1. Introduction
Purinergic P2X7 receptors (P2X7) are members of the P2X receptor family, a class of membrane proteins forming trimeric channels activated by extracellular ATP [
1]. They are expressed in different cell types, mainly in immune and glial cells. Following ATP gating, P2X7 initiates nonselective metal cation flux (Na
+, K
+ and Ca
2+), which in turn triggers distinct activation pathways such as the secretion of pro-inflammatory cytokines or modulation of cell death. Owing to its key role in multiple pathologies, including chronic inflammation, neurodegeneration, neuropathic pain, metabolic diseases, rheumatoid arthritis, Crohn’s disease and cancer, P2X7 has become a particularly relevant therapeutic target, sparking intense interest for drug development [
2].
Among the P2X family, P2X7 exhibits several hallmark features, including an elongated intracellular C-terminus, the necessity of unusually high concentrations of ATP for channel activation, and ATP-evoked current facilitation (also known as ATP sensitization) [
3]. This process is caused by repeated or prolonged ATP applications, which yield a leftward shift of agonist affinity [
4,
5], and is inhibited by cholesterol [
6,
7], an essential component of the cell membrane. Recent work has further indicated that this inhibition may be counteracted by the presence of palmitoylated cysteine residues located in the intracellular C-terminus of the P2X7, suggesting that lipid composition may represent a powerful means to allosterically regulate receptor function [
8].
In addition to ATP-evoked current facilitation, “macropore” formation or cell permeabilization is another hallmark of P2X7 [
3,
9]. First observed over forty years ago in mast cells [
10], this process was initially attributed to the “P2Z” receptor before being reclassified as the seventh member of the P2X family, P2X7, following its first cloning [
3]. Later confirmed in various other cell lines, including mouse RAW264 and human macrophages, microglia and HEK293 cells, across both recombinant and endogenous systems [
3,
11,
12,
13,
14,
15,
16], this phenomenon allows the passage of nanometer-sized molecules (<900 Da) across the cell membrane. Macropore formation is usually monitored by the cellular uptake of fluorescent dyes [
17], such as ethidium and YO-PRO-1, and the activation of this pathway eventually leads to cell death [
9]. Often confounded with the ATP-evoked current facilitation phenomenon [
18,
19], several hypotheses have been proposed to explain macropore formation, including possible interactions with the pore forming pannexin hemichannel [
20], but none have proved entirely satisfactory. One hypothesis that has remained prevalent until very recently was the so-called “pore dilation” theory [
21,
22], which postulated that ATP stimulation induces a progressive, time-dependent expansion of the pore, gradually increasing both current and large molecule permeability. This idea was appealing in its simplicity, but has been challenged by recent work [
23], and a new permeation mechanism supported by biochemical [
8], electrophysiological [
23,
24,
25,
26], photochemical [
26] and modeling [
23,
26] data has emerged, in which the passage of small cationic molecules, such as
N-methyl-
d-glucamine (NMDG
+) and spermidine, occurs immediately after ATP gating, via a non-dilated, open channel state. The main difference resides in the permeation rates, whereby NMDG
+ ions flow at a reduced rate compared to that of Na
+ [
26].
At first glance, this new permeation mechanism appears to reconcile most of the data [
27], but there remains an unanswered question as to how nanometer-sized molecules larger than NMDG
+ (195 Da), such as YO-PRO-1 (376 Da) or even larger dyes (e.g., YOYO-1, 763 Da) [
14], can directly permeate the P2X7 channel itself [
16,
28]. While reconstruction of an engineered P2X7, truncated at the N- and C-termini, into artificial liposomes supported the hypothesis that the pore itself is capable of YO-PRO-1 dye uptake, recent cryoelectron microscopy (cryo-EM) structures of ATP-bound, full-length rat P2X7 revealed that the open channel diameter measures only 5 Å, with no evidence of a dilated state [
29], an aperture that seems too narrow to allow the passage of nanometer-sized molecules. Given that the minimal cross-section of YO-PRO-1 is ~7 Å, it is therefore hard to conceive that cellular uptake of molecules similar to or larger than YO-PRO-1 occurs solely through the P2X7 open pore itself, suggesting that additional routes are possible [
16]. Given the importance of P2X7 in numerous pathologies, there is an urgent need to understand the molecular foundations of macropore formation and current facilitation, which undoubtedly remain two of the most enigmatic P2X features.
Recently, Ca
2+-activated Cl
− channels (CaCCs) have been suggested to constitute one such possible route. Pharmacological inhibition of these channels prevents dye uptake in different cell lines, including HEK293 cells [
30], human and mouse macrophages [
14,
30] as well as human microglia [
13]. In addition, a functional coupling between these channels and P2X7 has also been described in
Xenopus oocytes [
31]. In mouse macrophages, the transmembrane protein 16F (TMEM16F or Anoctamin-6) was identified as one of these CaCCs and proposed to mediate effects essential for innate immunity downstream of P2X7 [
30]. TMEM16F belongs to the TMEM16 family, a vast group of membrane proteins characterized by a high degree of functional diversity, including roles such as Ca
2+-activated Cl
− channels, Ca
2+-dependent phospholipid scramblases, and dual function non-selective ion channel/phospholipid scramblases [
32,
33,
34]. There are ten TMEM16 proteins (TMEM16A-K, excluding I) found in humans, and recent evidence suggests that TMEM16F functions as a dual Ca
2+-dependent phospholipid scramblase and Ca
2+-activated poorly selective (rather than purely Cl
−) ion channel. The scramblase activity of TMEM16F, which is its primary function, led to the suggestion that its activation by ATP-evoked P2X7 Ca
2+ flux mediates downstream intracellular signaling processes, resulting in outcomes such as membrane blebbing and apoptosis [
30]. However, the significance of TMEM16F to P2X7 features, such as macropore formation and current facilitation, remains to be elucidated.
In this work, we set out to test the hypothesis that P2X7 and TMEM16 form functional complexes in HEK293T cells and Xenopus oocytes. We combined mutagenesis techniques, genetic approaches, biochemical experiments, single-channel and whole-cell recordings, and cell imaging to show that these complexes have implications in both agonist-evoked current facilitation and macropore formation. Our data thus offer new insights into these enigmatic processes that have proved difficult to explain for 40 years.
3. Discussion
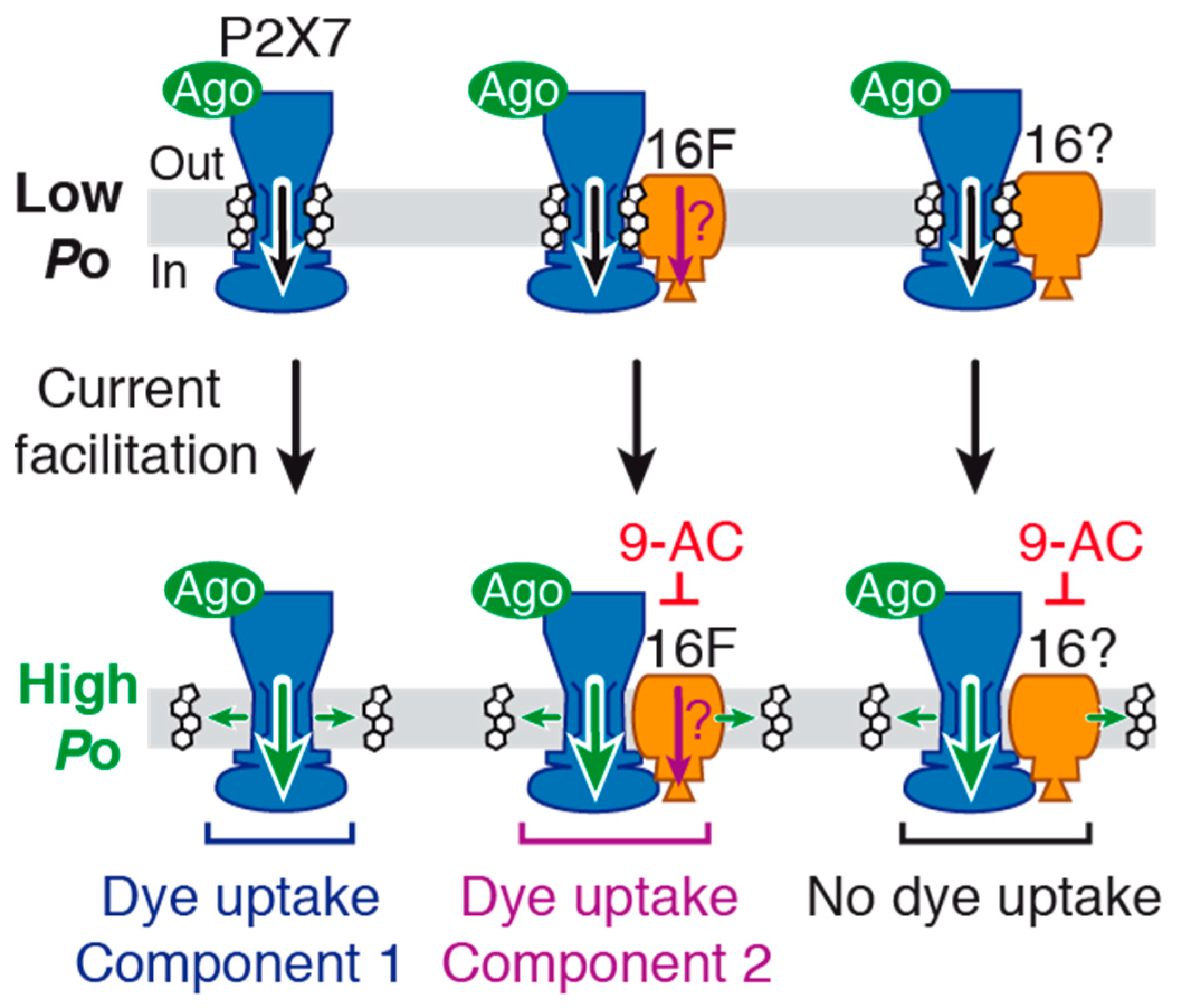
In this work, we shed new light on the molecular mechanisms underlying macropore formation and agonist-evoked current facilitation, two hallmarks of P2X7 that have remained enigmatic for several decades. We reveal that both phenomena implicate functional complexes formed between P2X7 and TMEM16 channels (
Figure 8). We also provide evidence that current facilitation does not stem from pore dilation. Indeed, our single-channel data reveal an increase of P2X7 open probability following current facilitation, but not the progressive appearance of a new conducting state.
In agreement with previous work [
30,
31], we confirm that P2X7 and TMEM16 channels are able to form functional complexes in both HEK293T and
Xenopus oocytes. In HEK293T cells, we identify the subtype TMEM16F as part of these functional complexes involved in dye uptake and show that the TMEM16F and P2X7 channels are located in proximity to one another. However, whether they directly interact through protein-protein contacts, or indirectly through Ca
2+ signaling requires further experimental testing. In addition, because all experiments described in this study were carried out in overexpressing systems, overexpressed P2X7 and TMEM16F may be misplaced in terms of cellular localization and show altered behavior that may not be exactly representative of the native interaction. Therefore, further work is also needed to demonstrate that this functional interaction is also relevant to endogenous proteins.
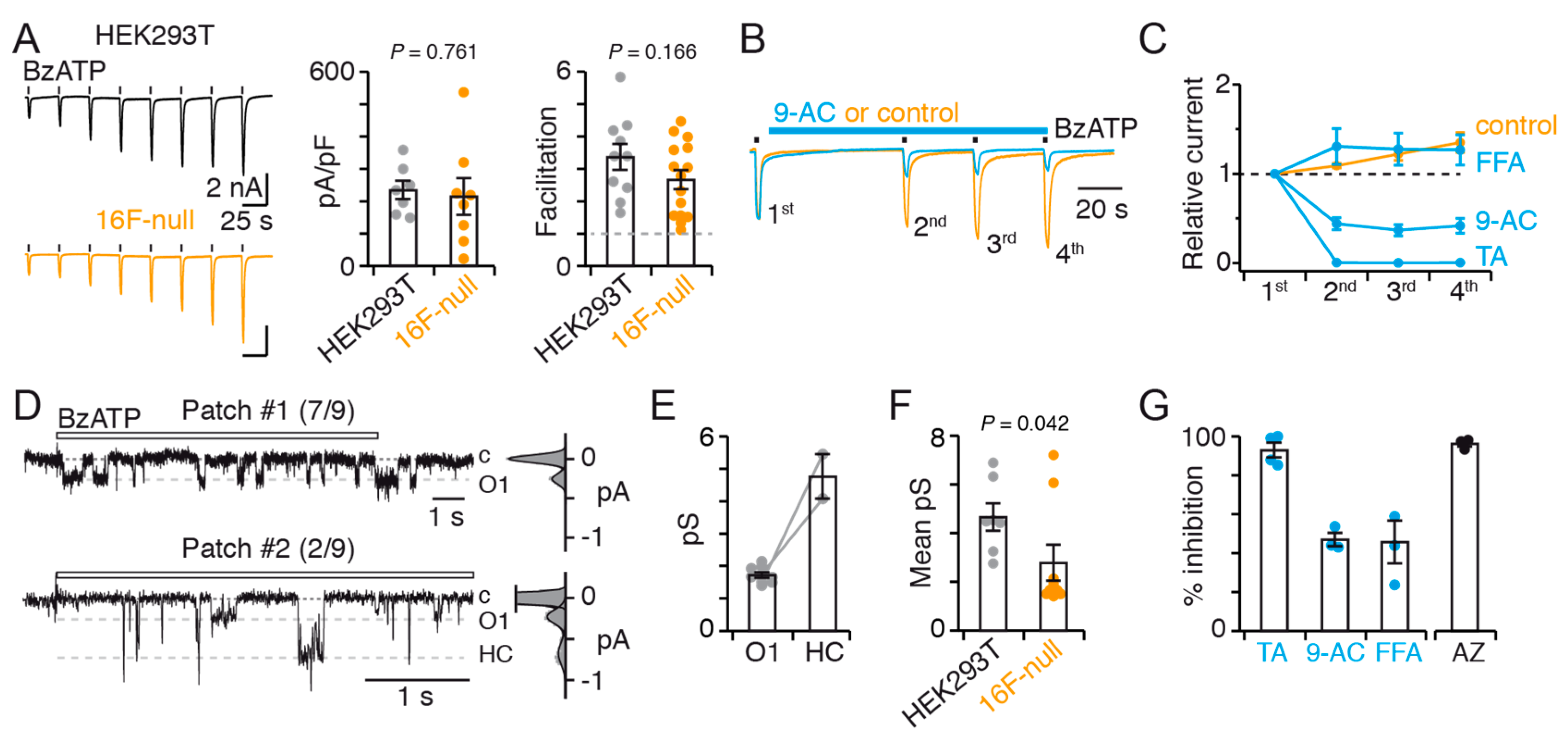
The first important finding of this study is that TMEM16F has important implications for both P2X7 expression and function. We show that TMEM16F influences P2X7 expression, suggesting that TMEM16F may act as a regulating element of P2X7 expression. Although the underlying mechanism is currently unknown and deserves additional studies, one possibility could be that TMEM16F regulates P2X7 transcriptional activity. This hypothesis is consistent with the fact that TMEM16F strongly reduces both total and cell-surface expression of P2X7. We also show that native TMEM16F contributes to the function of P2X7, through the occurrence of an O2 conducting state, that is not observed in the absence of TMEM16F. The significance of this state is currently unknown. One may argue that currents originating from the O2 state stem from TMEM16F channel, but this possibility seems unlikely because the activity of TMEM16F, which is an outwardly-rectifying channel, is expected to be minimal at negative potentials, as used in our protocols to record P2X7 activity [
44,
45]. Another possibility could be that the close physical proximity of TMEM16F to P2X7 somehow influences single-channel conductance characteristics.
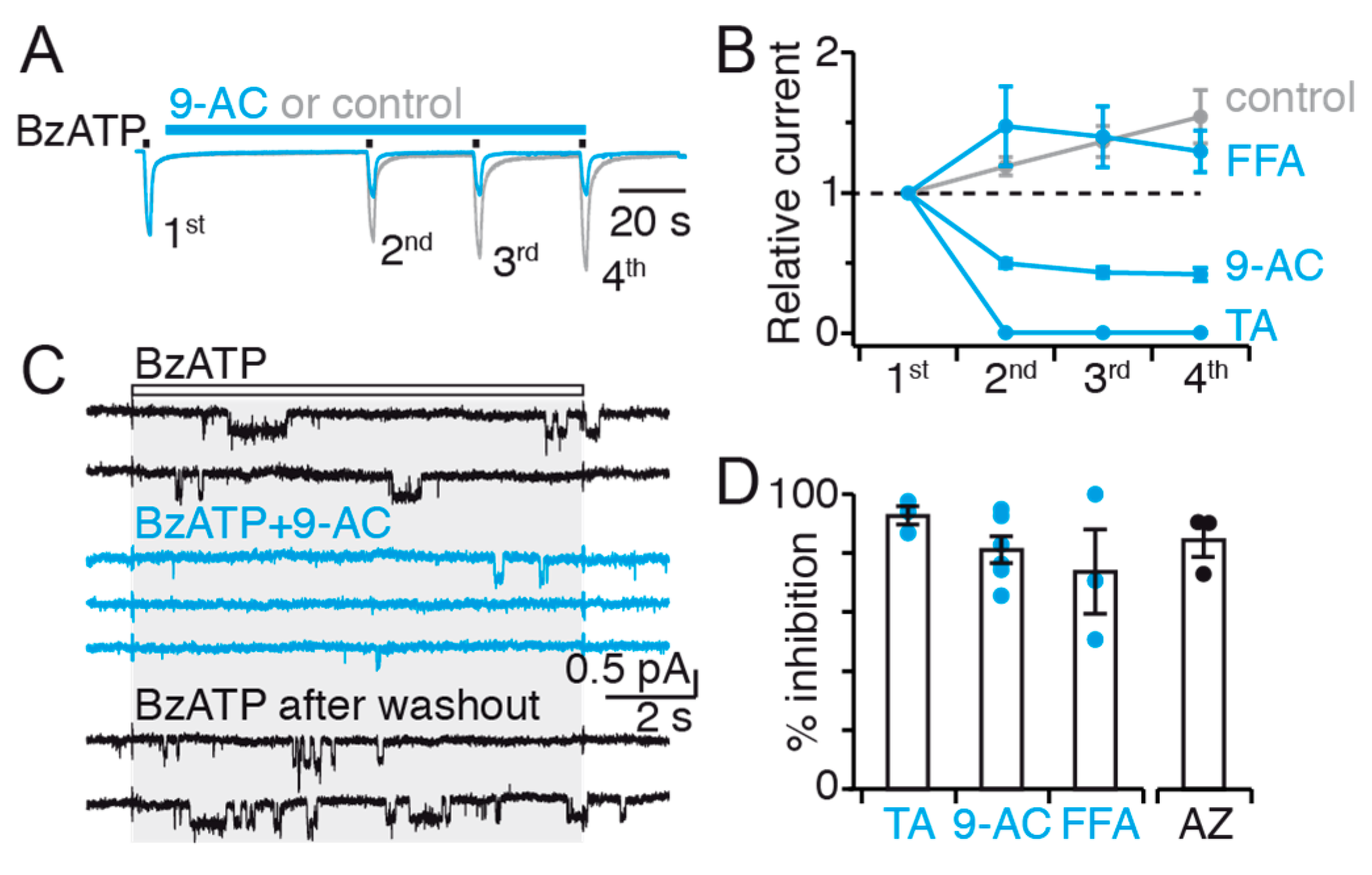
The lack of action of 9-AC on BzATP-induced P2X7 currents in CaCC-void Axolotl oocytes provides strong evidence that this inhibitor does not directly bind to P2X7. However, the fact that it inhibits P2X7 currents in both HEK293T cells and
Xenopus oocytes suggests instead that 9-AC binds to endogenous TMEM16 channels and inhibits P2X7 activity via an allosteric interaction, due to a close juxtaposition of the two channels (
Figure 8). Our data further suggest that 9-AC inhibits TMEM16F channels involved in dye uptake. Yet, the fact that 9-AC no longer inhibits dye uptake in 16F-null cells, while still inhibiting BzATP-evoked currents, suggests that another TMEM16 subtype is coupled to P2X7 ion channel function, including current facilitation, but not to dye uptake (
Figure 8). 9-AC is a non-selective inhibitor, and therefore additional experiments with more specific TMEM16 inhibitors are needed to identity this (these) subtype(s).
Our data also suggest that a significant fraction of P2X7 resides outside of functional complexes made with TMEM16F. We show that 9-AC does not abolish BzATP-evoked currents, even after a 1-min long application (
Figure 3A), thus raising the possibility that a fraction of “free” P2X7 channels (i.e., those not embedded in functional complexes) reside at the cell membrane (
Figure 8).
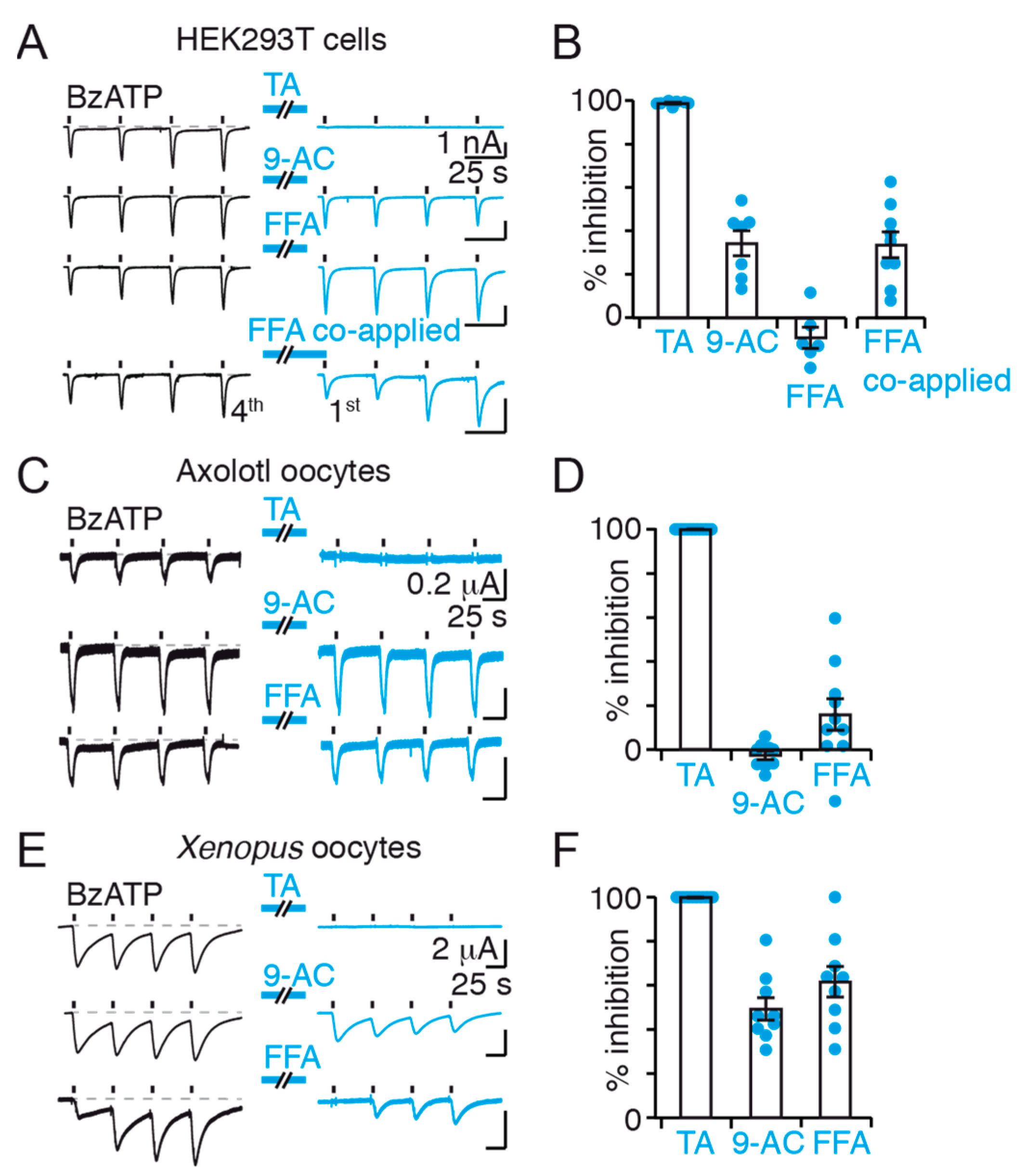
Conversely, the mode of action of FFA and TA remains unclear. Although FFA inhibition was observed in
Xenopus oocytes with no direct action on P2X7, its inhibitory effect was variable in HEK293T cells, and no definitive conclusion can be drawn. For TA, however, P2X7 currents were systematically abolished following application of the inhibitor, even in Axolotl oocytes, suggesting that it may either directly inhibit P2X7 channel or act non-specifically through a currently unknown mechanism. Regardless of the mechanism at play, this result is in apparent disagreement with previous work having found that co-application of TA and ATP to human microglia did not inhibit endogenous P2X7 channel activity [
13]. The reasons behind this discrepancy are unknown but may result from factors differing between recombinant and native systems. Finally, our data demonstrate that TA is not suitable for dye uptake experiments because of its fluorescence quenching properties, a finding that is also supported by a very recent study [
46].
The second important finding of the present work is that the functional interplay between P2X7 and members of the TMEM16 family largely contributes to current facilitation, with no obvious contribution of the TMEM16F subtype. This is supported by the fact that 9-AC strongly impairs BzATP-evoked current facilitation in both HEK293T and 16F-null cells. Interestingly, TMEM16 channels were also previously suggested to contribute to current facilitation in
Xenopus oocytes through a different mechanism [
31]. The authors suggested that current facilitation results in the secondary activation of TMEM16A Cl
− channels that follows P2X7 activation and subsequent Ca
2+ flux. However, we show that in HEK293T cells the mechanism underlying current facilitation results in a dramatic increase of P2X7 N
Po, with no evidence of the appearance of a new, secondary unitary conductance that would correspond to the activation of another channel. Therefore, it appears that different mechanisms underlying current facilitation are at play depending on the cell type. The two mechanisms can co-exist, but at present we do not know whether those TMEM16 Cl
− channels are active in our conditions.
In close agreement with previous data [
7,
8], our results suggest that plasma membrane cholesterol is a negative allosteric modulator of P2X7. It maintains the functional complex-embedded P2X7, and possibly “free” P2X7, in a low channel activity state in response to initial, brief agonist application. Longer or repeated agonist application then switches channels to higher activity states, which, in turn, underlies the observed increased current amplitudes and slowed deactivation kinetics. As suggested previously [
6,
8], we too propose that this effect is likely attributable to a progressive dissociation of cholesterol from channels (
Figure 8). Molecular mechanisms underlying agonist-evoked cholesterol dissociation remain poorly understood, but one possibility would be an activity-dependent increase of membrane mobility of complexes from high (e.g., lipid raft) to low cholesterol content [
47]. Whether cholesterol binds directly to P2X7, TMEM16 channels or the functional complex as a whole, remains to be determined. A recent study has demonstrated that cholesterol inhibits P2X7, most likely through interactions with membrane palmitoyl moieties attached to cysteine residues located in an intracellular juxtamembrane cysteine-rich domain of P2X7 [
8]. Supported by P2X7 cryo-EM structures [
29], one possibility is that cholesterol binding to those competing transmembrane sites interferes with P2X7 channel gating, most likely by stabilizing the closed channel state, and/or by destabilizing the open channel state.
The third important finding of this study is that large molecule permeability proceeds through at least two components in HEK293T cells (
Figure 8). The first one (component 1 in
Figure 8) likely entails a direct passage through the P2X7 pore, as suggested previously [
8]. Cryo-EM structures of P2X7 suggest the existence of a finite ~5 Å open channel diameter, with no evidence of larger sized pores [
29]. The minimal cross-section of YO-PRO-1 being ~7Å, which exceeds the 5 Å-outer limit of the P2X7 open pore, suggests that direct passage of YO-PRO-1 would occur at extremely low rates, as recently suggested in the case of P2X2, for which permeation rates would be largely below to 10
6 molecules per second per channel [
26]. We therefore suggest that molecules smaller than YO-PRO-1, such as NMDG
+, robustly diffuse through the P2X7 open pore, as revealed previously [
26], albeit at slower permeation rates than those of Na
+. However, for larger molecules, such as YOYO-1 (763 Da) or TO-TO-1 (894 Da) for which P2X7-dependent cell permeabilization has been described [
11,
14], direct permeation through the resolved P2X7 open channel structure seems challenging [
29]. Therefore, the contribution of a second, non-exclusive component (component 2 in
Figure 8) that involves TMEM16F channels (and perhaps other TMEM16 members) represents an alternative permeation mechanism of larger nanometer-sized molecules. Importantly, as this second component is linked to a weakly expressed TMEM16F (relative to P2X7), our data may explain why knockdown of TMEM16F by siRNAs, for which low expression levels can escape silencing, does not apparently affect P2X7 dye uptake in macrophages [
14] and HEK cells [
30]. Our data thus provide a possible explanation for previous data that have remained otherwise difficult to explain [
9,
10,
14,
16]. In light of our results showing that long agonist exposure increases P2X7 N
Po, we speculate that components 1 and 2 are boosted during long agonist exposure, and thus both contribute to the enhanced levels of large molecule cell membrane permeability observed in dye uptake experiments.
The precise permeation pathway underlying large molecule permeation of component 2 is currently unknown, but may involve the TMEM16F pore, due to its function as a poorly selective ion channel [
44,
48], enabling the passage of small organic cations such as NMDG
+ [
44]. Another possibility is that dye uptake shares the same mechanisms as those implicated in TMEM16F scramblase activity [
44,
45,
49]. Whatever the mechanism, the identification of the dye uptake pathway of component 2 merits further study.
In conclusion, our data reveal that P2X7 can form functional complexes with TMEM16 channels in HEK293T cells. Interestingly, a very recent work has also reported similar modulation of P2X7 activity with another channel, TMEM163 [
50], suggesting that P2X7 may functionally associate with different kinds of ion channel families. We propose that the functional complexes formed between P2X7 and TMEM16 channels act as a regulating hub, which, upon P2X7 activation and dependent on the surrounding membrane cholesterol level, orchestrates a hive of activity, eliciting not only channel gating (e.g., efflux of K
+ and Ca
2+ signaling), but also other cell-specific signaling, including membrane blebbing, interleukin release and phospholipid scrambling [
9]. Given the critical roles of P2X7 and TMEM16 channels in many diseases, this platform may be involved in important pathophysiological processes that lead, for example, to inflammation [
51] and mechanical allodynia [
52,
53], and may therefore be therapeutically important. Future work is needed to unravel its broader implications in disease.
4. Materials and Methods
4.1. Cell Culture and Transfection
HEK293T cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum, 1× GlutaMax, 100 units/mL penicillin and 100 μg/mL streptomycin (Gibco Life Technologies). For HEK293T hTMEM16F-null cells, this medium was further supplemented with 1 μg/mL puromycin dihydrochloride (Gibco Life Technologies).
Trypsin-treated cells were seeded onto 9–12 mm glass coverslips (VWR) in 35 mm dishes for patch-clamp and fluorescence experiments, or in 100 mm dishes for biochemical experiments, in both cases pre-treated with poly-l-lysine (Sigma Aldrich, St. Louis, MO, USA). Cells were incubated at 37 °C and in presence of 5% CO2.
Transfections were carried out using the calcium phosphate precipitation method. The cDNA encoding rat P2rx7 gene (ID Q64663) constructs and enhanced Green Fluorescent Protein construct (eGFP) were contained within pcDNA3.1(+) plasmids (Invitrogen). rP2X7-mScarlet was designed by tagging mScarlet (from gene synthesis) in the C-terminus of rP2X7 within a pcDNA3.1(+) vector. rTMEM16F cDNA was contained within a pCMV6 vector featuring a myc-DDK tag in the C-terminus (OriGene, RR212483). This myc-DDK tag containing the sequence DYKDDDDK can be recognized by anti-FLAG antibody. For whole-cell patch clamp experiments, cells were co-transfected with the rP2X7 construct (0.5–0.8 μg) and an eGFP (0.3 μg) which allowed to identify cells having undergone efficient transfection. For single-channel experiments, the quantity of transfected rP2X7 construct DNA was reduced to 0.01–0.08 μg. Cells were washed one day after transfection with PBS solution, and the medium replaced with fresh. For biochemical experiments, each 100 mm dish was either co-transfected with cDNAs encoding myc tagged rP2X7 (5 μg) and myc-DDK tagged rTMEM16F constructs (5 or 0.25 μg, as indicated) or transfected with each construct alone (10 μg). For dye uptake rescue experiments, cells were transfected with 2 μg myc tagged rP2X7 construct either alone or co-transfected with 1, 0.1, 0.01 or 0.002 μg myc tagged rTMEM16F. For co-localization by confocal microscopy, cells were transfected with 2 μg myc tagged rP2X7 and 0.1 μg myc tagged rTMEM16F.
4.2. Oocytes Preparation and Injection
Ovarian lobes were surgically removed from
Xenopus laevis and
Ambystoma mexicanum Axolotl females and oocytes were isolated as described [
54,
55]. After nuclear injection of 300 pg of rat P2X7 construct, oocytes were incubated in Barth’s solution containing 1.8 mM CaCl
2 and gentamycin (10 mg/mL) at 19 °C for 1–3 days before electrophysiological recordings.
4.3. Ethics Approval
All experimental procedures complied with official European guidelines for the care and use of laboratory animals (Directive 2010/63/UE) and were approved by the ethical committee of Bordeaux and French MESRI ministry. All animals were treated humanely and with regard for alleviation of suffering.
4.4. Molecular Biology
CRISPR/Cas9 method was carried out as previously described [
56,
57]. The oligonucleotide encoding the sgRNA sequence (5′-AATAGTACTCACAAACTCCG-3′), which targets exon 2 of
TMEM16F gene, containing BbsI overhangs was cloned into BbsI sites in pSpCas9(BB)-2A-Puro (Addgene plasmid ID:48139). The plasmid obtained was transfected into HEK293T using the calcium phosphate precipitation method. After 48 h of transfection, 1 μg/mL puromycin was applied to select cells for 72–96 h, with medium change every 24 h. Then, transfected cells were serial-diluted in 96-well plates to select for single-cell colonies. After 14–21 days, the single-cell colonies were expanded and screened for the absence of hTMEM16F. Verification was carried out by sequencing (Eurofins Genomics, see
Figure S3) and Western blot with the rabbit anti-hTMEM16F antibody (Sigma Aldrich, HPA038958) (see
Figure 5A).
4.5. Patch-Clamp Electrophysiology
Whole-cell recordings were carried out 24–48 h after transfection, and single-channel recordings 24 h after transfection. Patch pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus, Holliston, MA, USA) to yield resistances of 3–5 MΩ for whole-cell recordings, and 15–20 MΩ for single-channel recordings. For single-channel recordings, pipettes were coated with Sylgard 184 (Dow Corning Co., Midland, MI, USA) and fire polished before use. pH was verified for all solutions used, and if necessary, adjusted carefully using NaOH to pH 7.32–7.33. All solutions were maintained at approximately 300 mOsm.
Cells were voltage-clamped to −60 mV (whole-cell) or −120 mV (outside-out) using the EPC10 amplifier (HEKA, Reutlingen, Germany), and data were recorded with PATCHMASTER software. Data were acquired at 10 kHz and low-pass filtered at 2.9 kHz. Applications of agonist and/or inhibitor were carried out by perfusion, using three capillary tubes placed directly over the cell/patch of interest. These capillaries are displaced using the SF-77B Perfusion Fast Step system (Warner), ensuring solution exchange within 5–10 ms. For whole-cell recordings, Normal Extracellular Solution (NES) contained 140 mM NaCl, 2.8 mM KCl, 1 mM CaCl2, 0.1 mM MgCl2, 10 mM HEPES, 10 mM Glucose, pH 7.32–7.33. Patch pipettes contained 140 mM KCl, 5 mM MgCl2, 5 mM EGTA, 10 mM HEPES, adjusted to pH 7.3 with NaOH. For single-channel recordings, NES contained 147 mM NaCl, 2 mM KCl, 1 mM CaCl2, 0.1 mM MgCl2, 10 mM HEPES, 10 mM Glucose adjusted to pH 7.3 with NaOH. Intracellular solution contained 147 mM NaF, 10 mM HEPES and 10 mM EGTA, adjusted to pH 7.3.
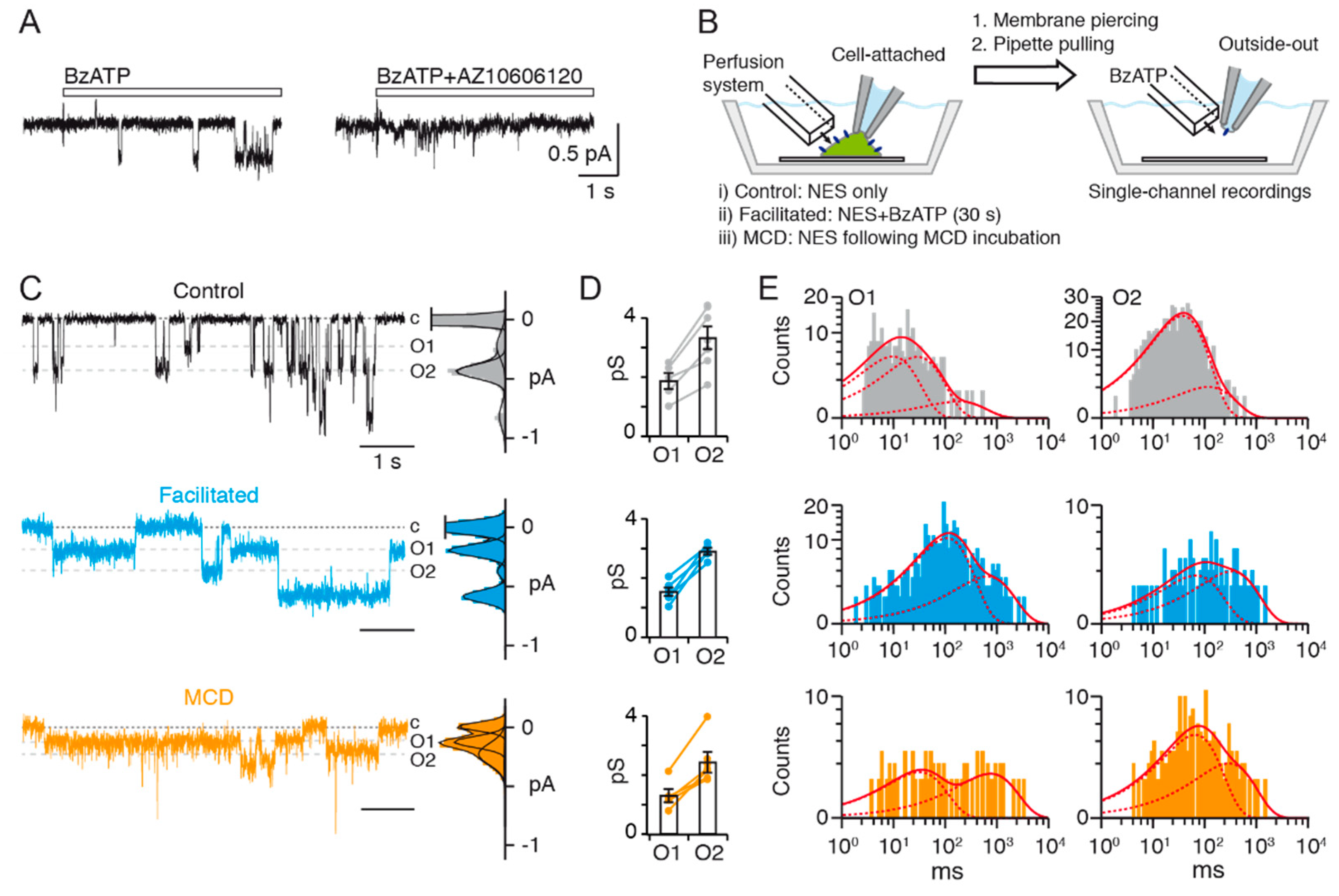
2′(3′)-O-(4-benzoylbenzoyl) adenosine 5′-triphosphate (BzATP) (triethylammonium salt, Sigma Aldrich) was used as P2X7 agonist at a concentration of 10 μM. TMEM16 inhibitors used were the following: tannic acid (TA) 20 μM, flufenamic acid (FFA) 100 μM, 9-anthracene-carboxylic acid (9-AC) 1 mM. Specific P2X7 antagonist AZ10606120 (dihydrochloride salt, Tocris) was used at a concentration of 1 μM. BzATP and TA were solubilized in NES. In the case of AZ10606120, FFA and 9-AC, concentrated stock solutions were firstly produced in DMSO, which were subsequently diluted in NES to achieve the desired working concentration in ≤0.1% DMSO. For control application (without inhibitors), 0.1% DMSO was added in NES. Owing to the facilitation effect produced by prolonged agonist exposure, only one cell was patched per coverslip, to ensure that receptors studied are indeed “naïve” upon the first agonist stimulation. For cells and patches treated by methyl-β-cyclodextrin (MCD), coverslips were incubated with 50 μL of 15 mM MCD (Sigma Aldrich) solubilized in DMEM for 15 min at 37 °C. This volume ensures complete coverage of the coverslip with MCD solution. Following incubation, MCD solution is removed by gentle washing with NES, before being used for electrophysiological experiments. In facilitated conditions, perfusion of BzATP (10 μM) was carried out for 30 s while in the cell-attached configuration, before piercing of the membrane and excising of the patch to the outside-out configuration, in which recordings were carried out.
4.6. Xenopus and Axolotl Oocyte Electrophysiology
TEVC recordings were performed as previously described [
54,
55]. Briefly, recordings were carried out at room temperature using glass pipettes (1–2 MΩ) filled with 3 M KCl solution to ensure reliable holding potentials. Oocytes were voltage-clamped at −60 mV and membrane currents were recorded with an OC-725B amplifier (Warner Instruments) and digitized at 1 kHz on a Power PC Macintosh G4 using Axograph X. Oocytes were perfused at a flow rate of 10–12 mL/min with Ringer solution, pH 7.4 containing in mM: 115 NaCl, 3 NaOH, 2 KCl, 1 CaCl
2, and 10 HEPES. BzATP and TMEM16 inhibitors were solubilized in Ringer solution and used at the same concentration as in patch-clamp experiments. Agonist and drugs were applied using a computer-driven valve system (Ala Scientific, Farmingdale, NY, USA).
4.7. Co-Immunoprecipitation, Cell Surface Biotinylation Assays and Western Blotting
For co-immunoprecipitation, non-transfected or transfected 16F-null cells with myc tagged rP2X7 and myc-DDK tagged rTMEM16F at a ratio of 1:0.05, were lysed by incubation with gentle agitation for 80 min at 4 °C with lysis buffer containing 20 mM HEPES, 100 mM NaCl, 1% Triton-X, 5 mM EDTA, Pierce Protease Inhibitor Tablets (Thermo Fischer, Waltham, MA, USA). After 10 min of centrifugation at 14,000 rpm the supernatant was collected. Input samples were collected at this stage. After an initial pre-clearing process of cell lysate with Protein G Sepharose Fast Flow resin (Sigma Aldrich, see manufacturer’s instructions), 5 μg anti-FLAG mouse antibody (Sigma Aldrich, F1804) was added to the cell lysate and left under gentle agitation for 2 h at 4 °C. Protein G Sepharose Fast Flow resin was then added and left under gentle agitation for 1 h at 4 °C. Following this, the resin was washed three times with washing buffer containing 20 mM HEPES, 500 mM NaCl, 1% Triton-X, 5 mM EDTA, Pierce Protease Inhibitor Tablets (Thermo Fischer), and two times with lysis buffer. The resin was then resuspended in NuPage LDS Sample Buffer x1 (Thermo Fischer) and 70 mM DTT, and boiled for 10 min. The supernatant was loaded onto NuPage Novex Bis-Tris 4%–12% gel (Thermo Fischer) and migrated in MOPS buffer. Transfer onto nitrocellulose membrane was carried out using the TransBlot Turbo system (BioRad) and the membrane blocked for 30 min in TPBS (PBS supplemented by 1% dried non-fat mik, 0.5% BSA, 0.05% Tween-20). The membrane was incubated overnight at 4 °C with TPBS containing either anti-c-myc mouse antibody, dilution 1:500 (Invitrogen, Thermo Fischer, reference 13–2500) or anti-β-actin mouse antibody, dilution 1:5000 (Sigma Aldrich, reference A5441). Three washes with TPBS were carried out, before a second incubation in TPBS containing HRP-conjugated sheep anti-mouse antibody, dilution 1:10,000 (GE Life Sciences, reference NA9310) for 1–2 h at room temperature. Three further washes with TPBS were carried out before revelation using Amersham ECL Select Western Blotting Detection Reagent (GE Life Sciences). Chemiluminescence was measured using the Amersham Imager 600. Images have been cropped for presentation; original uncut images are available in
Figure S5.
For cell surface biotinylation, transfected HEK293T and 16F-null cells were washed three times with PBS+ (PBS supplemented with 1 mM MgCl2 and 0.4 mM CaCl2, adjusted to pH 8.0), and incubated 30 min with 2 mM sNHS-SS-Biotin (Thermo Fisher, reference 21,331) in PBS+. Then, a washout step with PBS+ was carried out, before quenching any excess sNHS-SS-Biotin for 10 min with 20 mM Tris in PBS+. Three further washing steps with PBS were carried out before lysis step. Following lysis (as described above), neutravidin-agarose resin (Thermo Fisher, reference 29,200) was added to the cell lysate and incubated under gentle agitation overnight at 4 °C. Samples were then prepared as described for co-immunoprecipitation and loaded onto Mini-PROTEAN TGX 4–15% and migrated in TGS buffer. Western blotting was carried out as described above.
4.8. Immunocytochemistry
Transfected or non-transfected 16F-null cells were fixed 24 h post transfection in 4% PFA (PBS, 4% paraformaldehyde, pH 8.0) for 15 min at room temperature. Coverslips were then washed in PBS for 5 min and permeabilized for 30 min in block solution (PBS, 1% BSA, 0.1% Triton X-100). Coverslips were incubated at 4 °C overnight with primary anti-hTMEM16F rabbit antibody (Sigma Aldrich, HPA038958) diluted 1:500 in block solution. Coverslips were then washed in PBS and incubated with secondary fluorescent probe-conjugated antibody (goat anti-rabbit IgG H&L (Alexa Fluor® 488) (ab150077, Abcam, Cambridge, UK) diluted 1:500, and Hoechst stain (4 μg/μL) for 1 h at RT in block solution. Coverslips were then mounted using mounting reagent (Prolong Gold antifade, P36930, Invitrogen) and allowed to dry overnight prior to imaging with Leica SPE.
4.9. Fluorescence Microscopy
Video fluorescence microscopy measurements were carried out using a Leica FW4000 and 40× objective (Platform of Quantitative Imagery, Faculté de Pharmacie, Université de Strasbourg). Acquisition of fluorescence images was carried out at an interval of every 5 s using the software MetaMorph (Molecular Devices). The experiment was divided into two acquisition periods; an initial acquisition of 10 min where cells were incubated in NES/YO-PRO-1 solution where YO-PRO-1 is at a concentration of 10 μM (iodide salt, Thermo Fischer). The solution was then gently exchanged for a NES/YO-PRO-1 solution containing BzATP and inhibitors (or BzATP alone) for a second acquisition period of 15 min. This second solution containing inhibitors was prepared immediately prior to application, and concentrations used are the same as those used for electrophysiology. Cells were maintained at 37 °C during measurements.
Confocal imaging was captured with Leica SPE using oil immersion objective: 63×, Numerical Aperture 1.4. Excitation (λexc) and emission (λem) wavelengths were as follows: Hoechst (λexc = 364 nm; λem = 430–481 nm), Alexa 488 (λexc = 495 nm; λem = 500–600 nm) and mScarlet (λexc = 561 nm; λem = 648–708 nm).
4.10. Fluorescence Spectroscopy
Fluorescence spectra were obtained on SAFAS Xenius Spectrophotometer. Measurements were made in quartz cuvettes. A 1 mL solution was excited at 480 nm. Solutions contained NES supplemented with 10 μM YO-PRO-1 alone (−dsDNA in
Figure 4D), or 10 μM YO-PRO-1 complexed to 29 μg dsDNA (+dsDNA), or 10 μM YO-PRO-1 complexed to 29 μg dsDNA with inhibitors (+dsDNA +inhibitors). Inhibitor concentrations used were the same as those used for electrophysiology. The bandwidth for excitation and emission was 10 nm.
4.11. Data Analysis
For electrophysiological data analysis, FitMaster (HEKA Electronics v2 × 73 × 1) and Igor Pro (WaveMetrics, v6.32A) were used. Experiments were repeated several times, over at least two independent transfections, with precise details given in figure legends. The extent of facilitation (as in
Figure 7A) was determined by calculating the fold difference between the 8th agonist-evoked current and initial agonist-evoked current in a series of 8 repeated applications. The current density (as in
Figure 7A) was calculated as the pA/pF of the 8th agonist-evoked current in a series of 8 repeated applications.
For single channel data analysis, TAC and TACFit (Bruxton) were used. Data were re-filtered offline to give a final f
c of 100 Hz. Dead time (t
d) was set to 1.8 ms and transitions shorter than t
d were ignored. Transitions longer than t
d were accepted as events. Single-channel current amplitudes were determined by all points histograms fitted to Gaussian distributions, using maximum likelihood methods:
where
f(
I) is the total probability density of a given amplitude value
I,
Ai is the
ith channel amplitude, σ
i is the standard deviation of the
ith channel amplitude, and
ai is the fraction of the data represented by the
ith amplitude. Conductance was determined by dividing current amplitudes by the holding potential (−120 mV).
Only patches featuring three or fewer channels were analyzed, and analysis of stacked events resulting from simultaneous channel openings was less than 5% of the total events analyzed. The number of channels present within a given patch can be determined by visual inspection of the maximum number of coinciding stacking events. These stacked openings are designated as such, in order to verify the proportion that they represent of the total events analyzed. Assuming N independent and identical channels in the patch, each with a probability
Po of being open, the probability that
k channels open simultaneously is given by the binomial distribution:
If one assumes that the O2 state results in the simultaneous opening of two O1 states (k = 2), then its probability of occurring is equal to 0.0003, taking N = 3 (we selected only patches containing three or fewer channels for analysis) and Po = 0.01 (0.03/3 = 0.01 with NPo experimentally determined as 0.03 for O1 in control conditions). This theoretical value is largely below the actual value of 0.05 we determined for the O2 state (that is 0.14/3 ≈ 0.05 with NPo = 0.14 for O2 state in control conditions), suggesting that O2 did not originate from stackings of double O1 openings.
For control conditions, we recorded from 7 different outside-out patches, in which 1518 events were analyzed. Each patch contained between 1 to 15 sweeps. For facilitated conditions, we recorded from 5 outside-out patches, in which 754 events were analyzed (ranged between 3 to 19 sweeps). For MCD-treated cells, we recorded single-channel currents from 7 outside-out patches excised from cells that were pre-treated with MCD, and in which 396 events were analyzed (ranged between 1 to 13 sweeps).
Detection of events was carried out by using 50% of the single-channel current amplitude as the detection threshold. Dwell time histograms were fitted by the minimum number of exponential functions according to the following equation:
where
t is the time,
τi is the time constant, and
ai is the relative area (the sum of all areas is equal to (1). The mean open time for a selected state is given by:
For conductance analysis, the same patches were analyzed as in mean open time analysis, with the addition of supplementary patches. These supplementary patches contained too many stacked events to be considered for mean open time analysis but remain suitable for unitary conductance analysis.
For analysis of percentage inhibition of rP2X7 single-channel currents by CaCC inhibitors, a weighting is established of open-channel activity compared to closed-channel activity (within the 10 s period of drug application, illustrated in
Figure 3C by grey shading). This weighting is calculated for recordings in patches exposed solely to BzATP, and then for subsequent recordings of the same patch exposed to BzATP and CaCC inhibitor co-applications. In the case of patches which did not exhibit instant inhibition, BzATP and CaCC inhibitor co-applications were repeated until a steady-state level of inhibition was achieved. These steady-state inhibitions were then averaged. A percentage inhibition is therefore calculated for each patch, which are subsequently averaged.
Deactivation currents (decay currents when BzATP is removed) were fit by single exponentials according to the following equation:
where
t is the time,
τd is time constant of deactivation, and A is the current amplitude.
For fluorescence experiment analysis, Fiji and Igor Pro were used. For co-localization analysis Pearson’s correlation coefficient was measured using Coloc2 in ImageJ.
4.12. Statistical Analysis
Data are reported as mean values ± SEM. All experiments were replicated over a minimum of 2 transfections and 3 cells. For electrophysiological data, exact data points are shown as dots within histograms, and for fluorescence data, the number of cells is shown in brackets. For statistical analysis, GraphPad Prism software (v8.0.2) was used. The normality of data distribution was tested before selection of an appropriate test of statistical significance and p < 0.05 was considered as significant difference. For electrophysiological and Western blot data, Mann-Whitney or two-tailed (paired or unpaired, when indicated) Student’s t-test was used. For fluorescence measurements, one-way Kruskal-Wallis analysis of variance test followed by Dunn’s multiple comparison post-hoc test or Mann-Whitney test was employed, as indicated in figure legends.
4.13. Data Availability
All data and associated protocols for this study are available in the main text and Supporting information. Materials may be requested to T.G.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







