
Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis


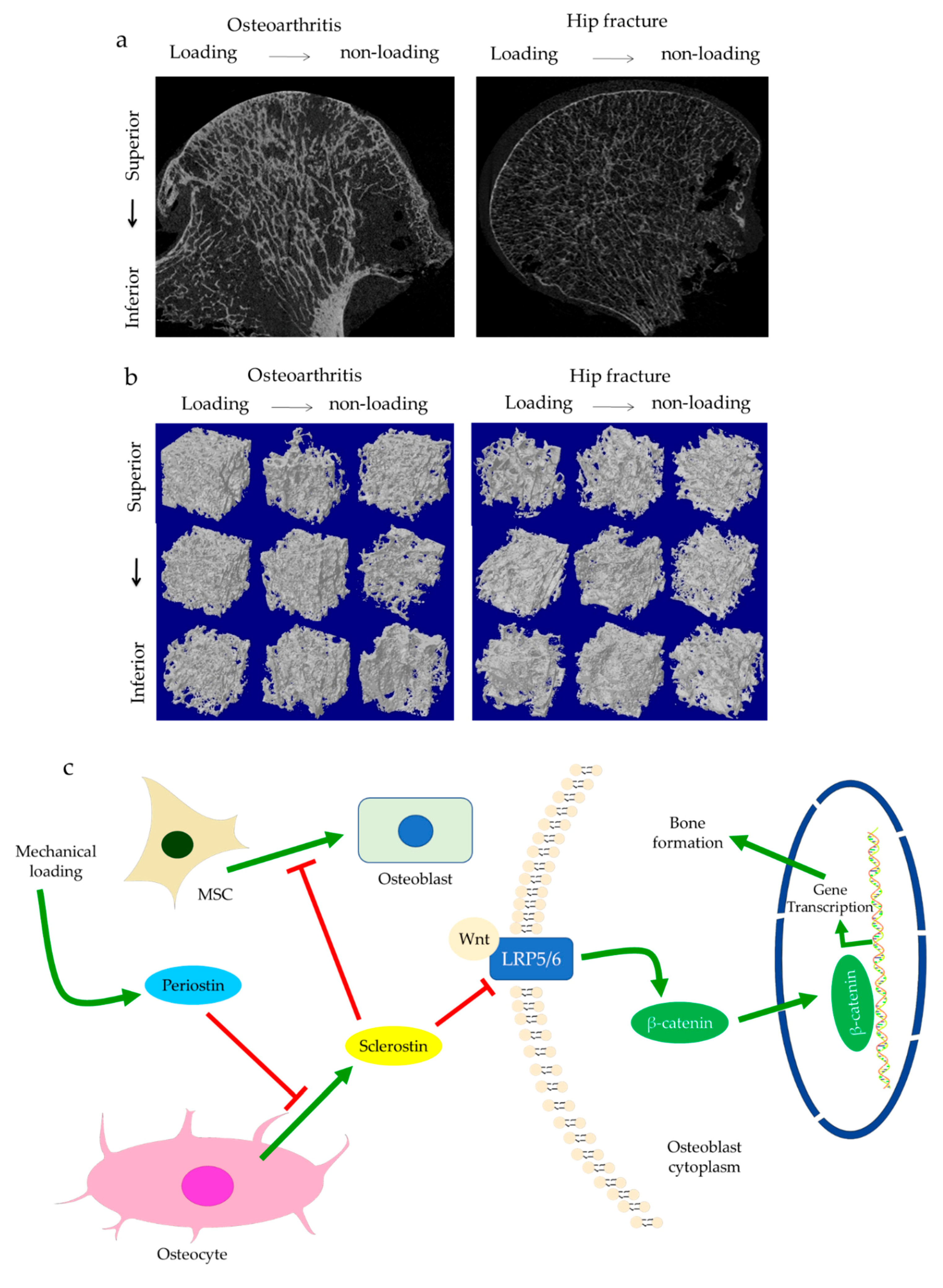{kind=link}
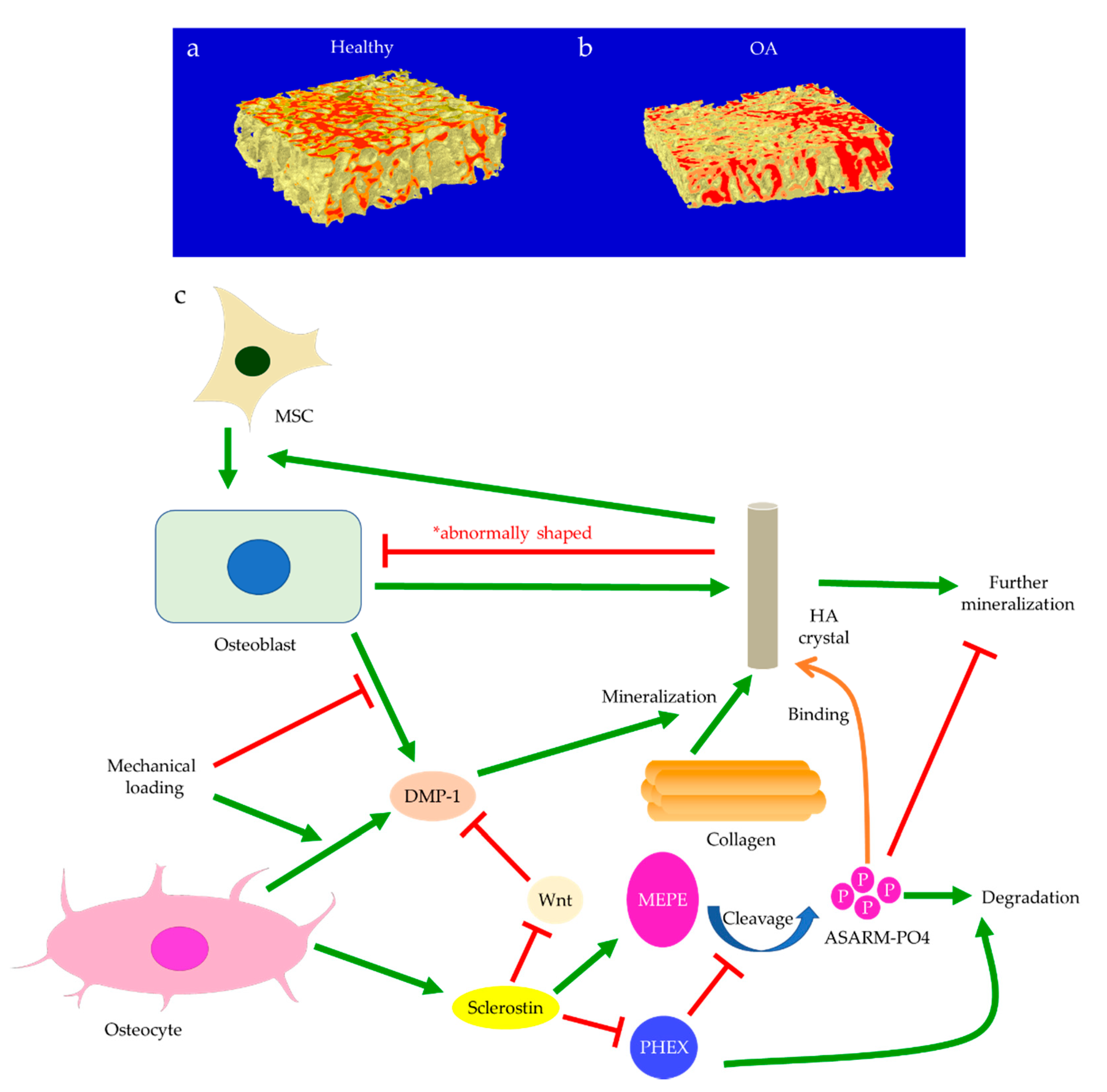{kind=link}
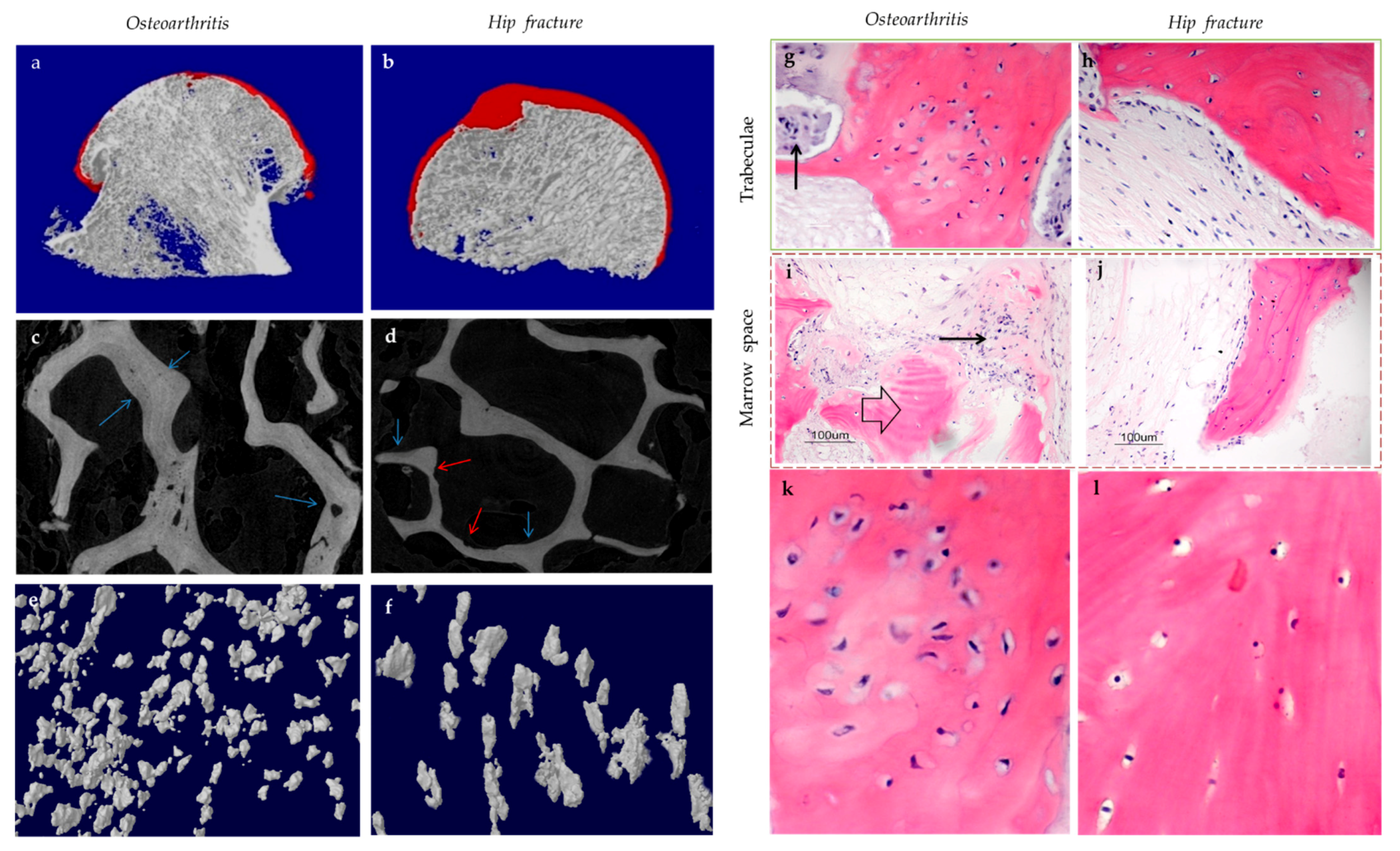{kind=link}
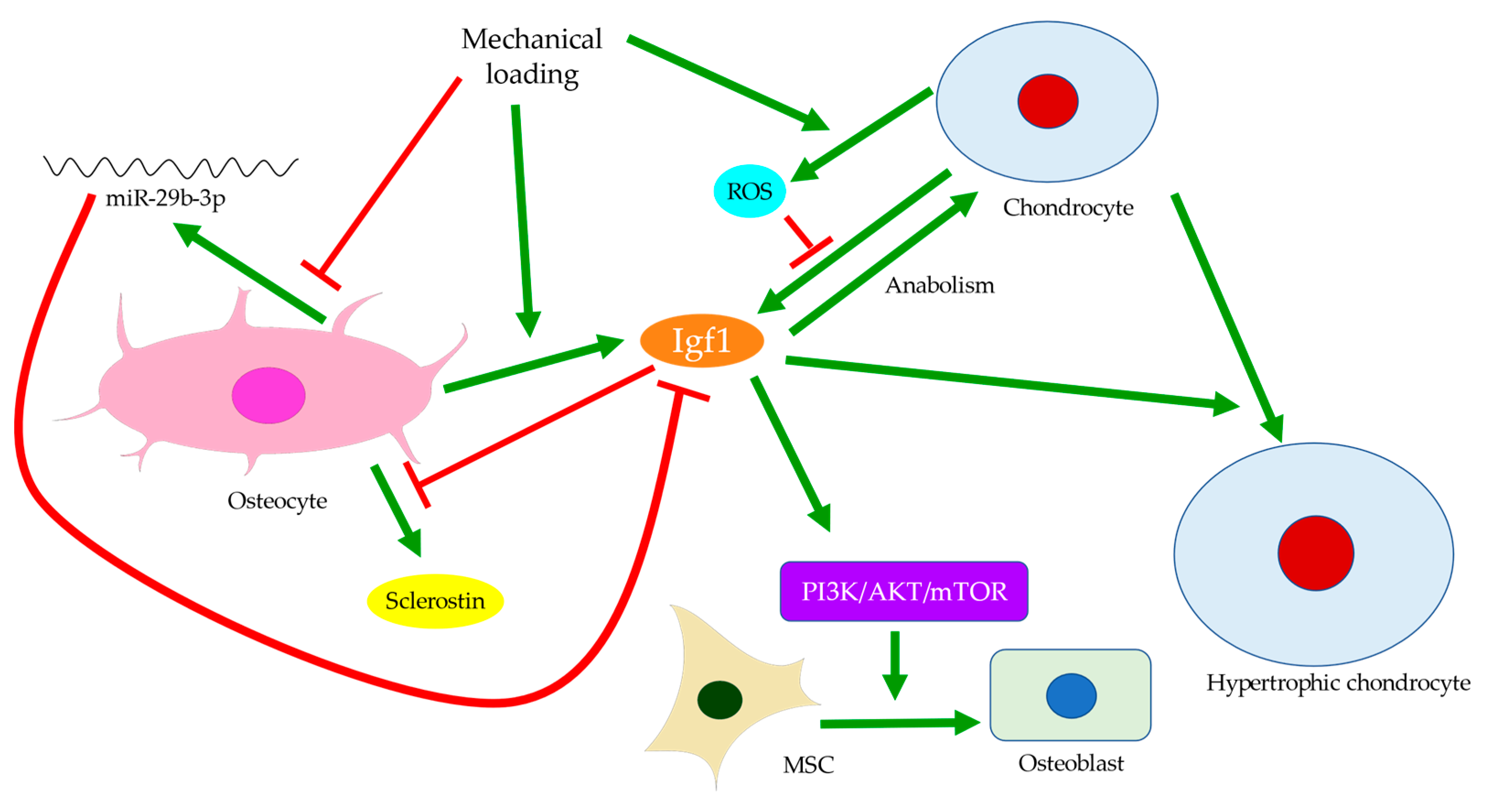{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Disturbances of Subchondral Bone
Crosstalk with Cartilage Degeneration
3. Osteocyte Dysfunction
3.1. Osteocyte Morphology
3.2. Regulation of Osteoblasts
3.3. Regulation of Mesenchymal Progenitors
3.4. Regulation of Cartilage Tissues
3.5. Regulation of Osteoclasts
4. Allometry
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kendzerska, T.; Juni, P.; King, L.K.; Croxford, R.; Stanaitis, I.; Hawker, G.A. The longitudinal relationship between hand, hip and knee osteoarthritis and cardiovascular events: A population-based cohort study. Osteoarthr. Cartil. 2017, 25, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Brandt, K.D.; Dieppe, P.; Radin, E. Etiopathogenesis of osteoarthritis. Med. Clin. N. Am. 2009, 93, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Gerstenfeld, L.; Bishop, G.; Davis, A.D.; Mason, Z.D.; Einhorn, T.A.; Maciewicz, R.A.; Newham, P.; Foster, M.; Jackson, S.; et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res. Ther. 2009, 11, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K.; Nango, N.; Kubota, S.; Okazaki, N.; Taguchi, K.; Osaki, M.; Ito, M. Relationship between microstructure and degree of mineralization in subchondral bone of osteoarthritis: A synchrotron radiation µCT study. J. Bone Miner. Res. 2012, 27, 1511–1517. [Google Scholar] [CrossRef]
- Coats, A.M.; Zioupos, P.; Aspden, R.M. Material properties of subchondral bone from patients with osteoporosis or osteoarthritis by microindentation testing and electron probe microanalysis. Calcif. Tissue Int. 2003, 73, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Couchourel, D.; Aubry, I.; Delalandre, A.; Lavigne, M.; Martel-Pelletier, J.; Pelletier, J.-P.; Lajeunesse, D. Altered mineralization of human osteoarthritic osteoblasts is attributable to abnormal type I collagen production. Arthritis Rheumatol. 2009, 60, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Bettica, P.; Cline, G.; Hart, D.J.; Meyer, J.; Spector, T.D. Evidence for increased bone resorption in patients with progressive knee osteoarthritis: Longitudinal results from the Chingford study. Arthritis Rheumatol. 2002, 46, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Xiao, H.; Liao, J.; Zhou, N.; Zhao, C.; Xu, W.; Xu, W.; Liang, X.; Huang, W. Osteocyte TGFβ1-Smad2/3 is positively associated with bone turnover parameters in subchondral bone of advanced osteoarthritis. Int. J. Mol. Med. 2020, 46, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lin, S.; Liu, Y.; Yuan, B.; Harris, S.E.; Feng, J.Q. Dmp1 null mice develop a unique osteoarthritis-like phenotype. Int. J. Biol. Sci. 2016, 12, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, N.; Standley, K.N.; Bianchi, E.N.; Stadelmann, V.; Foti, M.; Conway, S.J.; Ferrari, S.L. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J. Biol. Chem. 2009, 284, 35939–35950. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Sun, X.; Ma, J.; Ma, X.; Xing, G.; Wang, Y.; Sun, L.; Wang, J.; Li, F.; Li, Y. Involvement of periostin-sclerostin-Wnt/β-catenin signaling pathway in the prevention of neurectomy-induced bone loss by naringin. Biochem. Biophys. Res. Commun. 2015, 468, 587–593. [Google Scholar] [CrossRef]
- Funck-Brentano, T.; Bouaziz, W.; Marty, C.; Geoffroy, V.; Hay, E.; Cohen-Solal, M. Dkk-1-Mediated inhibition of Wnt signaling in bone ameliorates osteoarthritis in mice. Arthritis Rheumatol. 2014, 66, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, C.; Ferretti, M. The osteocyte: From “prisoner” to “orchestrator”. J. Funct. Morphol. Kinesiol. 2021, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.D.; Nevitt, M.C.; Wildy, K.; Barrow, K.D.; Harris, F.; Felson, D.; Peterfy, C.; Visser, M.; Harris, T.B.; Wang, B.W.E.; et al. The relationship of antiresorptive drug use to structural findings and symptoms of knee osteoarthritis. Arthritis Rheumatol. 2004, 50, 3516–3525. [Google Scholar] [CrossRef]
- Ding, M.; Danielsen, C.C.; Hvid, I. The Effects of bone remodeling inhibition by alendronate on three-dimensional microarchitecture of subchondral bone tissues in guinea pig primary osteoarthrosis. Calcif. Tissue Int. 2008, 82, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayami, T.; Pickarski, M.; Wesolowski, G.A.; Mclane, J.; Bone, A.; Destefano, J.; Rodan, G.A.; Duong, L.T. The role of subchondral bone remodeling in osteoarthritis: Reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheumatol. 2004, 50, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Neutzsky-Wulff, A.V.; Bonewald, L.F.; Karsdal, M.A. Local communication on and within bone controls bone remodeling. Bone 2009, 44, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Lajeunesse, D.; Reboul, P. Subchondral bone in osteoarthritis: A biologic link with articular cartilage leading to abnormal remodeling. Curr. Opin. Rheumatol. 2003, 15, 628–633. [Google Scholar] [CrossRef]
- Massicotte, F.; Aubry, I.; Martel-Pelletier, J.; Pelletier, J.-P.; Fernandes, J.; Lajeunesse, D. Abnormal insulin-like growth factor 1 signaling in human osteoarthritic subchondral bone osteoblasts. Arthritis Res. Ther. 2006, 8, R177. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.; Deberg, M.A.; Bellahcène, A.; Castronovo, V.; Msika, P.; Delcour, J.P.; Crielaard, J.M.; Henrotin, Y.E. Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheumatol. 2008, 58, 442–455. [Google Scholar] [CrossRef]
- Hemmatian, H.; Bakker, A.D.; Klein-Nulend, J.; van Lenthe, G.H. Aging, osteocytes, and mechanotransduction. Curr. Osteoporos. Rep. 2017, 15, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Ten Dijke, P.; Krause, C.; de Gorter, D.J.J.; Löwik, C.W.G.M.; van Bezooijen, R.L. Osteocyte-Derived sclerostin inhibits bone formation: Its role in bone morphogenetic protein and Wnt signaling. J. Bone Jt. Surg. 2008, 90, 31–35. [Google Scholar] [CrossRef]
- Van Bezooijen, R.L.; Roelen, B.A.J.; Visser, A.; van der Wee-Pals, L.; de Wilt, E.; Karperien, M.; Hamersma, H.; Papapoulos, S.E.; ten Dijke, P.; Löwik, C.W.G.M. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 2004, 199, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. Osteocytes: A proposed multifunctional bone cell. J. Musculoskelet. Neuronal Interact. 2002, 2, 239–241. [Google Scholar]
- Li, J.; Xue, J.; Jing, Y.; Wang, M.; Shu, R.; Xu, H.; Xue, C.; Feng, J.; Wang, P.; Bai, D. SOST deficiency aggravates osteoarthritis in mice by promoting sclerosis of subchondral bone. Biomed. Res. Int. 2019, 2019, 7623562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappard, C.; Peyrin, F.; Bonnassie, A.; Lemineur, G.; Brunet-Imbault, B.; Lespessailles, E.; Benhamou, C.L. Subchondral bone micro-architectural alterations in osteoarthritis: A synchrotron micro-computed tomography study. Osteoarthr. Cartil. 2006, 14, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K.; Ito, M.; Osaki, M.; Uetani, M.; Shindo, H. In vivo structural analysis of subchondral trabecular bone in osteoarthritis of the hip using multi-detector row CT. Osteoarthr. Cartil. 2011, 19, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Jaiprakash, A.; Prasadam, I.; Feng, J.Q.; Liu, Y.; Crawford, R.; Xiao, Y. Phenotypic characterization of osteoarthritic osteocytes from the sclerotic zones: A possible pathological role in subchondral bone sclerosis. Int. J. Biol. Sci. 2012, 8, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Jiang, X.; Dai, Z.; Guo, X.; Weng, T.; Wang, J.; Li, Y.; Feng, G.; Gao, X.; He, L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/β-Catenin signaling. J. Bone Miner. Res. 2009, 24, 1651–1661. [Google Scholar] [CrossRef]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Ma, X.; Wei, Y.; Tong, W.; Tower, R.J.; Chandra, A.; Wang, L.; Sun, Z.; Yang, Z.; Badar, F.; et al. Loading-Induced reduction in sclerostin as a mechanism of subchondral bone plate sclerosis in mouse knee joints during late-stage osteoarthritis. Arthritis Rheumatol. 2018, 70, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Ko, F.C.; Dragomir, C.; Plumb, D.A.; Goldring, S.R.; Wright, T.M.; Goldring, M.B.; van der Meulen, M.C.H. In vivo cyclic compression causes cartilage degeneration and subchondral bone changes in mouse tibiae. Arthritis Rheumatol. 2013, 65, 1569–1578. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, N.; Garnero, P.; Ferrari, S. Periostin action in bone. Mol. Cell. Endocrinol. 2016, 432, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Tajika, Y.; Moue, T.; Ishikawa, S.; Asano, K.; Okumo, T.; Takagi, H.; Hisamitsu, T. Influence of periostin on synoviocytes in knee osteoarthritis. In Vivo 2017, 31, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Chijimatsu, R.; Kunugiza, Y.; Taniyama, Y.; Nakamura, N.; Tomita, T.; Yoshikawa, H. Expression and pathological effects of periostin in human osteoarthritis cartilage. BMC Musculoskelet. Disord. 2015, 16, 215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Fang, H.; Chen, Y.; Shen, J.; Lu, H.; Zeng, C.; Ren, J.; Zeng, H.; Li, Z.; Chen, S.; et al. Gene expression analyses of subchondral bone in early experimental osteoarthritis by microarray. PLoS ONE 2012, 7, e32356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnilä, M.A.J.; Thevenot, J.; Aho, O.-M.; Tiitu, V.; Rautiainen, J.; Kauppinen, S.; Nieminen, M.T.; Pritzker, K.; Valkealahti, M.; Lehenkari, P.; et al. Association between subchondral bone structure and osteoarthritis histopathological grade. J. Orthop. Res. 2017, 35, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Odgaard, A.; Danielsen, C.C.; Hvid, I. Mutual associations among microstructural, physical and mechanical properties of human cancellous bone. J. Bone Jt. Surg. Br. 2002, 84, 900–907. [Google Scholar] [CrossRef]
- Wang, T.; Wen, C.Y.; Yan, C.H.; Lu, W.W.; Chiu, K.Y. Spatial and temporal changes of subchondral bone proceed to microscopic articular cartilage degeneration in guinea pigs with spontaneous osteoarthritis. Osteoarthr. Cartil. 2013, 21, 574–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, S.J.; Izuhara, K.; Kudo, Y.; Litvin, J.; Markwald, R.; Ouyang, G.; Arron, J.R.; Holweg, C.T.; Kudo, A. The role of periostin in tissue remodeling across health and disease. Cell. Mol. Life. Sci. 2014, 71, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Spatz, J.M.; Ellman, R.; Cloutier, A.M.; Louis, L.; van Vliet, M.; Suva, L.J.; Dwyer, D.; Stolina, M.; Ke, H.Z.; Bouxsein, M.L. Sclerostin antibody inhibits skeletal deterioration due to reduced mechanical loading. J. Bone Miner. Res. 2013, 28, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Gingery, A.; Subramaniam, M.; Pitel, K.S.; Li, X.; Ke, H.Z.; Turner, R.T.; Iwaniec, U.T.; Hawse, J.R. Sclerostin antibody treatment rescues the osteopenic bone phenotype of TGFβ inducible early gene-1 knockout female mice. J. Cell Physiol. 2020, 235, 5679–5688. [Google Scholar] [CrossRef]
- Mazur, C.M.; Woo, J.J.; Yee, C.S.; Fields, A.J.; Acevedo, C.; Bailey, K.N.; Kaya, S.; Fowler, T.W.; Lotz, J.C.; Dang, A.; et al. Osteocyte dysfunction promotes osteoarthritis through MMP13-dependent suppression of subchondral bone homeostasis. Bone Res. 2019, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Rowe, P.S.N. Regulation of bone-renal mineral and energy metabolism: The PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 61–86. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Rios, H.F.; Myers, E.R.; Lu, Y.; Feng, J.Q.; Boskey, A.L. DMP1 depletion decreases bone mineralization in vivo: An FTIR imaging analysis. J. Bone Miner. Res. 2005, 20, 2169–2177. [Google Scholar] [CrossRef]
- Baranyay, F.J.; Wang, Y.; Wluka, A.E.; English, D.R.; Giles, G.G.; Sullivan, R.O.; Cicuttini, F.M. Association of bone marrow lesions with knee structures and risk factors for bone marrow lesions in the knees of clinically healthy, community-based adults. Semin. Arthritis Rheum. 2007, 37, 112–118. [Google Scholar] [CrossRef]
- Davies-Tuck, M.L.; Wluka, A.E.; Forbes, A.; Wang, Y.; English, D.R.; Giles, G.G.; O’Sullivan, R.; Cicuttini, F.M. Development of bone marrow lesions is associated with adverse effects on knee cartilage while resolution is associated with improvement--a potential target for prevention of knee osteoarthritis: A longitudinal study. Arthritis Res. Ther. 2010, 12, R10. [Google Scholar] [CrossRef] [Green Version]
- Dore, D.; Martens, A.; Quinn, S.; Ding, C.; Winzenberg, T.; Zhai, G.; Pelletier, J.-P.; Martel-Pelletier, J.; Abram, F.; Cicuttini, F.; et al. Bone marrow lesions predict site-specific cartilage defect development and volume loss: A prospective study in older adults. Arthritis Res. Ther. 2010, 12, R222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornaat, P.R.; Kloppenburg, M.; Sharma, R.; Botha-Scheepers, S.A.; Le Graverand, M.-P.H.; Coene, L.N.J.E.M.; Bloem, J.L.; Watt, I. Bone marrow edema-like lesions change in volume in the majority of patients with osteoarthritis; associations with clinical features. Eur. Radiol. 2007, 17, 3073–3078. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, K.; Ramachandran, A.; Hao, J.; He, G.; Park, K.W.; Cho, M.; George, A. Dual functional roles of dentin matrix protein 1. Implications in biomineralization and gene transcription by activation of intracellular Ca2+ store. J. Biol. Chem. 2003, 278, 17500–17508. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lin, J.; Shao, J.; Zuo, Q.; Wang, S.; Wolff, A.; Nguyen, D.T.; Rintoul, L.; Du, Z.; Gu, Y.; et al. Aberrant activation of Wnt signaling pathway altered osteocyte mineralization. Bone 2019, 127, 324–333. [Google Scholar] [CrossRef]
- Gluhak-Heinrich, J.; Ye, L.; Bonewald, L.F.; Feng, J.Q.; MacDougall, M.; Harris, S.E.; Pavlin, D. Mechanical loading stimulates dentin matrix protein 1 (DMP1) expression in osteocytes in vivo. J. Bone Miner. Res. 2003, 18, 807–817. [Google Scholar] [CrossRef]
- Ding, M.; Odgaard, A.; Hvid, I. Changes in the three-dimensional microstructure of human tibial cancellous bone in early osteoarthritis. J. Bone Jt. Surg. Br. 2003, 85, 906–912. [Google Scholar] [CrossRef]
- Brown, S.J.; Pollintine, P.; Powell, D.E.; Davie, M.W.J.; Sharp, C.A. Regional differences in mechanical and material properties of femoral head cancellous bone in health and osteoarthritis. Calcif. Tissue Int. 2002, 71, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Cicuttini, F.; Jones, G. Tibial subchondral bone size and knee cartilage defects: Relevance to knee osteoarthritis. Osteoarthr. Cartil. 2007, 15, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xuan, S.; Bouxsein, M.L.; von Stechow, D.; Akeno, N.; Faugere, M.C.; Malluche, H.; Zhao, G.; Rosen, C.J.; Efstratiadis, A.; et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J. Biol. Chem. 2002, 277, 44005–44012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, K.L.; Oh, S.; Sung, Y.; Dasari, R.R.; Kirschner, M.W.; Tabin, C.J. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 2013, 495, 375–378. [Google Scholar] [CrossRef]
- Sheng, M.H.; Lau, K.H.; Baylink, D.J. Role of osteocyte-derived insulin-like growth factor I in developmental growth, modeling, remodeling, and regeneration of the bone. J. Bone Metab. 2014, 21, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Wang, Y.; Bikle, D.D. IGF-1 signaling mediated cell-specific skeletal mechano-transduction. J. Orthop. Res. 2018, 36, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Chubinskaya, S.; Hakimiyan, A.; Pacione, C.; Yanke, A.; Rappoport, L.; Aigner, T.; Rueger, D.C.; Loeser, R.F. Synergistic effect of IGF-1 and OP-1 on matrix formation by normal and OA chondrocytes cultured in alginate beads. Osteoarthr. Cartil. 2007, 15, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeser, R.F. Molecular mechanisms of cartilage destruction: Mechanics, inflammatory mediators, and aging collide. Arthritis Rheumatol. 2006, 54, 1357–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Adams, J.; Leddy, H.A.; McNulty, A.L.; O’Conor, C.J.; Guilak, F. The mechanobiology of articular cartilage: Bearing the burden of osteoarthritis. Curr. Rheumatol. Rep. 2014, 16, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plumb, M.S.; Treon, K.; Aspden, R.M. Competing regulation of matrix biosynthesis by mechanical and IGF-1 signalling in elderly human articular cartilage in vitro. Biochim. Biophys. Acta 2006, 1760, 762–767. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Yu, Y.E.; Zhang, X.; Watts, T.; Zhou, B.; Wang, J.; Wang, T.; Zhao, W.; Chiu, K.Y.; et al. Subchondral trabecular rod loss and plate thickening in the development of osteoarthritis. J. Bone Miner. Res. 2018, 33, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, T.; Hagino, H.; Okano, T.; Enokida, M.; Teshima, R. Role of subchondral bone in osteoarthritis development: A comparative study of two strains of guinea pigs with and without spontaneously occurring osteoarthritis. Arthritis Rheumatol. 2007, 56, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Fukuda, K.; Yamazaki, K.; Hashimoto, K.; Ueda, H.; Mori, S.; Hamanishi, C. Cyclic compression loaded on cartilage explants enhances the production of reactive oxygen species. J. Rheumatol. 2007, 34, 556–562. [Google Scholar]
- Wolff, K.J.; Ramakrishnan, P.S.; Brouillette, M.J.; Journot, B.J.; McKinley, T.O.; Buckwalter, J.A.; Martin, J.A. Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J. Orthop. Res. 2013, 31, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youlten, S.E.; Kemp, J.P.; Logan, J.G.; Ghirardello, E.J.; Sergio, C.M.; Dack, M.R.G.; Guilfoyle, S.E.; Leitch, V.D.; Butterfield, N.C.; Komla-Ebri, D.; et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat. Commun. 2021, 12, 2444. [Google Scholar] [CrossRef]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of sost/sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.F.; Saunders, M.M.; Shingle, D.L.; Cimbala, J.M.; Zhou, Z.; Donahue, H.J. Mechanically stimulated osteocytes regulate osteoblastic activity via gap junctions. Am. J. Physiol. Cell Physiol. 2007, 292, C545–C552. [Google Scholar] [CrossRef] [Green Version]
- Staines, K.A.; Poulet, B.; Farquharson, C.; Pitsillides, A.A. The sclerostin and MEPE axis in the development of osteoarthritis. Osteoarthr. Cartil. 2013, 21. [Google Scholar] [CrossRef] [Green Version]
- Morse, A.; Schindeler, A.; McDonald, M.M.; Kneissel, M.; Kramer, I.; Little, D.G. Sclerostin antibody augments the anabolic bone formation response in a mouse model of mechanical tibial loading. J. Bone Miner. Res. 2018, 33, 486–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein-Nulend, J.; Bacabac, R.G.; Bakker, A.D. Mechanical loading and how it affects bone cells: The role of the osteocyte cytoskeleton in maintaining our skeleton. Eur. Cells Mater. 2012, 24, 278–291. [Google Scholar] [CrossRef]
- Lyons, J.S.; Joca, H.C.; Law, R.A.; Williams, K.M.; Kerr, J.P.; Shi, G.; Khairallah, R.J.; Martin, S.S.; Konstantopoulos, K.; Ward, C.W.; et al. Microtubules tune mechanotransduction through NOX2 and TRPV4 to decrease sclerostin abundance in osteocytes. Sci. Signal. 2017, 10, eaan5748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Fu, X.; Ma, J.; Lin, M.; Zhang, P.; Wang, Y.; Yan, Q.; Tao, C.; Liu, W.; Tang, B.; et al. Kindlin-2 mediates mechanotransduction in bone by regulating expression of Sclerostin in osteocytes. Commun. Biol. 2021, 4, 402. [Google Scholar] [CrossRef] [PubMed]
- Rabelo, G.D.; vom Scheidt, A.; Klebig, F.; Hemmatian, H.; Citak, M.; Amling, M.; Busse, B.; Jähn, K. Multiscale bone quality analysis in osteoarthritic knee joints reveal a role of the mechanosensory osteocyte network in osteophytes. Sci. Rep. 2020, 10, 673. [Google Scholar] [CrossRef] [Green Version]
- Intemann, J.; De Gorter, D.J.J.; Naylor, A.J.; Dankbar, B.; Wehmeyer, C. Importance of osteocyte-mediated regulation of bone remodelling in inflammatory bone disease. Swiss Med. Wkly. 2020, 150, w20187. [Google Scholar] [CrossRef]
- Prideaux, M.; Loveridge, N.; Pitsillides, A.A.; Farquharson, C. Extracellular matrix mineralization promotes E11/gp38 glycoprotein expression and drives osteocytic differentiation. PLoS ONE 2012, 7, e36786. [Google Scholar] [CrossRef] [Green Version]
- Ilas, D.C.; Churchman, S.M.; Baboolal, T.; Giannoudis, P.V.; Aderinto, J.; McGonagle, D.; Jones, E. The simultaneous analysis of mesenchymal stem cells and early osteocytes accumulation in osteoarthritic femoral head sclerotic bone. Rheumatology 2019, 58, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Van Hove, R.P.; Nolte, P.A.; Vatsa, A.; Semeins, C.M.; Salmon, P.L.; Smit, T.H.; Klein-Nulend, J. Osteocyte morphology in human tibiae of different bone pathologies with different bone mineral density—Is there a role for mechanosensing? Bone 2009, 45, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Hemmatian, H.; Jalali, R.; Semeins, C.M.; Hogervorst, J.M.A.; van Lenthe, G.H.; Klein-Nulend, J.; Bakker, A.D. Mechanical loading differentially affects osteocytes in fibulae from lactating mice compared to osteocytes in virgin mice: Possible role for lacuna size. Calcif. Tissue Int. 2018, 103, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Yuan, B.; Qin, C.; Cao, Z.; Xie, Y.; Dallas, S.L.; McKee, M.D.; Drezner, M.K.; Bonewald, L.F.; Feng, J.Q. The biological function of DMP-1 in osteocyte maturation is mediated by its 57-kDa C-terminal fragment. J. Bone Miner. Res. 2011, 26, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacabac, R.G.; Mizuno, D.; Schmidt, C.F.; MacKintosh, F.C.; Van Loon, J.J.W.A.; Klein-Nulend, J.; Smit, T.H. Round versus flat: Bone cell morphology, elasticity, and mechanosensing. J. Biomech. 2008, 41, 1590–1598. [Google Scholar] [CrossRef]
- Kim, J.; Ishikawa, K.; Sunaga, J.; Adachi, T. Uniaxially fixed mechanical boundary condition elicits cellular alignment in collagen matrix with induction of osteogenesis. Sci. Rep. 2021, 11, 9009. [Google Scholar] [CrossRef]
- Kerschnitzki, M.; Wagermaier, W.; Roschger, P.; Seto, J.; Shahar, R.; Duda, G.N.; Mundlos, S.; Fratzl, P. The organization of the osteocyte network mirrors the extracellular matrix orientation in bone. J. Struct. Biol. 2011, 173, 303–311. [Google Scholar] [CrossRef]
- Van Oers, R.F.M.; Wang, H.; Bacabac, R.G. Osteocyte shape and mechanical loading. Curr. Osteoporos. Rep. 2015, 13, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.H. Skeletal adaptation to mechanical loading. Clin. Rev. Bone Miner. Metab. 2008, 5, 181–194. [Google Scholar] [CrossRef]
- Wittkowske, C.; Reilly, G.C.; Lacroix, D.; Perrault, C.M. In vitro bone cell models: Impact of fluid shear stress on bone formation. Front Bioeng. Biotechnol. 2016, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Flynn, B.P.; Bhole, A.P.; Saeidi, N.; Liles, M.; Dimarzio, C.A.; Ruberti, J.W. Mechanical strain stabilizes reconstituted collagen fibrils against enzymatic degradation by mammalian collagenase matrix metalloproteinase 8 (MMP-8). PLoS ONE 2010, 5, e12337. [Google Scholar] [CrossRef] [Green Version]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen fibrillogenesis: Fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008, 20, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Kormpakis, I.; Havlioglu, N.; Ominsky, M.S.; Galatz, L.M.; Thomopoulos, S. Sclerostin antibody treatment enhances rotator cuff tendon-to-bone healing in an animal model. J. Bone Jt. Surg. Am. 2017, 99, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, N.; Gineyts, E.; Ammann, P.; Conway, S.J.; Garnero, P.; Ferrari, S. Periostin deficiency increases bone damage and impairs injury response to fatigue loading in adult mice. PLoS ONE 2013, 8, e78347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wei, W.; Chi, M.; Wan, Y.; Li, X.; Qi, M.; Zhou, Y. FOXO1 Mediates advanced glycation end products induced mouse osteocyte-like MLO-Y4 cell apoptosis and dysfunctions. J. Diabetes Res. 2019, 2019, 6757428. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Hulley, P. The osteocyte—A novel endocrine regulator of body phosphate homeostasis. Maturitas 2010, 67, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R. The osteocyte: Key player in regulating bone turnover. RMD Open 2015, 1, e000049. [Google Scholar] [CrossRef]
- Rony, L.; Perrot, R.; Hubert, L.; Chappard, D. Osteocyte staining with rhodamine in osteonecrosis and osteoarthritis of the femoral head. Microsc. Res. Tech. 2019, 82, 2072–2078. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Arozamena, J.; García-Renedo, R.; García-Ibarbia, C.; Pascual-Carra, M.A.; González-Macías, J.; Riancho, J.A. Osteocyte deficiency in hip fractures. Calcif. Tissue Int. 2011, 89, 327. [Google Scholar] [CrossRef]
- Duan, W.R.; Garner, D.S.; Williams, S.D.; Funckes-Shippy, C.L.; Spath, I.S.; Blomme, E.A. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J. Pathol. 2003, 199, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, J.; Hoemann, C.D.; Lascau-Coman, V.; Ouyang, W.; McKee, M.D.; Shive, M.S.; Buschmann, M.D. Drilling and microfracture lead to different bone structure and necrosis during bone-marrow stimulation for cartilage repair. J. Orthop. Res. 2009, 27, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Andreev, D.; Liu, M.; Weidner, D.; Kachler, K.; Faas, M.; Grüneboom, A.; Schlötzer-Schrehardt, U.; Muñoz, L.E.; Steffen, U.; Grötsch, B.; et al. Osteocyte necrosis triggers osteoclast-mediated bone loss through macrophage-inducible C-type lectin. J. Clin. Investig. 2020, 130, 4811–4830. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yang, S.; Shao, J.; Li, Y.-P. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front. Biosci. 2007, 12, 3068–3092. [Google Scholar] [CrossRef] [Green Version]
- Sawakami, K.; Robling, A.G.; Ai, M.; Pitner, N.D.; Liu, D.; Warden, S.J.; Li, J.; Maye, P.; Rowe, D.W.; Duncan, R.L.; et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J. Biol. Chem. 2006, 281, 23698–23711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruotti, N.; Corrado, A.; Cantatore, F.P. Osteoblast role in osteoarthritis pathogenesis. J. Cell. Physiol. 2017, 232, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Zoch, M.L.; Clemens, T.L.; Riddle, R.C. New insights into the biology of osteocalcin. Bone 2016, 82, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Kim, D.J.; Shen, J.; Zou, Z.; O’Keefe, R.J. Runx2 plays a central role in osteoarthritis development. J. Orthop. Translat. 2020, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Wang, Y.; Gao, J.; Yan, Z.; Li, Z.; Zou, X.; Li, Y.; Wang, J.; Guo, Y. miR-29b-3p regulated osteoblast differentiation via regulating IGF-1 secretion of mechanically stimulated osteocytes. Cell. Mol. Biol. Lett. 2019, 24, 11. [Google Scholar] [CrossRef] [Green Version]
- D’Emic, M.D.; Benson, R.B.J. Measurement, variation, and scaling of osteocyte lacunae: A case study in birds. Bone 2013, 57, 300–310. [Google Scholar] [CrossRef]
- Teti, A.; Zallone, A. Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone 2009, 44, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Grunmeier, O., 3rd; D’Emic, M.D. Scaling of statically derived osteocyte lacunae in extant birds: Implications for palaeophysiological reconstruction. Biol. Lett. 2019, 15, 20180837. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, M.; Palumbo, C. Static osteogenesis versus dynamic osteogenesis: A comparison between two different types of bone formation. Appl. Sci. 2021, 11, 2025. [Google Scholar] [CrossRef]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. N. Y. Acad. Sci. 2010, 1192, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhang, J.; Wu, J.; Guan, Y.; Weng, Y.; Shang, P. Oscillatory fluid flow elicits changes in morphology, cytoskeleton and integrin-associated molecules in MLO-Y4 cells, but not in MC3T3-E1 cells. Biol. Res. 2012, 45, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef] [Green Version]
- Perez-Campo, F.M.; Santurtun, A.; Garcia-Ibarbia, C.; Pascual, M.A.; Valero, C.; Garces, C.; Sanudo, C.; Zarrabeitia, M.T.; Riancho, J.A. Osterix and RUNX2 are transcriptional regulators of sclerostin in human bone. Calcif. Tissue Int. 2016, 99, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Raguin, E.; Shahar, R. High resolution 3D structures of mineralized tissues in health and disease. Nat. Rev. Endocrinol. 2021, 17, 307–316. [Google Scholar] [CrossRef]
- Arnold, A.; Dennison, E.; Kovacs, C.S.; Mannstadt, M.; Rizzoli, R.; Brandi, M.L.; Clarke, B.; Thakker, R.V. Hormonal regulation of biomineralization. Nat. Rev. Endocrinol. 2021, 17, 261–275. [Google Scholar] [CrossRef]
- Atkins, G.J.; Rowe, P.S.; Lim, H.P.; Welldon, K.J.; Ormsby, R.; Wijenayaka, A.R.; Zelenchuk, L.; Evdokiou, A.; Findlay, D.M. Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J. Bone Miner. Res. 2011, 26, 1425–1436. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.L.; Khor, K.A.; Sui, J.J.; Zhang, J.H.; Chen, W.N. Protein expression profiles in osteoblasts in response to differentially shaped hydroxyapatite nanoparticles. Biomaterials 2009, 30, 5385–5391. [Google Scholar] [CrossRef]
- Karperien, M.; Roelen, B.A.J.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Hierck, B.P.; DeRuiter, M.C.; Meijer, D.; Gibbs, S. Tissue formation during embryogenesis. In Tissue Engineering, 2nd ed.; Blitterswijk, C.A.V., De Boer, J., Eds.; Academic Press: Oxford, UK, 2015; pp. 67–109. [Google Scholar]
- Koelling, S.; Kruegel, J.; Irmer, M.; Path, J.R.; Sadowski, B.; Miro, X.; Miosge, N. Migratory chondrogenic progenitor cells from repair tissue during the later stages of human osteoarthritis. Cell Stem Cell 2009, 4, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Grogan, S.P.; Miyaki, S.; Asahara, H.; D’Lima, D.D.; Lotz, M.K. Mesenchymal progenitor cell markers in human articular cartilage: Normal distribution and changes in osteoarthritis. Arthritis Res. Ther. 2009, 11, R85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoudis, P.V.; Goff, T.; Roshdy, T.; Jones, E.; McGonagle, D. Does mobilisation and transmigration of mesenchymal stem cells occur after trauma? Injury 2010, 41, 1099–1102. [Google Scholar] [CrossRef]
- Jones, E.A.; Crawford, A.; English, A.; Henshaw, K.; Mundy, J.; Corscadden, D.; Chapman, T.; Emery, P.; Hatton, P.; McGonagle, D. Synovial fluid mesenchymal stem cells in health and early osteoarthritis: Detection and functional evaluation at the single-cell level. Arthritis Rheumatol. 2008, 58, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Tang, S.Y.; Nguyen, D.; Alliston, T. Load regulates bone formation and sclerostin expression through a TGFβ-dependent mechanism. PLoS ONE 2013, 8, e53813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherifi, C.; Monteagudo, S.; Lories, R.J. Promising targets for therapy of osteoarthritis: A review on the Wnt and TGF-β signalling pathways. Ther. Adv. Musculoskelet. Dis. 2021, 13. [Google Scholar] [CrossRef]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Lories, R.J.; Luyten, F.P. The bone-cartilage unit in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 43–49. [Google Scholar] [CrossRef]
- Dozza, B.; Lesci, I.G.; Duchi, S.; Della Bella, E.; Martini, L.; Salamanna, F.; Falconi, M.; Cinotti, S.; Fini, M.; Lucarelli, E.; et al. When size matters: Differences in demineralized bone matrix particles affect collagen structure, mesenchymal stem cell behavior, and osteogenic potential. J. Biomed. Mater. Res. A 2017, 105, 1019–1033. [Google Scholar] [CrossRef]
- Roohani-Esfahani, S.-I.; Nouri-Khorasani, S.; Lu, Z.; Appleyard, R.; Zreiqat, H. The influence hydroxyapatite nanoparticle shape and size on the properties of biphasic calcium phosphate scaffolds coated with hydroxyapatite–PCL composites. Biomaterials 2010, 31, 5498–5509. [Google Scholar] [CrossRef] [PubMed]
- Zandi, M.; Mirzadeh, H.; Mayer, C.; Urch, H.; Eslaminejad, M.B.; Bagheri, F.; Mivehchi, H. Biocompatibility evaluation of nano-rod hydroxyapatite/gelatin coated with nano-HAp as a novel scaffold using mesenchymal stem cells. J. Biomed. Mater. Res. A 2010, 92A, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Bodhak, S.; Bose, S.; Bandyopadhyay, A. Role of surface charge and wettability on early stage mineralization and bone cell–materials interactions of polarized hydroxyapatite. Acta Biomater. 2009, 5, 2178–2188. [Google Scholar] [CrossRef] [PubMed]
- Bertazzo, S.; Zambuzzi, W.F.; Campos, D.D.P.; Ogeda, T.L.; Ferreira, C.V.; Bertran, C.A. Hydroxyapatite surface solubility and effect on cell adhesion. Colloids Surf. B Biointerfaces 2010, 78, 177–184. [Google Scholar] [CrossRef]
- Lin, L.; Chow, K.L.; Leng, Y. Study of hydroxyapatite osteoinductivity with an osteogenic differentiation of mesenchymal stem cells. J. Biomed. Mater. Res. A 2009, 89A, 326–335. [Google Scholar] [CrossRef]
- Burr, D.B.; Radin, E.L. Microfractures and microcracks in subchondral bone: Are they relevant to osteoarthrosis? Rheum. Dis. Clin. N. Am. 2003, 29, 675–685. [Google Scholar] [CrossRef]
- Ruscitto, A.; Morel, M.M.; Shawber, C.J.; Reeve, G.; Lecholop, M.K.; Bonthius, D.; Yao, H.; Embree, M.C. Evidence of vasculature and chondrocyte to osteoblast transdifferentiation in craniofacial synovial joints: Implications for osteoarthritis diagnosis and therapy. FASEB J. 2020, 34, 4445–4461. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, P.; Mohan, S. The art of building bone: Emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, M.; Witzmann, C.; Koch, M.; Lang, S.; Kerschbaum, M.; Baumann, F.; Krutsch, W.; Docheva, D.; Alt, V.; Pfeifer, C. Attenuation of hypertrophy in human MSCs via treatment with a retinoic acid receptor inverse agonist. Int. J. Mol. Sci. 2020, 21, 1444. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, P.; Xing, W.; Cheng, S.; Mohan, S. Epiphyseal bone formation occurs via thyroid hormone regulation of chondrocyte to osteoblast transdifferentiation. Sci. Rep. 2017, 7, 10432. [Google Scholar] [CrossRef]
- Dong, Y.-F.; Soung, D.Y.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J. Cell Physiol. 2006, 208, 77–86. [Google Scholar] [CrossRef]
- Chan, B.Y.; Fuller, E.S.; Russell, A.K.; Smith, S.M.; Smith, M.M.; Jackson, M.T.; Cake, M.A.; Read, R.A.; Bateman, J.F.; Sambrook, P.N.; et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthr. Cartil. 2011, 19, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Ma, L.; Wu, L.; Jin, Q. Wnt-β-Catenin signaling pathway inhibition by sclerostin may protect against degradation in healthy but not osteoarthritic cartilage. Mol. Med. Rep. 2017, 15, 2423–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Kumagai, K.; Imai, S.; Miyatake, K.; Saito, T. Sclerostin is upregulated in the early stage of chondrogenic differentiation, but not required in endochondral ossification in vitro. PLoS ONE 2018, 13, e0201839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’Brien, C.A. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.; David, V.; Laurence, J.S.; Schwarz, P.M.; Lafer, E.M.; Hedge, A.M.; Rowe, P.S. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology 2008, 149, 1757–1772. [Google Scholar] [CrossRef] [Green Version]
- Pathak, J.L.; Bravenboer, N.; Luyten, F.P.; Verschueren, P.; Lems, W.F.; Klein-Nulend, J.; Bakker, A.D. Mechanical loading reduces inflammation-induced human osteocyte-to-osteoclast communication. Calcif. Tissue Int. 2015, 97, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Bakker, A.D.; Everts, V.; Klein-Nulend, J. Inhibition of osteoclastogenesis by mechanically loaded osteocytes: Involvement of MEPE. Calcif. Tissue Int. 2010, 87, 461–468. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Temiyasathit, S.; Lee, P.; Kim, C.H.; Tummala, P.; Yao, W.; Kingery, W.; Malone, A.M.; Kwon, R.Y.; Jacobs, C.R. Osteocytes as mechanosensors in the inhibition of bone resorption due to mechanical loading. Bone 2008, 42, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, O.D.; Herman, B.C.; Laudier, D.M.; Majeska, R.J.; Sun, H.B.; Schaffler, M.B. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 2012, 50, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, L.I.; Gortazar, A.R.; Davis, H.M.; Condon, K.W.; Gabilondo, H.; Maycas, M.; Allen, M.R.; Bellido, T. Inhibition of osteocyte apoptosis prevents the increase in osteocytic receptor activator of nuclear factor κB ligand (RANKL) but does not stop bone resorption or the loss of bone induced by unloading. J. Biol. Chem. 2015, 290, 18934–18942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crockett, J.C.; Schutze, N.; Tosh, D.; Jatzke, S.; Duthie, A.; Jakob, F.; Rogers, M.J. The matricellular protein CYR61 inhibits osteoclastogenesis by a mechanism independent of αvβ3 and αvβ5. Endocrinology 2007, 148, 5761–5768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botter, S.M.; van Osch, G.J.V.M.; Clockaerts, S.; Waarsing, J.H.; Weinans, H.; van Leeuwen, J.P.T.M. Osteoarthritis induction leads to early and temporal subchondral plate porosity in the tibial plateau of mice: An in vivo microfocal computed tomography study. Arthritis Rheumatol. 2011, 63, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Bjorklund, M. Cellular allometry of mitochondrial functionality establishes the optimal cell size. Dev. Cell 2016, 39, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, T.P.; Pessa, H.K.; Caldez, M.J.; Fuhrer, T.; Diril, M.K.; Sauer, U.; Kaldis, P.; Bjorklund, M. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr. Biol. 2014, 24, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, T.P.; Bjorklund, M. Mitochondrial function and cell size: An allometric relationship. Trends Cell Biol. 2017, 27, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Caldez, M.J.; Kaldis, P.; Björklund, M. Cell size control—A mechanism for maintaining fitness and function. BioEssays 2017, 39, 1700058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsyth, C.B.; Cole, A.; Murphy, G.; Bienias, J.L.; Im, H.-J.; Loeser, R.F., Jr. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1118–1124. [Google Scholar] [CrossRef] [Green Version]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appel, H.; Ruiz-Heiland, G.; Listing, J.; Zwerina, J.; Herrmann, M.; Mueller, R.; Haibel, H.; Baraliakos, X.; Hempfing, A.; Rudwaleit, M.; et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthritis Rheumatol. 2009, 60, 3257–3262. [Google Scholar] [CrossRef] [PubMed]


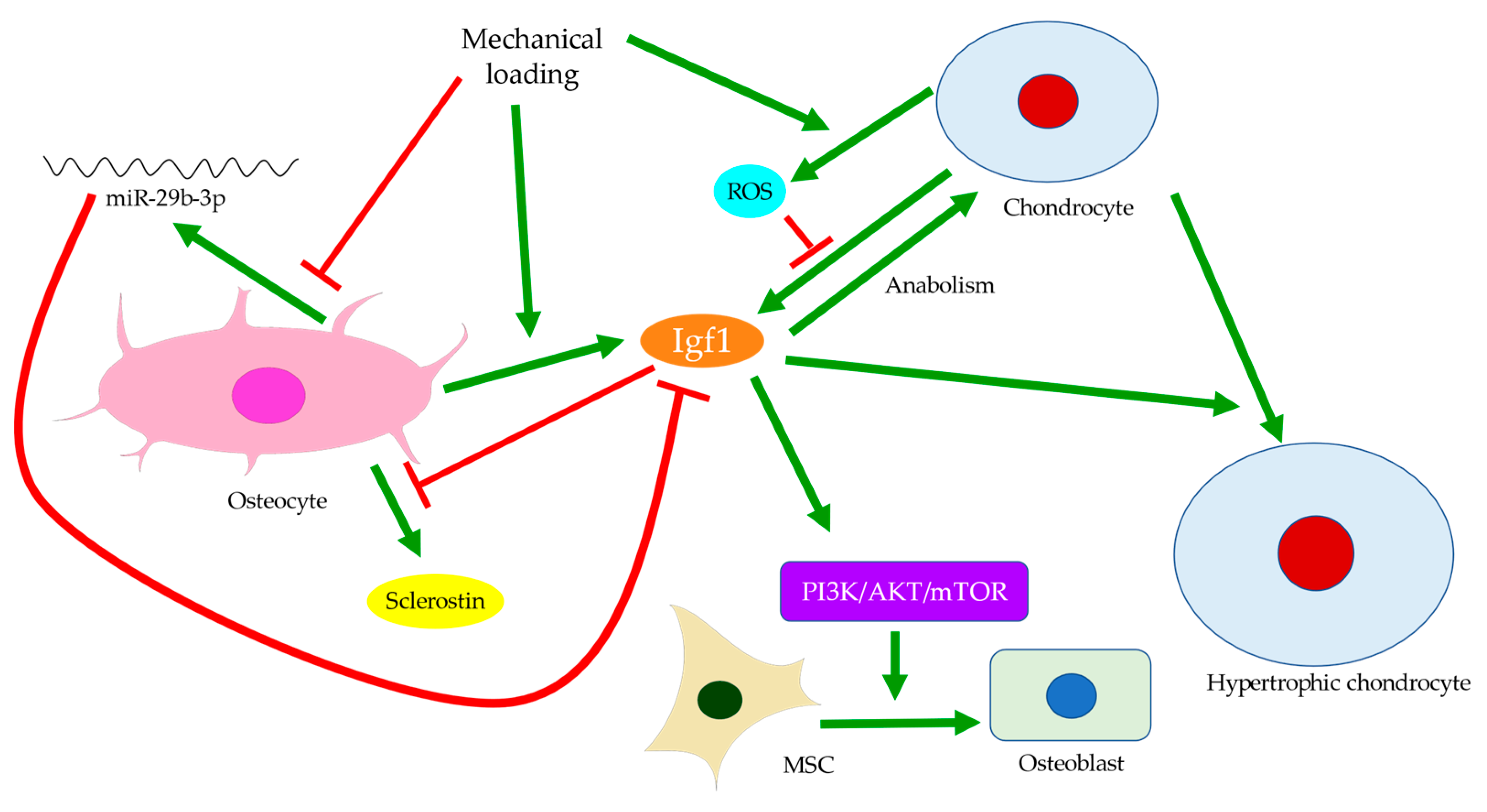

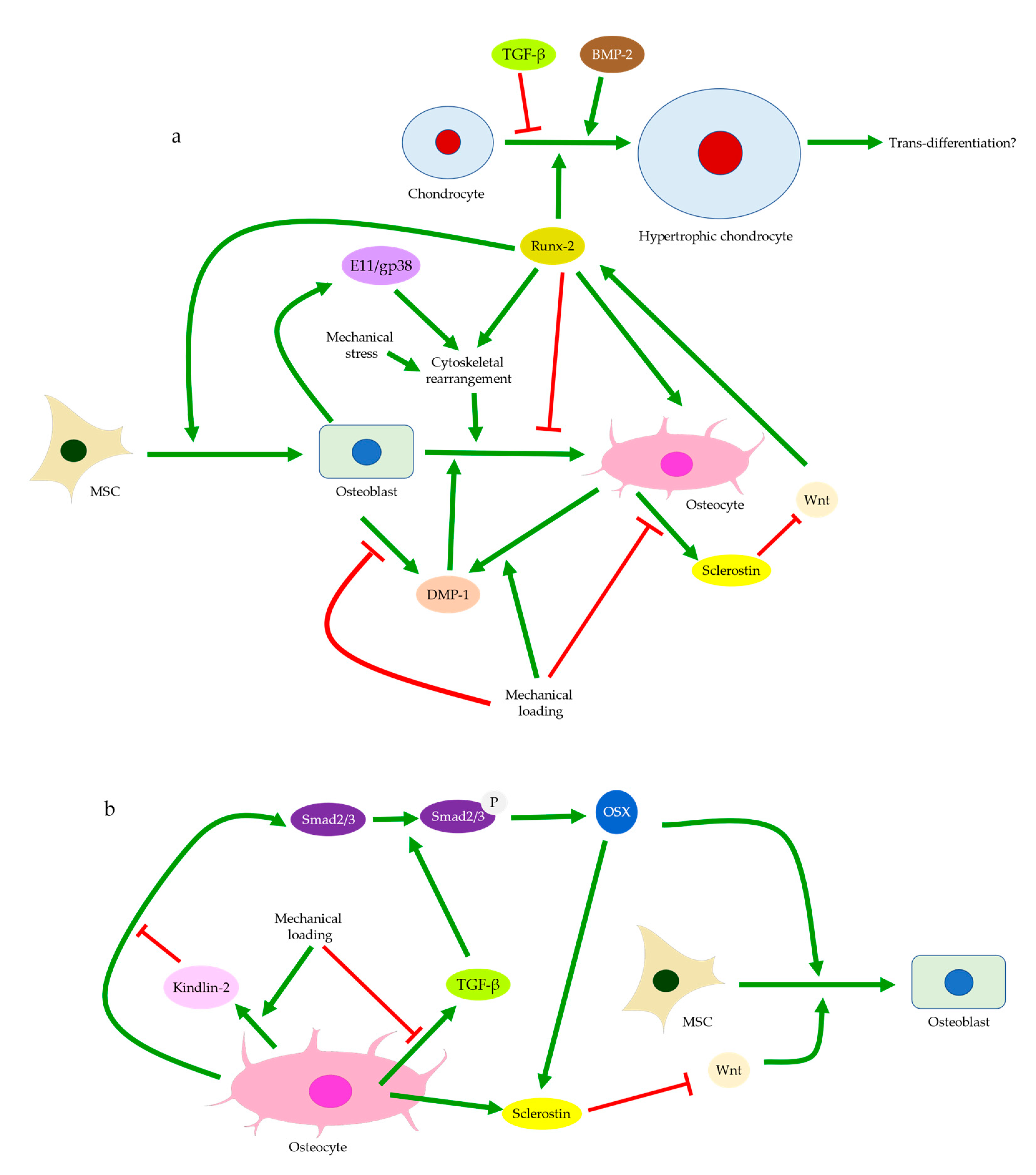

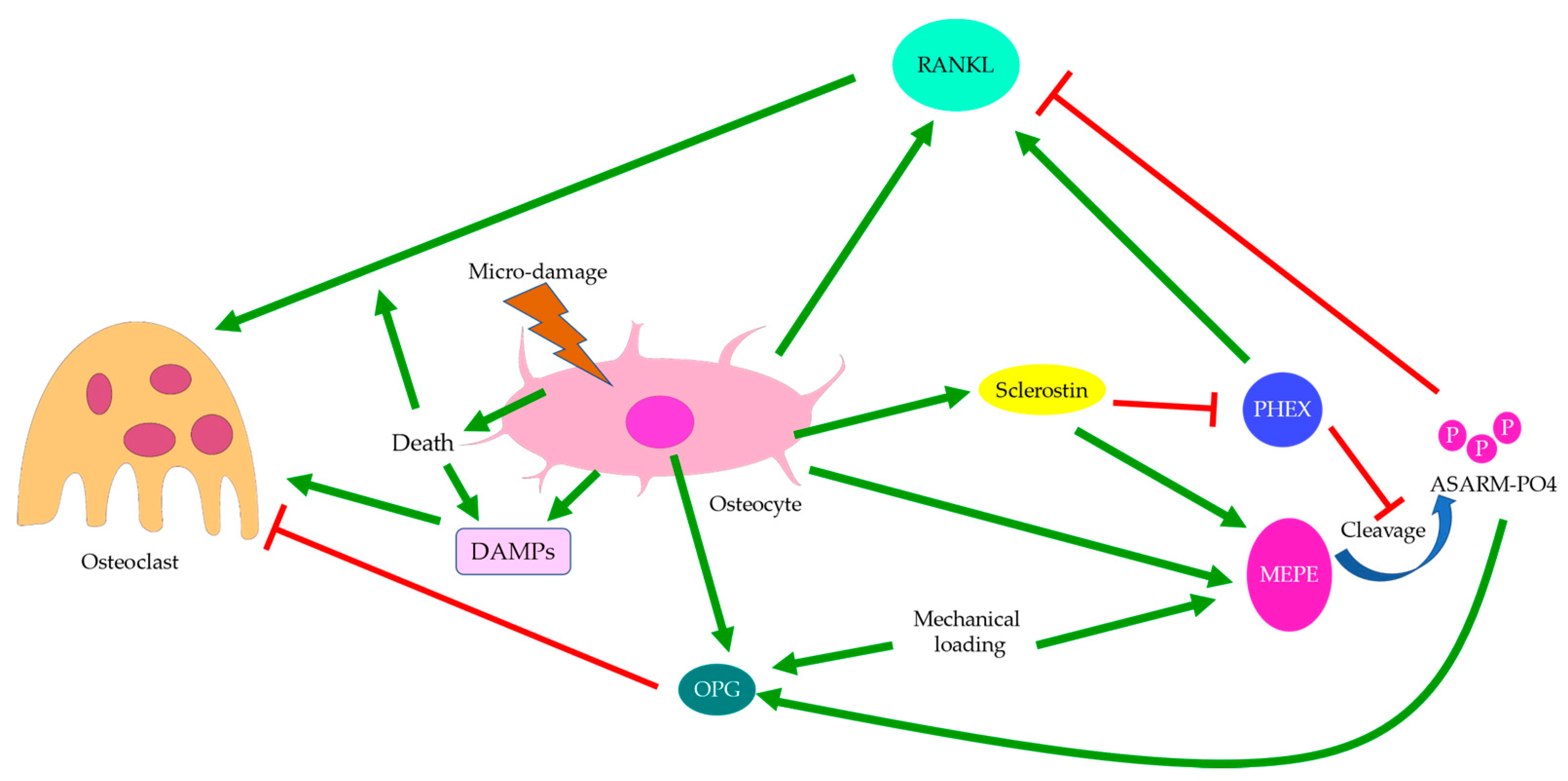
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Wen, C. Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 6522. https://doi.org/10.3390/ijms22126522
Zhang L, Wen C. Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis. International Journal of Molecular Sciences. 2021; 22(12):6522. https://doi.org/10.3390/ijms22126522
Chicago/Turabian StyleZhang, Lanlan, and Chunyi Wen. 2021. "Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis" International Journal of Molecular Sciences 22, no. 12: 6522. https://doi.org/10.3390/ijms22126522
APA StyleZhang, L., & Wen, C. (2021). Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis. International Journal of Molecular Sciences, 22(12), 6522. https://doi.org/10.3390/ijms22126522