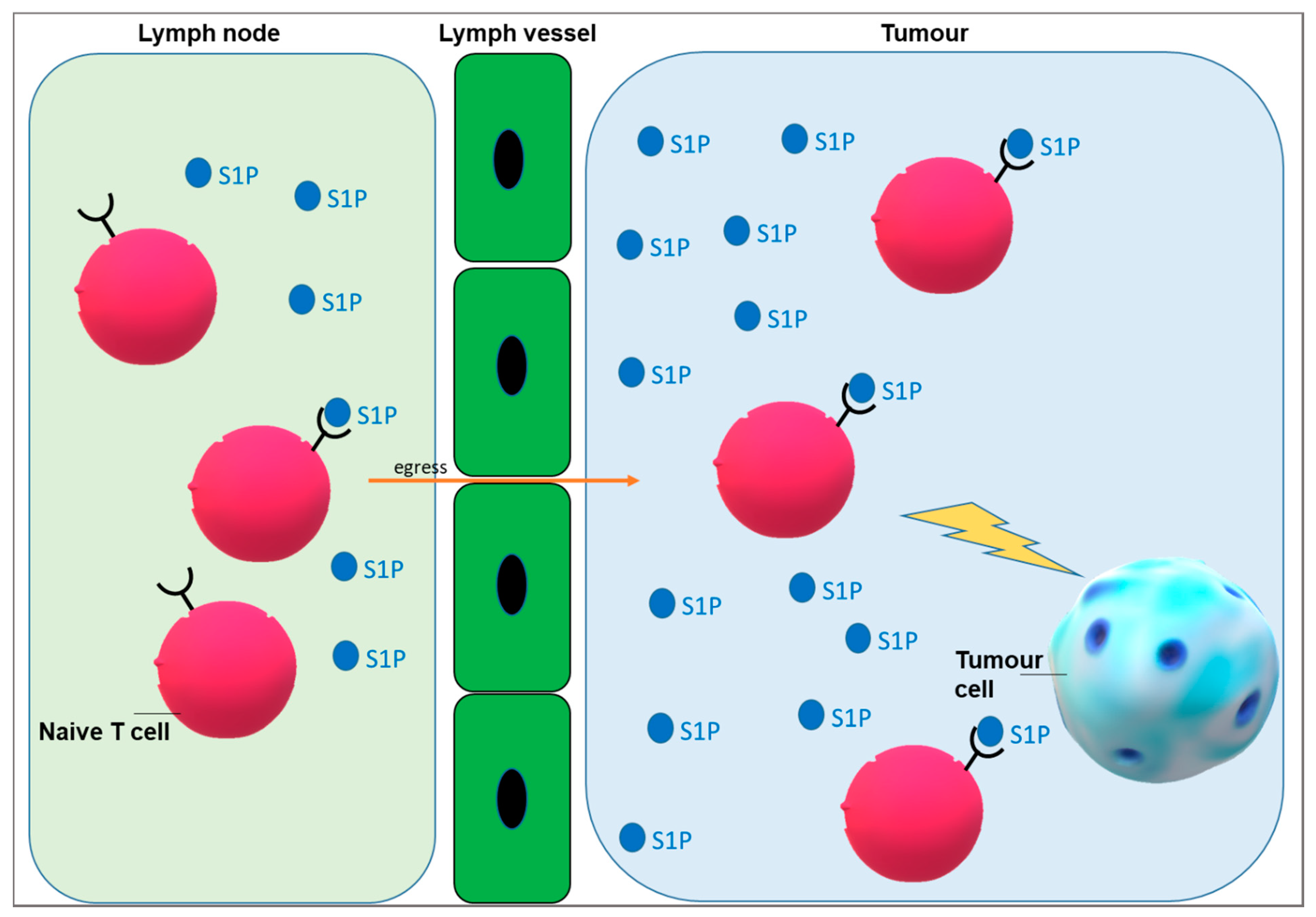
3. Targeting S1P Signalling to Improve Immunotherapy
4. Employment of Glycosphingolipids in Immunotherapy against Cancer
4.1. Creating Vaccines against Cancer
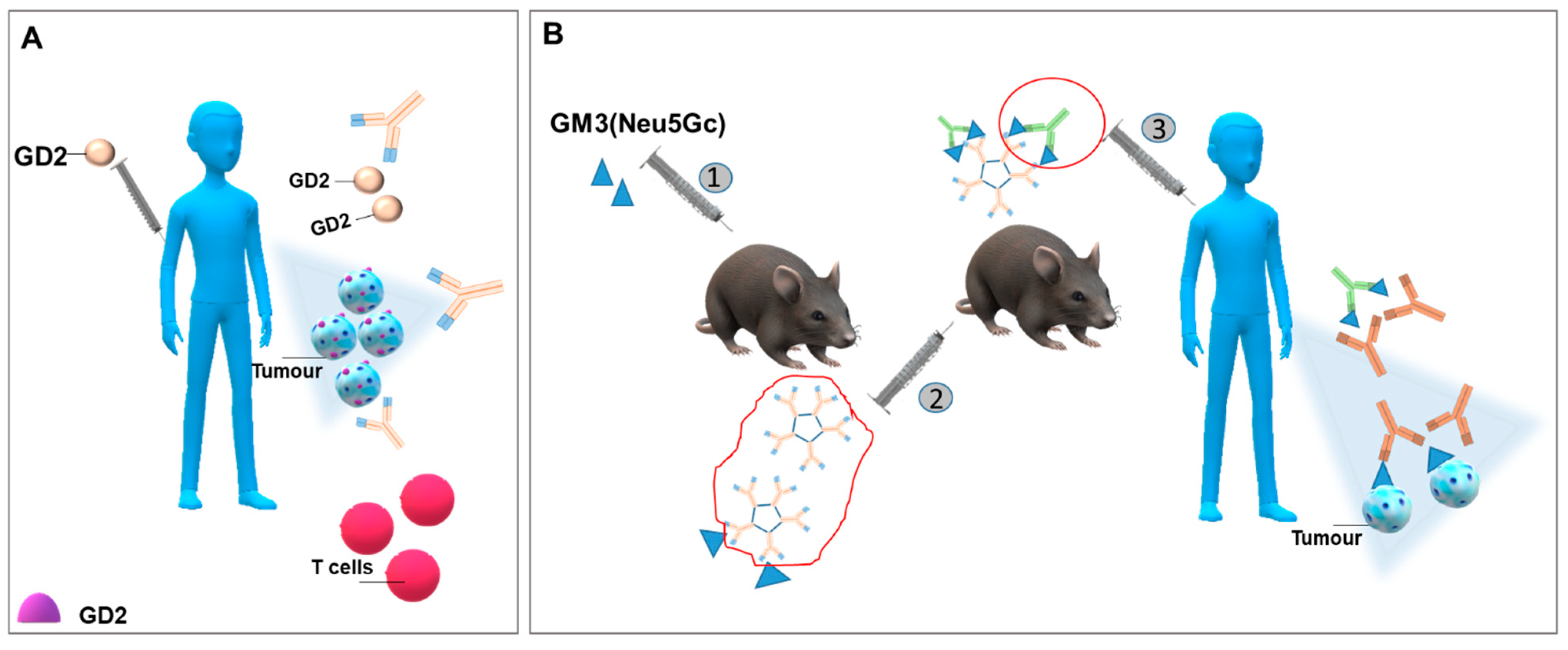
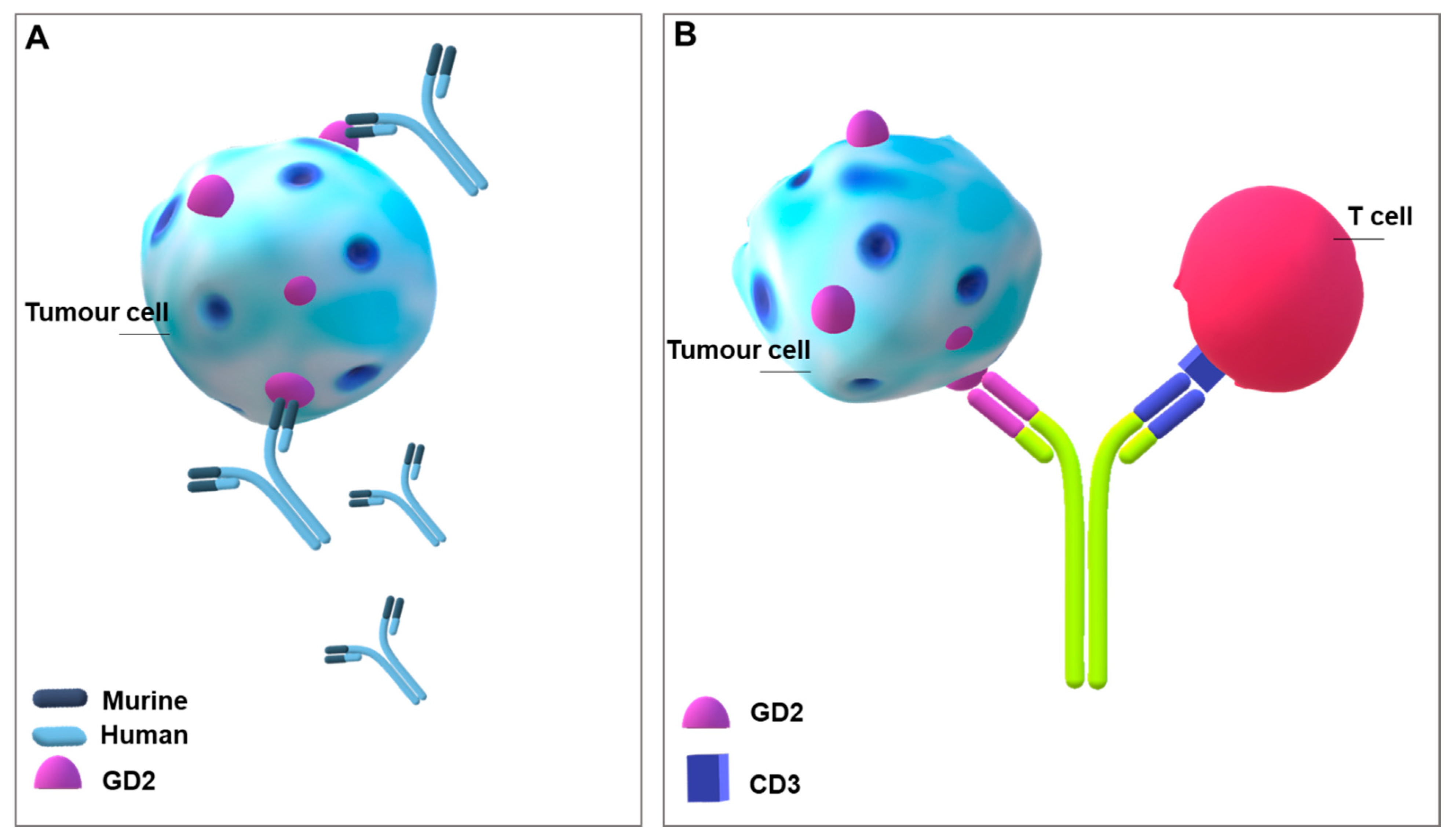
4.2. Generating Antibodies against Glycosphingolipids Expressed by Cancer Cells
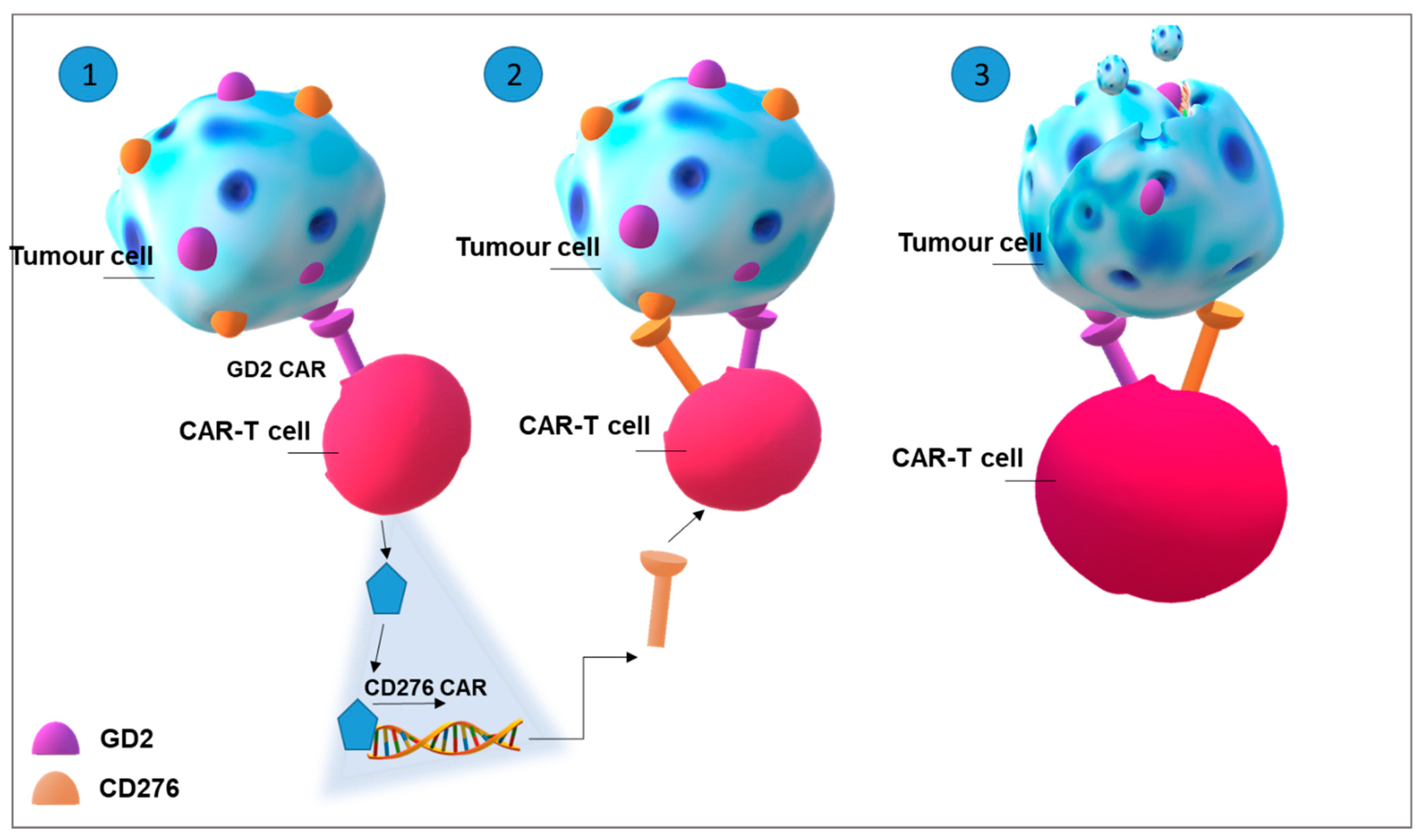
4.3. Improving CAR-T Cell Therapy in Solid Tumours: Glycosphingolipids as Targets
5. Perspectives and Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muenst, S.; Läubli, H.; Soysal, S.D.; Zippelius, A.; Tzankov, A.; Hoeller, S. The immune system and cancer evasion strategies: Therapeutic concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Acebes-Fernández, V.; Landeria-Viñuela, A.; Juanes-Velasco, P.; Hernández, A.P.; Otazo-Perez, A.; Manzano-Román, R.; Gongora, R.; Fuentes, M. Nanomedicine and onco-immunotherapy: From the bench to bedside to biomarkers. Nanomaterials 2020, 10, 1274. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chen, L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef]
- Furuya, H.; Shimizu, Y.; Kawamori, T. Sphingolipids in cancer. Cancer Metastasis Rev. 2011, 30, 567–576. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohmi, Y.; Ohkawa, Y.; Bhuiyan, R.H.; Zhang, P.; Tajima, O.; Hashimoto, N.; Hamamura, K. New era of research on cancer-associated glycosphingolipids. Cancer Sci. 2019, 110, 1544–1551. [Google Scholar] [CrossRef]
- Durrant, L.G.; Noble, P.; Spendlove, I. Immunology in the clinic review series; focus on cancer: Glycolipids as targets for tumour immunotherapy. Clin. Exp. Immunol. 2012, 167, 206–215. [Google Scholar] [CrossRef]
- Schauer, R.; Kamerling, J.P. Exploration of the sialic acid world. Adv. Carbohydr. Chem. Biochem. 2018, 75, 1–213. [Google Scholar] [CrossRef] [PubMed]
- Gahmberg, C.G.; Hakomori, S.I. Altered growth behavior of malignant cells associated with changes in externally labeled glycoprotein and glycolipid. Proc. Natl. Acad. Sci. USA 1973, 70, 3329–3333. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv. Cancer Res. 1989, 52, 257–331. [Google Scholar] [CrossRef]
- Hakomori, S. Glycosylation defining cancer malignancy: New wine in an old bottle. Proc. Natl. Acad. Sci. USA 2002, 99, 10231–10233. [Google Scholar] [CrossRef] [PubMed]
- Marquina, G.; Waki, H.; Fernandez, L.E.; Kon, K.; Carr, A.; Valiente, O.; Perez, R.; Ando, S. Gangliosides expressed in human breast cancer. Cancer Res. 1996, 56, 5165–5171. [Google Scholar]
- Kojima, N.; Hakomori, S. Specific interaction between gangliotriaosylceramide (Gg3) and sialosyllactosylceramide (GM3) as a basis for specific cellular recognition between lymphoma and melanoma cells. J. Biol. Chem. 1989, 264, 20159–20162. [Google Scholar] [CrossRef]
- Kawamura, S.; Ohyama, C.; Watanabe, R.; Satoh, M.; Saito, S.; Hoshi, S.; Gasa, S.; Orikasa, S. Glycolipid composition in bladder tumor: A crucial role of GM3 ganglioside in tumor invasion. Int. J. Cancer 2001, 94, 343–347. [Google Scholar] [CrossRef]
- Watanabe, R.; Ohyama, C.; Aoki, H.; Takahashi, T.; Satoh, M.; Saito, S.; Hoshi, S.; Ishii, A.; Saito, M.; Arai, Y. Ganglioside G(M3) overexpression induces apoptosis and reduces malignant potential in murine bladder cancer. Cancer Res. 2002, 62, 3850–3854. [Google Scholar]
- Prinetti, A.; Aureli, M.; Illuzzi, G.; Prioni, S.; Nocco, V.; Scandroglio, F.; Gagliano, N.; Tredici, G.; Rodriguez-Menendez, V.; Chigorno, V.; et al. GM3 synthase overexpression results in reduced cell motility and in caveolin-1 upregulation in human ovarian carcinoma cells. Glycobiology 2010, 20, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Prinetti, A.; Cao, T.; Illuzzi, G.; Prioni, S.; Aureli, M.; Gagliano, N.; Tredici, G.; Rodriguez-Menendez, V.; Chigorno, V.; Sonnino, S. A glycosphingolipid/caveolin-1 signaling complex inhibits motility of human ovarian carcinoma cells. J. Biol. Chem. 2011, 286, 40900–40910. [Google Scholar] [CrossRef]
- Chang, F.; Li, R.; Ladisch, S. Shedding of gangliosides by human medulloblastoma cells. Exp. Cell Res. 1997, 234, 341–346. [Google Scholar] [CrossRef]
- Ladisch, S.; Li, R.; Olson, E. Ceramide structure predicts tumor ganglioside immunosuppressive activity. Proc. Natl. Acad. Sci. USA 1994, 91, 1974–1978. [Google Scholar] [CrossRef]
- Ladisch, S.; Chang, F.; Li, R.; Cogen, P.; Johnson, D. Detection of medulloblastoma and astrocytoma-associated ganglioside GD3 in cerebrospinal fluid. Cancer Lett. 1997, 120, 71–78. [Google Scholar] [CrossRef]
- Valentino, L.A.; Ladisch, S. Circulating tumor gangliosides enhance platelet activation. Blood 1994, 83, 2872–2877. [Google Scholar] [CrossRef]
- Kanoh, H.; Nitta, T.; Go, S.; Inamori, K.I.; Veillon, L.; Nihei, W.; Fujii, M.; Kabayama, K.; Shimoyama, A.; Fukase, K.; et al. Homeostatic and pathogenic roles of GM3 ganglioside molecular species in TLR4 signaling in obesity. EMBO J. 2020, 39, e101732. [Google Scholar] [CrossRef]
- Prinetti, A.; Prioni, S.; Loberto, N.; Aureli, M.; Nocco, V.; Illuzzi, G.; Mauri, L.; Valsecchi, M.; Chigorno, V.; Sonnino, S. Aberrant glycosphingolipid expression and membrane organization in tumor cells: Consequences on tumor-host interactions. Adv. Exp. Med. Biol. 2011, 705, 643–667. [Google Scholar] [CrossRef] [PubMed]
- Prinetti, A.; Prioni, S.; Loberto, N.; Aureli, M.; Chigorno, V.; Sonnino, S. Regulation of tumor phenotypes by caveolin-1 and sphingolipid-controlled membrane signaling complexes. Biochim. Biophys. Acta 2008, 1780, 585–596. [Google Scholar] [CrossRef]
- Prinetti, A.; Loberto, N.; Chigorno, V.; Sonnino, S. Glycosphingolipid behaviour in complex membranes. Biochim. Biophys. Acta 2009, 1788, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.J.; Nakayama, K.; Hikita, T.; Handa, K.; Hakomori, S.I. Epidermal growth factor receptor tyrosine kinase is modulated by GM3 interaction with N-linked GlcNAc termini of the receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 18987–18991. [Google Scholar] [CrossRef]
- Sonnino, S.; Prinetti, A. Lipids and membrane lateral organization. Front. Physiol. 2010, 1. [Google Scholar] [CrossRef]
- Sonnino, S.; Prinetti, A. Membrane domains and the “lipid raft” concept. Curr. Med. Chem. 2013, 20, 4–21. [Google Scholar] [PubMed]
- Iwabuchi, K.; Prinetti, A.; Sonnino, S.; Mauri, L.; Kobayashi, T.; Ishii, K.; Kaga, N.; Murayama, K.; Kurihara, H.; Nakayama, H.; et al. Involvement of very long fatty acid-containing lactosylceramide in lactosylceramide-mediated superoxide generation and migration in neutrophils. Glycoconj. J. 2008, 25, 357–374. [Google Scholar] [CrossRef]
- Nakayama, H.; Yoshizaki, F.; Prinetti, A.; Sonnino, S.; Mauri, L.; Takamori, K.; Ogawa, H.; Iwabuchi, K. Lyn-coupled LacCer-enriched lipid rafts are required for CD11b/CD18-mediated neutrophil phagocytosis of nonopsonized microorganisms. J. Leukoc. Biol. 2008, 83, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Kurihara, H.; Morita, Y.S.; Kinoshita, T.; Mauri, L.; Prinetti, A.; Sonnino, S.; Yokoyama, N.; Ogawa, H.; Takamori, K.; et al. Lipoarabinomannan binding to lactosylceramide in lipid rafts is essential for the phagocytosis of mycobacteria by human neutrophils. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, K.; Uchiyama, M.; Yamochi, T.; Nishibashi, K.; Hosono, O.; Takahashi, N.; Kina, S.; Tanaka, H.; Lin, X.; Dang, N.H.; et al. Caveolin-1 triggers T-cell activation via CD26 in association with CARMA1. J. Biol. Chem. 2007, 282, 10117–10131. [Google Scholar] [CrossRef] [PubMed]
- Waddington, K.E.; Jury, E.C. Manipulating membrane lipid profiles to restore T-cell function in autoimmunity. Biochem. Soc. Trans. 2015, 43, 745–751. [Google Scholar] [CrossRef]
- Cheng, P.C.; Dykstra, M.L.; Mitchell, R.N.; Pierce, S.K. A role for lipid rafts in B cell antigen receptor signaling and antigen targeting. J. Exp. Med. 1999, 190, 1549–1560. [Google Scholar] [CrossRef]
- Hamilton, V.T.; Stone, D.M.; Cantor, G.H. Translocation of the B cell receptor to lipid rafts is inhibited in B cells from BLV-infected, persistent lymphocytosis cattle. Virology 2003, 315, 135–147. [Google Scholar] [CrossRef][Green Version]
- Park, Y.K.; Lee, J.W.; Ko, Y.G.; Hong, S.; Park, S.H. Lipid rafts are required for efficient signal transduction by CD1d. Biochem. Biophys. Res. Commun. 2005, 327, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, M.L.; Cherukuri, A.; Pierce, S.K. Floating the raft hypothesis for immune receptors: Access to rafts controls receptor signaling and trafficking. Traffic 2001, 2, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.I.; Ramadas, R.A.; Medoff, B.D. The role of CARMA1 in T cells. Crit. Rev. Immunol. 2013, 33, 219–243. [Google Scholar] [CrossRef]
- D’Aprile, C.; Prioni, S.; Mauri, L.; Prinetti, A.; Grassi, S. Lipid rafts as platforms for sphingosine 1-phosphate metabolism and signalling. Cell. Signal. 2021, 80, 109929. [Google Scholar] [CrossRef] [PubMed]
- Giussani, P.; Tringali, C.; Riboni, L.; Viani, P.; Venerando, B. Sphingolipids: Key regulators of apoptosis and pivotal players in cancer drug resistance. Int. J. Mol. Sci. 2014, 15, 4356–4392. [Google Scholar] [CrossRef]
- Giussani, P.; Bassi, R.; Anelli, V.; Brioschi, L.; De Zen, F.; Riccitelli, E.; Caroli, M.; Campanella, R.; Gaini, S.M.; Viani, P.; et al. Glucosylceramide synthase protects glioblastoma cells against autophagic and apoptotic death induced by temozolomide and Paclitaxel. Cancer Investig. 2012, 30, 27–37. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Takuwa, Y.; Okamoto, Y.; Yoshioka, K.; Takuwa, N. Sphingosine-1-phosphate signaling in physiology and diseases. Biofactors 2012, 38, 329–337. [Google Scholar] [CrossRef]
- Riccitelli, E.; Giussani, P.; Di Vito, C.; Condomitti, G.; Tringali, C.; Caroli, M.; Galli, R.; Viani, P.; Riboni, L. Extracellular sphingosine-1-phosphate: A novel actor in human glioblastoma stem cell survival. PLoS ONE 2013, 8, e68229. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705. [Google Scholar] [CrossRef]
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Muñoz, A. Role of bioactive sphingolipids in physiology and pathology. Essays Biochem. 2020, 64, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Yang, H.; Hsieh, C.Y.; Sun, W.; Chang, C.C.; Schroeder, J.J. Evaluation of chemotherapeutic and cancer-protective properties of sphingosine and C2-ceramide in a human breast stem cell derived carcinogenesis model. Int. J. Oncol. 2019, 54, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Chen, T.; Liang, X.; Duan, J.; Qian, S.; Qiao, K.; Zhang, L.; Liu, Y.; Wang, J. C24-ceramide drives gallbladder cancer progression through directly targeting phosphatidylinositol 5-phosphate 4-kinase type-2 gamma to facilitate mammalian target of rapamycin signaling activation. Hepatology 2021, 73, 692–712. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Loberto, N.; Schiumarini, D.; Samarani, M.; Mancini, G.; Tamanini, A.; Lippi, G.; Dechecchi, M.C.; Bassi, R.; Giussani, P.; et al. Sphingolipids role in the regulation of inflammatory response: From leukocyte biology to bacterial infection. J. Leukoc. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.M.; Del Poeta, M. Sphingosine-1-phosphate receptors and innate immunity. Cell. Microbiol. 2018, 20, e12836. [Google Scholar] [CrossRef] [PubMed]
- Gadiyar, V.; Lahey, K.C.; Calianese, D.; Devoe, C.; Mehta, D.; Bono, K.; Desind, S.; Davra, V.; Birge, R.B. Cell death in the tumor microenvironment: Implications for cancer immunotherapy. Cells 2020, 9, 2207. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Bohn, E.; Kröber, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef]
- Peter, C.; Waibel, M.; Keppeler, H.; Lehmann, R.; Xu, G.; Halama, A.; Adamski, J.; Schulze-Osthoff, K.; Wesselborg, S.; Lauber, K. Release of lysophospholipid ‘find-me’ signals during apoptosis requires the ATP-binding cassette transporter A1. Autoimmunity 2012, 45, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhou, Y.; Friis, T.; Beagley, K.; Xiao, Y. S1P-S1PR1 signaling: The “Sphinx” in osteoimmunology. Front. Immunol. 2019, 10, 1409. [Google Scholar] [CrossRef]
- Veny, M.; Fernández-Clotet, A.; Panés, J. Controlling leukocyte trafficking in IBD. Pharmacol. Res. 2020, 159, 105050. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zamora-Pineda, J.; Degagné, E.; Saba, J.D. S1P lyase regulation of thymic egress and oncogenic inflammatory signaling. Mediat. Inflamm. 2017, 2017, 7685142. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Bryant, R.V.; Travis, S. Interfering with leukocyte trafficking in Crohn’s disease. Best Pract. Res. Clin. Gastroenterol. 2019, 38, 101617. [Google Scholar] [CrossRef]
- Don-Doncow, N.; Zhang, Y.; Matuskova, H.; Meissner, A. The emerging alliance of sphingosine-1-phosphate signalling and immune cells: From basic mechanisms to implications in hypertension. Br. J. Pharmacol. 2019, 176, 1989–2001. [Google Scholar] [CrossRef]
- Chakraborty, P.; Vaena, S.G.; Thyagarajan, K.; Chatterjee, S.; Al-Khami, A.; Selvam, S.P.; Nguyen, H.; Kang, I.; Wyatt, M.W.; Baliga, U.; et al. Pro-survival lipid sphingosine-1-phosphate metabolically programs T cells to limit anti-tumor activity. Cell Rep. 2019, 28, 1879–1893.e1877. [Google Scholar] [CrossRef]
- Liu, G.; Yang, K.; Burns, S.; Shrestha, S.; Chi, H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat. Immunol. 2010, 11, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef]
- Panneer Selvam, S.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D.; et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 8, ra58. [Google Scholar] [CrossRef]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef]
- Parham, K.A.; Zebol, J.R.; Tooley, K.L.; Sun, W.Y.; Moldenhauer, L.M.; Cockshell, M.P.; Gliddon, B.L.; Moretti, P.A.; Tigyi, G.; Pitson, S.M.; et al. Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-γ that regulates neoangiogenesis. FASEB J. 2015, 29, 3638–3653. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Mauri, L.; Prioni, S.; Cabitta, L.; Sonnino, S.; Prinetti, A.; Giussani, P. Sphingosine 1-phosphate receptors and metabolic enzymes as druggable targets for brain diseases. Front. Pharmacol. 2019, 10, 807. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Yu, J.Y.; Yin, D.; Patwardhan, G.A.; Gupta, V.; Hirabayashi, Y.; Holleran, W.M.; Giuliano, A.E.; Jazwinski, S.M.; Gouaze-Andersson, V.; et al. A role for ceramide in driving cancer cell resistance to doxorubicin. FASEB J. 2008, 22, 2541–2551. [Google Scholar] [CrossRef]
- Pan, Q.; Li, Q.; Liu, S.; Ning, N.; Zhang, X.; Xu, Y.; Chang, A.E.; Wicha, M.S. Concise review: Targeting cancer stem cells using immunologic approaches. Stem Cells 2015, 33, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Juin, S.K.; Majumdar, S. Cancer stem cells and ceramide signaling: The cutting edges of immunotherapy. Mol. Biol. Rep. 2020, 47, 8101–8111. [Google Scholar] [CrossRef]
- Oskouian, B.; Saba, J.D. Cancer treatment strategies targeting sphingolipid metabolism. Adv. Exp. Med. Biol. 2010, 688, 185–205. [Google Scholar] [PubMed]
- Ghosh, S.; Jawed, J.J.; Halder, K.; Banerjee, S.; Chowdhury, B.P.; Saha, A.; Juin, S.K.; Majumdar, S.B.; Bose, A.; Baral, R.; et al. TNFα mediated ceramide generation triggers cisplatin induced apoptosis in B16F10 melanoma in a PKCδ independent manner. Oncotarget 2018, 9, 37627–37646. [Google Scholar] [CrossRef]
- Ghosh, S.; Juin, S.K.; Nandi, P.; Majumdar, S.B.; Bose, A.; Baral, R.; Sil, P.C.; Majumdar, S. PKCζ mediated anti-proliferative effect of C2 ceramide on neutralization of the tumor microenvironment and melanoma regression. Cancer Immunol. Immunother. 2020, 69, 611–627. [Google Scholar] [CrossRef]
- von Wenckstern, H.; Zimmermann, K.; Kleuser, B. The role of the lysophospholipid sphingosine 1-phosphate in immune cell biology. Arch. Immunol. Ther. Exp. 2006, 54, 239–251. [Google Scholar] [CrossRef]
- Weigert, A.; Johann, A.M.; von Knethen, A.; Schmidt, H.; Geisslinger, G.; Brüne, B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 2006, 108, 1635–1642. [Google Scholar] [CrossRef]
- Weigert, A.; Tzieply, N.; von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.I.; Campos, L.E.; Castro, M.G.; Aladhami, A.; Oskeritzian, C.A.; Alvarez, S.E. Sphingosine-1 phosphate: A new modulator of immune plasticity in the tumor microenvironment. Front. Oncol. 2016, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Lagadari, M.; Lehmann, K.; Ziemer, M.; Truta-Feles, K.; Berod, L.; Idzko, M.; Barz, D.; Kamradt, T.; Maghazachi, A.A.; Norgauer, J. Sphingosine-1-phosphate inhibits the cytotoxic activity of NK cells via Gs protein-mediated signalling. Int. J. Oncol. 2009, 34, 287–294. [Google Scholar]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Riboni, L.; Abdel Hadi, L.; Navone, S.E.; Guarnaccia, L.; Campanella, R.; Marfia, G. Sphingosine-1-phosphate in the tumor microenvironment: A signaling hub regulating cancer hallmarks. Cells 2020, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- Vermaelen, K. Vaccine strategies to improve anti-cancer cellular immune responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Wu, D.; Ragupathi, G.; Lu, S.; Williams, L.; Hwu, W.J.; Johnson, D.; Livingston, P.O. Sequential immunization of melanoma patients with GD3 ganglioside vaccine and anti-idiotypic monoclonal antibody that mimics GD3 ganglioside. Clin. Cancer Res. 2004, 10, 4717–4723. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Suciu, S.; Rutkowski, P.; Marsden, J.; Santinami, M.; Corrie, P.; Aamdal, S.; Ascierto, P.A.; Patel, P.M.; Kruit, W.H.; et al. Adjuvant ganglioside GM2-KLH/QS-21 vaccination versus observation after resection of primary tumor > 1.5 mm in patients with stage II melanoma: Results of the EORTC 18961 randomized phase III trial. J. Clin. Oncol. 2013, 31, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Sait, S.; Modak, S. Anti-GD2 immunotherapy for neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Osorio, M.; Gracia, E.; Reigosa, E.; Hernandez, J.; de la Torre, A.; Saurez, G.; Perez, K.; Viada, C.; Cepeda, M.; Carr, A.; et al. Effect of vaccination with N-glycolyl GM3/VSSP vaccine by subcutaneous injection in patients with advanced cutaneous melanoma. Cancer Manag. Res. 2012, 4, 341–345. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neninger, E.; Díaz, R.M.; de la Torre, A.; Rives, R.; Díaz, A.; Saurez, G.; Gabri, M.R.; Alonso, D.F.; Wilkinson, B.; Alfonso, A.M.; et al. Active immunotherapy with 1E10 anti-idiotype vaccine in patients with small cell lung cancer: Report of a phase I trial. Cancer Biol. Ther. 2007, 6, 145–150. [Google Scholar] [CrossRef]
- Alfonso, S.; Diaz, R.M.; de la Torre, A.; Santiesteban, E.; Aguirre, F.; Pérez, K.; Rodríguez, J.L.; Barroso, M.e.C.; Hernández, A.M.; Toledo, D.; et al. 1E10 anti-idiotype vaccine in non-small cell lung cancer: Experience in stage IIIb/IV patients. Cancer Biol. Ther. 2007, 6, 1847–1852. [Google Scholar] [CrossRef]
- Gajdosik, Z. Racotumomab—A novel anti-idiotype monoclonal antibody vaccine for the treatment of cancer. Drugs Today 2014, 50, 301–307. [Google Scholar] [CrossRef]
- Uskent, N.; Ayla, S.; Molinas Mandel, N.; Ozkan, M.; Teomete, M.; Baloglu, H.; Aydıncer, C.; Yergok, H.; Dogan, E.; Berk, B.; et al. Prognostic significance of tumor tissue NeuGcGM3 ganglioside expression in patients receiving racotumomab immunotherapy. J. Oncol. 2020, 2020, 1360431. [Google Scholar] [CrossRef] [PubMed]
- Guthmann, M.D.; Castro, M.A.; Cinat, G.; Venier, C.; Koliren, L.; Bitton, R.J.; Vázquez, A.M.; Fainboim, L. Cellular and humoral immune response to N-Glycolyl-GM3 elicited by prolonged immunotherapy with an anti-idiotypic vaccine in high-risk and metastatic breast cancer patients. J. Immunother. 2006, 29, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Palomo, A.G.; Medinilla, A.L.; Segatori, V.; Barroso, M.D.C.; Blanco, R.; Gabri, M.R.; Pérez, A.C.; Monzón, K.L. Synergistic potentiation of the anti-metastatic effect of anti EGFR mAb by its combination with immunotherapies targeting the ganglioside NGcGM3. Oncotarget 2018, 9, 24069–24080. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Y.; Wang, Z.; Yu, F.; Li, M.; Zhu, H.; Wang, K.; Meng, M.; Zhao, W. The adjuvant of α-Galactosylceramide presented by gold nanoparticles enhances antitumor immune responses of MUC1 antigen-based tumor vaccines. Int. J. Nanomed. 2021, 16, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Affandi, A.J.; Grabowska, J.; Olesek, K.; Lopez Venegas, M.; Barbaria, A.; Rodríguez, E.; Mulder, P.P.G.; Pijffers, H.J.; Ambrosini, M.; Kalay, H.; et al. Selective tumor antigen vaccine delivery to human CD169+ antigen-presenting cells using ganglioside-liposomes. Proc. Natl. Acad. Sci. USA 2020, 117, 27528–27539. [Google Scholar] [CrossRef]
- Kholodenko, I.V.; Kalinovsky, D.V.; Doronin, I.I.; Deyev, S.M.; Kholodenko, R.V. Neuroblastoma origin and therapeutic targets for immunotherapy. J. Immunol. Res. 2018, 2018, 7394268. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Mora, J. Dinutuximab for the treatment of pediatric patients with high-risk neuroblastoma. Expert Rev. Clin. Pharmacol. 2016, 9, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Ly, S.; Anand, V.; El-Dana, F.; Nguyen, K.; Cai, Y.; Cai, S.; Piwnica-Worms, H.; Tripathy, D.; Sahin, A.A.; Andreeff, M.; et al. Anti-GD2 antibody dinutuximab inhibits triple-negative breast tumor growth by targeting GD2+ breast cancer stem-like cells. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Burki, T.K. hu3F8 for resistant or recurrent neuroblastoma. Lancet Oncol. 2018, 19, e583. [Google Scholar] [CrossRef]
- Federico, S.M.; McCarville, M.B.; Shulkin, B.L.; Sondel, P.M.; Hank, J.A.; Hutson, P.; Meagher, M.; Shafer, A.; Ng, C.Y.; Leung, W.; et al. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin. Cancer Res. 2017, 23, 6441–6449. [Google Scholar] [CrossRef]
- Furman, W.L.; Federico, S.M.; McCarville, M.B.; Shulkin, B.L.; Davidoff, A.M.; Krasin, M.J.; Sahr, N.; Sykes, A.; Wu, J.; Brennan, R.C.; et al. A phase II trial of Hu14.18K322A in combination with induction chemotherapy in children with newly diagnosed high-risk neuroblastoma. Clin. Cancer Res. 2019, 25, 6320–6328. [Google Scholar] [CrossRef]
- Cheng, M.; Ahmed, M.; Xu, H.; Cheung, N.K. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int. J. Cancer 2015, 136, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Ruf, P.; Schäfer, B.; Eissler, N.; Mocikat, R.; Hess, J.; Plöscher, M.; Wosch, S.; Suckstorff, I.; Zehetmeier, C.; Lindhofer, H. Ganglioside GD2-specific trifunctional surrogate antibody Surek demonstrates therapeutic activity in a mouse melanoma model. J. Transl. Med. 2012, 10, 219. [Google Scholar] [CrossRef]
- Cheng, M.; Santich, B.H.; Xu, H.; Ahmed, M.; Huse, M.; Cheung, N.K. Successful engineering of a highly potent single-chain variable-fragment (scFv) bispecific antibody to target disialoganglioside (GD2) positive tumors. Oncoimmunology 2016, 5, e1168557. [Google Scholar] [CrossRef]
- Cavdarli, S.; Delannoy, P.; Groux-Degroote, S. O-acetylated gangliosides as targets for cancer immunotherapy. Cells 2020, 9, 741. [Google Scholar] [CrossRef] [PubMed]
- Fleurence, J.; Cochonneau, D.; Fougeray, S.; Oliver, L.; Geraldo, F.; Terme, M.; Dorvillius, M.; Loussouarn, D.; Vallette, F.; Paris, F.; et al. Targeting and killing glioblastoma with monoclonal antibody to O-acetyl GD2 ganglioside. Oncotarget 2016, 7, 41172–41185. [Google Scholar] [CrossRef] [PubMed]
- Fleurence, J.; Bahri, M.; Fougeray, S.; Faraj, S.; Vermeulen, S.; Pinault, E.; Geraldo, F.; Oliver, L.; Véziers, J.; Marquet, P.; et al. Impairing temozolomide resistance driven by glioma stem-like cells with adjuvant immunotherapy targeting O-acetyl GD2 ganglioside. Int. J. Cancer 2020, 146, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T cells in solid tumors: Blueprints for building effective therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef]
- Rossig, C.; Kailayangiri, S.; Jamitzky, S.; Altvater, B. Carbohydrate targets for CAR T cells in solid childhood cancers. Front. Oncol. 2018, 8, 513. [Google Scholar] [CrossRef]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef]
- Andersch, L.; Radke, J.; Klaus, A.; Schwiebert, S.; Winkler, A.; Schumann, E.; Grunewald, L.; Zirngibl, F.; Flemmig, C.; Jensen, M.C.; et al. CD171- and GD2-specific CAR-T cells potently target retinoblastoma cells in preclinical in vitro testing. BMC Cancer 2019, 19, 895. [Google Scholar] [CrossRef]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- Bocca, P.; Di Carlo, E.; Caruana, I.; Emionite, L.; Cilli, M.; De Angelis, B.; Quintarelli, C.; Pezzolo, A.; Raffaghello, L.; Morandi, F.; et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology 2017, 7, e1378843. [Google Scholar] [CrossRef]
- Charan, M.; Dravid, P.; Cam, M.; Audino, A.; Gross, A.C.; Arnold, M.A.; Roberts, R.D.; Cripe, T.P.; Pertsemlidis, A.; Houghton, P.J.; et al. GD2-directed CAR-T cells in combination with HGF-targeted neutralizing antibody (AMG102) prevent primary tumor growth and metastasis in Ewing sarcoma. Int. J. Cancer 2020, 146, 3184–3195. [Google Scholar] [CrossRef]
- Chulanetra, M.; Morchang, A.; Sayour, E.; Eldjerou, L.; Milner, R.; Lagmay, J.; Cascio, M.; Stover, B.; Slayton, W.; Chaicumpa, W.; et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 2020, 10, 674–687. [Google Scholar] [PubMed]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.Z.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol. Res. 2018, 6, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, B.; Muthugounder, S.; Jambon, S.; Tibbetts, R.; Hung, L.; Bassiri, H.; Hogarty, M.D.; Barrett, D.M.; Shimada, H.; Asgharzadeh, S. Preclinical assessment of the efficacy and specificity of GD2-B7H3 SynNotch CAR-T in metastatic neuroblastoma. Nat. Commun. 2021, 12, 511. [Google Scholar] [CrossRef]
- Fleurence, J.; Fougeray, S.; Bahri, M.; Cochonneau, D.; Clémenceau, B.; Paris, F.; Heczey, A.; Birklé, S. Targeting O-acetyl-GD2 ganglioside for cancer immunotherapy. J. Immunol. Res. 2017, 5604891. [Google Scholar] [CrossRef]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Carrié, L.; Virazels, M.; Dufau, C.; Montfort, A.; Levade, T.; Ségui, B.; Andrieu-Abadie, N. New insights into the role of sphingolipid metabolism in melanoma. Cells 2020, 9, 1967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Springfield, R.; Chen, S.; Li, X.; Feng, X.; Moshirian, R.; Yang, R.; Yuan, W. α-GalCer and iNKT cell-based cancer immunotherapy: Realizing the therapeutic potentials. Front. Immunol. 2019, 10, 1126. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Shimizu, K.; Hasaba, S.; Date, M. Induction of specific adaptive immune responses by immunization with newly designed artificial glycosphingolipids. Sci. Rep. 2019, 9, 18803. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Structure |
|---|---|
| Sphingosine 1-phosphate (S1P) |  |
| Ceramide (Cer) |  |
| N-Acetylneuraminic acid (Neu5Ac) |  |
| N-glycolylneuraminic acid (Neu5Gc) |  |
| αGalactosylceramide (αGalCer) |  |
| Lactosylceramide (LacCer) |  |
| GM3 (Neu5Ac) |  |
| GD3 |  |
| GM2 |  |
| GD2 |  |
| GM1 |  |
| FucosylGM1 |  |
| Globo-H |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giussani, P.; Prinetti, A.; Tringali, C. The Role of Sphingolipids in Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 6492. https://doi.org/10.3390/ijms22126492
Giussani P, Prinetti A, Tringali C. The Role of Sphingolipids in Cancer Immunotherapy. International Journal of Molecular Sciences. 2021; 22(12):6492. https://doi.org/10.3390/ijms22126492
Chicago/Turabian StyleGiussani, Paola, Alessandro Prinetti, and Cristina Tringali. 2021. "The Role of Sphingolipids in Cancer Immunotherapy" International Journal of Molecular Sciences 22, no. 12: 6492. https://doi.org/10.3390/ijms22126492
APA StyleGiussani, P., Prinetti, A., & Tringali, C. (2021). The Role of Sphingolipids in Cancer Immunotherapy. International Journal of Molecular Sciences, 22(12), 6492. https://doi.org/10.3390/ijms22126492