
Early Effects of the Soluble Amyloid β25-35 Peptide in Rat Cortical Neurons: Modulation of Signal Transduction Mediated by Adenosine and Group I Metabotropic Glutamate Receptors

 ,
,  and
and
Abstract
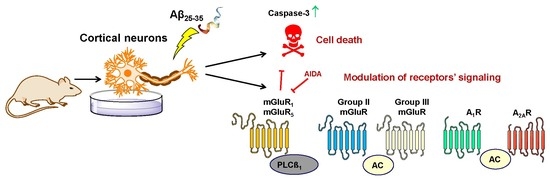
1. Introduction
2. Results
2.1. Characterization of Aβ-Induced Toxicity on Cortical Neurons
2.2. Aβ25-35 Exposure Effect on the Density and Affinity of Metabotropic Glutamate Receptors
2.3. Aβ25-35 Effect on Group I Metabotropic Glutamate Receptors’ Gene Expression and Protein Levels
2.4. Modulation of PLC β1 Signalling Pathway after Aβ25-35 Exposure
2.5. Effect of Aβ25-35 Exposure on Metabotropic Glutamate Receptors/Adenylyl Cyclase Pathway
2.6. Effect of Aβ25-35 Exposure on Adenosine A1 and A2A Receptors
2.7. Effect of Aβ25-35 Exposure on Adenosine Receptor Gene Expression
2.8. Effect of Aβ25-35 Exposure on Adenosine Receptors-Mediated Adenylyl Cyclase Activity
2.9. Effect of Aβ25-35 Exposure on the Expression of Genes Coding for CREB and CREM
2.10. Effect of Group I Metabotropic Glutamate and Adenosine A1 and A2A Receptors Ligands on Aβ25-35 Induced Toxicity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Primary Culture of Cortical Neurons
4.3. Drug Treatments and Evaluation of Cell Death
4.4. Caspase 3 Activity Assay
4.5. Quantification of Metabotropic Glutamate and Adenosine Receptors in Intact Cells by Radioligand Binding Assay
4.6. Extracellular Targeting of mGluR1, mGluR5 and mGluR2,3 by Immunochemistry
4.7. PLCβ1 Immunocytochemistry
4.8. Microscopy Imaging
4.9. Determination of Phospholipase C Activity
4.10. Determination of Adenylyl Cyclase Activity
4.11. Preparation of Total RNA and cDNA
4.12. Quantitative Real-Time RT-PCR Analysis
4.13. Protein Determination
4.14. Statistics and Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selkoe, D.J. Clearing the brain’s amyloid cobwebs. Neuron 2001, 32, 177–180. [Google Scholar] [CrossRef]
- Huber, G.; Martin, J.R.; Loffler, J.; Moreau, J.L. Involvement of amyloid precursor protein in memory formation in the rat: An indirect antibody approach. Brain Res. 1993, 603, 348–352. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef]
- Plant, L.D.; Boyle, J.P.; Smith, I.F.; Peers, C.; Pearson, H.A. The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J. Neurosci. 2003, 23, 5531–5535. [Google Scholar] [CrossRef]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. Beta-amyloid monomers are neuroprotective. J. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef]
- Savory, J.; Ghribi, O.; Herman, M.M. Is amyloid beta-peptide neurotoxic or neuroprotective and what is its role in the binding of metal ions? Neurobiol. Aging 2002, 23, 1089–1092. [Google Scholar] [CrossRef]
- Copani, A. The underexplored question of beta-amyloid monomers. Eur. J. Pharmacol. 2017, 817, 71–75. [Google Scholar] [CrossRef]
- Gouras, G.K.; Tampellini, D.; Takahashi, R.H.; Capetillo-Zarate, E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 119, 523–541. [Google Scholar] [CrossRef]
- Pike, C.J.; Burdick, D.; Walencewicz, A.J.; Glabe, C.G.; Cotman, C.W. Neurodegeneration induced by beta-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 1993, 13, 1676–1687. [Google Scholar] [CrossRef]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and neurotoxic effects of amyloid beta protein: Reversal by tachykinin neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Y.G.; Marlatt, M.W.; Smith, M.A.; Kosenko, E.A. Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: Evidence for Abeta(25-35). Exp. Neurol. 2010, 221, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Bhute, S.; Sarmah, D.; Datta, A.; Rane, P.; Shard, A.; Goswami, A.; Borah, A.; Kalia, K.; Dave, K.R.; Bhattacharya, P. Molecular Pathogenesis and Interventional Strategies for Alzheimer’s Disease: Promises and Pitfalls. ACS Pharmacol. Transl. Sci. 2020, 3, 472–488. [Google Scholar] [CrossRef]
- Ayton, S.; Bush, A.I. Beta-amyloid: The known unknowns. Ageing Res. Rev. 2021, 65, 101212. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Ribeiro, F.M.; Vieira, L.B.; Pires, R.G.; Olmo, R.P.; Ferguson, S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 2017, 115, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Battaglia, G.; Sortino, M.A.; Spampinato, S.; Molinaro, G.; Copani, A.; Nicoletti, F.; Bruno, V. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: Still a hot topic? Neurochem. Int. 2012, 61, 559–565. [Google Scholar] [CrossRef]
- Jakaria, M.; Park, S.Y.; Haque, M.E.; Karthivashan, G.; Kim, I.S.; Ganesan, P.; Choi, D.K. Neurotoxic Agent-Induced Injury in Neurodegenerative Disease Model: Focus on Involvement of Glutamate Receptors. Front. Mol. Neurosci. 2018, 11, 307. [Google Scholar] [CrossRef]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef]
- Canas, P.M.; Simoes, A.P.; Rodrigues, R.J.; Cunha, R.A. Predominant loss of glutamatergic terminal markers in a beta-amyloid peptide model of Alzheimer’s disease. Neuropharmacology 2014, 76 Pt A, 51–56. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; AP, I.J.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Dunwiddie, T.V.; Masino, S.A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef]
- Albasanz, J.L.; Perez, S.; Barrachina, M.; Ferrer, I.; Martin, M. Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease. Brain Pathol. 2008, 18, 211–219. [Google Scholar] [CrossRef]
- Cunha, R.A. Neuroprotection by adenosine in the brain: From A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal. 2005, 1, 111–134. [Google Scholar] [CrossRef] [PubMed]
- Leon-Navarro, D.A.; Albasanz, J.L.; Martin, M. Functional Cross-Talk between Adenosine and Metabotropic Glutamate Receptors. Curr. Neuropharmacol. 2019, 17, 422–437. [Google Scholar] [CrossRef]
- Albasanz, J.L.; Dalfo, E.; Ferrer, I.; Martin, M. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer’s disease and dementia with Lewy bodies correlates with stage of Alzheimer’s-disease-related changes. Neurobiol. Dis. 2005, 20, 685–693. [Google Scholar] [CrossRef]
- Castillo, C.A.; Albasanz, J.L.; León, D.; Jordán, J.; Pallás, M.; Camins, A.; Martín, M. Age-related expression of adenosine receptors in brain from the senescence-accelerated mouse. Exp. Gerontol. 2009, 44, 453–461. [Google Scholar] [CrossRef]
- Sanchez-Melgar, A.; Albasanz, J.L.; Pallas, M.; Martin, M. Resveratrol Differently Modulates Group I Metabotropic Glutamate Receptors Depending on Age in SAMP8 Mice. ACS Chem. Neurosci. 2020, 11, 1770–1780. [Google Scholar] [CrossRef]
- Rhee, S.G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001, 70, 281–312. [Google Scholar] [CrossRef]
- Fagiani, F.; Lanni, C.; Racchi, M.; Govoni, S. (Dys)regulation of Synaptic Activity and Neurotransmitter Release by beta-Amyloid: A Look Beyond Alzheimer’s Disease Pathogenesis. Front. Mol. Neurosci. 2021, 14, 635880. [Google Scholar] [CrossRef]
- Iloun, P.; Hooshmandi, E.; Gheibi, S.; Kashfi, K.; Ghasemi, R.; Ahmadiani, A. Roles and Interaction of the MAPK Signaling Cascade in Abeta25-35-Induced Neurotoxicity Using an Isolated Primary Hippocampal Cell Culture System. Cell Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Chen, N.; Wang, J.; He, Y.; Xu, Y.; Zhang, Y.; Gong, Q.; Yu, C.; Gao, J. Trilobatin Protects Against Abeta25-35-Induced Hippocampal HT22 Cells Apoptosis Through Mediating ROS/p38/Caspase 3-Dependent Pathway. Front. Pharmacol. 2020, 11, 584. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Cieslak, M.; Wojtczak, A. Role of purinergic receptors in the Alzheimer’s disease. Purinergic Signal. 2018, 14, 331–344. [Google Scholar] [CrossRef]
- Burnstock, G.; Krugel, U.; Abbracchio, M.P.; Illes, P. Purinergic signalling: From normal behaviour to pathological brain function. Prog. Neurobiol. 2011, 95, 229–274. [Google Scholar] [CrossRef]
- Stone, T.W.; Ceruti, S.; Abbracchio, M.P. Adenosine receptors and neurological disease: Neuroprotection and neurodegeneration. Handb. Exp. Pharmacol. 2009, 535–587. [Google Scholar] [CrossRef]
- Albasanz, J.L.; Castillo, C.A.; Barrachina, M.; Ferrer, I.; Martín, M. Modulation of Signal Transduction Pathways in Senescence-Accelerated Mice P8 Strain: A Useful Tool for Alzheimer’s Disease Research. In The Clinical Spectrum of Alzheimer’s Disease -The Charge toward Comprehensive Diagnostic and Therapeutic Strategies; InTechOpen: London, UK, 2011. [Google Scholar]
- Srivastava, A.; Das, B.; Yao, A.Y.; Yan, R. Metabotropic Glutamate Receptors in Alzheimer’s Disease Synaptic Dysfunction: Therapeutic Opportunities and Hope for the Future. J. Alzheimers Dis. 2020, 78, 1345–1361. [Google Scholar] [CrossRef]
- Nicoletti, F.; Orlando, R.; Di Menna, L.; Cannella, M.; Notartomaso, S.; Mascio, G.; Iacovelli, L.; Matrisciano, F.; Fazio, F.; Caraci, F.; et al. Targeting mGlu Receptors for Optimization of Antipsychotic Activity and Disease-Modifying Effect in Schizophrenia. Front. Psychiatry 2019, 10, 49. [Google Scholar] [CrossRef]
- Hovelso, N.; Sotty, F.; Montezinho, L.P.; Pinheiro, P.S.; Herrik, K.F.; Mork, A. Therapeutic potential of metabotropic glutamate receptor modulators. Curr. Neuropharmacol. 2012, 10, 12–48. [Google Scholar] [CrossRef]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Castillo, C.A.; Leon, D.A.; Ballesteros-Yanez, I.; Iglesias, I.; Martin, M.; Albasanz, J.L. Glutamate differently modulates metabotropic glutamate receptors in neuronal and glial cells. Neurochem. Res. 2010, 35, 1050–1063. [Google Scholar] [CrossRef]
- Ashe, K.H.; Zahs, K.R. Probing the biology of Alzheimer’s disease in mice. Neuron 2010, 66, 631–645. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Partridge, V.; Leon, W.C.; Canneva, F.; Allard, S.; Arvanitis, D.N.; Vercauteren, F.; Houle, D.; Ducatenzeiler, A.; Klein, W.L.; et al. Transgenic mice as a model of pre-clinical Alzheimer’s disease. Curr. Alzheimer. Res. 2011, 8, 4–23. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Kowalewski, J.M.; Renner, M.; Bousset, L.; Koulakoff, A.; Melki, R.; Giaume, C.; Triller, A. Beta-amyloid and ATP-induced diffusional trapping of astrocyte and neuronal metabotropic glutamate type-5 receptors. Glia 2013, 61, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef]
- Kumar, A.; Dhull, D.K.; Mishra, P.S. Therapeutic potential of mGluR5 targeting in Alzheimer’s disease. Front. Neurosci. 2015, 9, 215. [Google Scholar] [CrossRef]
- Hu, N.W.; Nicoll, A.J.; Zhang, D.; Mably, A.J.; O’Malley, T.; Purro, S.A.; Terry, C.; Collinge, J.; Walsh, D.M.; Rowan, M.J. mGlu5 receptors and cellular prion protein mediate amyloid-beta-facilitated synaptic long-term depression in vivo. Nat. Commun. 2014, 5, 3374. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Esseltine, J.L.; DeVries, R.A.; Cregan, S.P.; Ferguson, S.S. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 2014, 7, 40. [Google Scholar] [CrossRef]
- Ostapchenko, V.G.; Beraldo, F.H.; Guimaraes, A.L.; Mishra, S.; Guzman, M.; Fan, J.; Martins, V.R.; Prado, V.F.; Prado, M.A. Increased prion protein processing and expression of metabotropic glutamate receptor 1 in a mouse model of Alzheimer’s disease. J. Neurochem. 2013, 127, 415–425. [Google Scholar] [CrossRef]
- Casley, C.S.; Lakics, V.; Lee, H.G.; Broad, L.M.; Day, T.A.; Cluett, T.; Smith, M.A.; O’Neill, M.J.; Kingston, A.E. Up-regulation of astrocyte metabotropic glutamate receptor 5 by amyloid-beta peptide. Brain Res. 2009, 1260, 65–75. [Google Scholar] [CrossRef]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef]
- Haas, L.T.; Salazar, S.V.; Kostylev, M.A.; Um, J.W.; Kaufman, A.C.; Strittmatter, S.M. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain 2016, 139, 526–546. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid beta oligomers (AbetaOs) in Alzheimer’s disease. J. Neural. Transm. (Vienna) 2018, 125, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Foster, D.J.; Conn, P.J. Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef]
- Pshenichkin, S.; Dolinska, M.; Klauzinska, M.; Luchenko, V.; Grajkowska, E.; Wroblewski, J.T. Dual neurotoxic and neuroprotective role of metabotropic glutamate receptor 1 in conditions of trophic deprivation—Possible role as a dependence receptor. Neuropharmacology 2008, 55, 500–508. [Google Scholar] [CrossRef]
- Battaglia, G.; Bruno, V.; Pisani, A.; Centonze, D.; Catania, M.V.; Calabresi, P.; Nicoletti, F. Selective blockade of type-1 metabotropic glutamate receptors induces neuroprotection by enhancing gabaergic transmission. Mol. Cell Neurosci. 2001, 17, 1071–1083. [Google Scholar] [CrossRef]
- Landucci, E.; Boscia, F.; Gerace, E.; Scartabelli, T.; Cozzi, A.; Moroni, F.; Mannaioni, G.; Pellegrini-Giampietro, D.E. Involvement of endocannabinoid signaling in the neuroprotective effects of subtype 1 metabotropic glutamate receptor antagonists in models of cerebral ischemia. Int. Rev. Neurobiol. 2009, 85, 337–350. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.E. The distinct role of mGlu1 receptors in post-ischemic neuronal death. Trends Pharmacol. Sci. 2003, 24, 461–470. [Google Scholar] [CrossRef]
- Lesage, A.; Steckler, T. Metabotropic glutamate mGlu1 receptor stimulation and blockade: Therapeutic opportunities in psychiatric illness. Eur. J. Pharmacol. 2010, 639, 2–16. [Google Scholar] [CrossRef]
- Bruno, V.; Ksiazek, I.; Battaglia, G.; Lukic, S.; Leonhardt, T.; Sauer, D.; Gasparini, F.; Kuhn, R.; Nicoletti, F.; Flor, P.J. Selective blockade of metabotropic glutamate receptor subtype 5 is neuroprotective. Neuropharmacology 2000, 39, 2223–2230. [Google Scholar] [CrossRef]
- Masilamoni, G.J.; Bogenpohl, J.W.; Alagille, D.; Delevich, K.; Tamagnan, G.; Votaw, J.R.; Wichmann, T.; Smith, Y. Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain 2011, 134, 2057–2073. [Google Scholar] [CrossRef]
- Morin, N.; Morissette, M.; Gregoire, L.; Gomez-Mancilla, B.; Gasparini, F.; Di Paolo, T. Chronic treatment with MPEP, an mGlu5 receptor antagonist, normalizes basal ganglia glutamate neurotransmission in L-DOPA-treated parkinsonian monkeys. Neuropharmacology 2013, 73, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Ulivieri, M.; Volpi, C.; Gargaro, M.; Fallarino, F. Targeting metabotropic glutamate receptors for the treatment of neuroinflammation. Curr. Opin. Pharmacol. 2018, 38, 16–23. [Google Scholar] [CrossRef]
- Tyszkiewicz, J.P.; Yan, Z. Beta-Amyloid peptides impair PKC-dependent functions of metabotropic glutamate receptors in prefrontal cortical neurons. J. Neurophysiol. 2005, 93, 3102–3111. [Google Scholar] [CrossRef] [PubMed]
- Chalimoniuk, M.; Strosznajder, J.B. Aging modulates nitric oxide synthesis and cGMP levels in hippocampus and cerebellum. Effects of amyloid beta peptide. Mol. Chem. Neuropathol. 1998, 35, 77–95. [Google Scholar] [CrossRef]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef]
- Dalfo, E.; Albasanz, J.L.; Martin, M.; Ferrer, I. Abnormal metabotropic glutamate receptor expression and signaling in the cerebral cortex in diffuse Lewy body disease is associated with irregular alpha-synuclein/phospholipase C (PLCbeta1) interactions. Brain Pathol. 2004, 14, 388–398. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef]
- Atkinson, P.J.; Young, K.W.; Ennion, S.J.; Kew, J.N.; Nahorski, S.R.; Challiss, R.A. Altered expression of G(q/11alpha) protein shapes mGlu1 and mGlu5 receptor-mediated single cell inositol 1,4,5-trisphosphate and Ca(2+) signaling. Mol. Pharmacol. 2006, 69, 174–184. [Google Scholar] [CrossRef]
- Castillo, C.A.; Leon, D.; Ruiz, M.A.; Albasanz, J.L.; Martin, M. Modulation of adenosine A1 and A2A receptors in C6 glioma cells during hypoxia: Involvement of endogenous adenosine. J. Neurochem. 2008, 105, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, F.; Tang, M.; Zhao, Y.; Wang, X. Amyloid beta and tau are involved in sleep disorder in Alzheimer’s disease by orexin A and adenosine A(1) receptor. Int. J. Mol. Med. 2019, 43, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Londzin, P.; Zamora, M.; Kakol, B.; Taborek, A.; Folwarczna, J. Potential of Caffeine in Alzheimer’s Disease-A Review of Experimental Studies. Nutrients 2021, 13, 537. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25-35)-induced cognitive deficits in mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Porciuncula, L.O.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Neuroprotection by caffeine and adenosine A2A receptor blockade of beta-amyloid neurotoxicity. Br. J. Pharmacol. 2003, 138, 1207–1209. [Google Scholar] [CrossRef]
- Keshavarz, M.; Farrokhi, M.R.; Amiri, A. Caffeine Neuroprotective Mechanism Against beta-Amyloid Neurotoxicity in SHSY5Y Cell Line: Involvement of Adenosine, Ryanodine, and N-Methyl-D-Aspartate Receptors. Adv. Pharm. Bull. 2017, 7, 579–584. [Google Scholar] [CrossRef]
- Giunta, S.; Andriolo, V.; Castorina, A. Dual blockade of the A1 and A2A adenosine receptor prevents amyloid beta toxicity in neuroblastoma cells exposed to aluminum chloride. Int. J. Biochem. Cell Biol. 2014, 54, 122–136. [Google Scholar] [CrossRef]
- Goel, R.; Bhat, S.A.; Hanif, K.; Nath, C.; Shukla, R. Angiotensin II Receptor Blockers Attenuate Lipopolysaccharide-Induced Memory Impairment by Modulation of NF-kappaB-Mediated BDNF/CREB Expression and Apoptosis in Spontaneously Hypertensive Rats. Mol. Neurobiol. 2018, 55, 1725–1739. [Google Scholar] [CrossRef]
- Zimbone, S.; Monaco, I.; Giani, F.; Pandini, G.; Copani, A.G.; Giuffrida, M.L.; Rizzarelli, E. Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Sanders, O.; Rajagopal, L. Phosphodiesterase Inhibitors for Alzheimer’s Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale. J. Alzheimers Dis. Rep. 2020, 4, 185–215. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Mandelkow, E.; Selkoe, D.J. Alzheimer disease in 2020. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Canter, R.G.; Penney, J.; Tsai, L.H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Dar, K.B.; Bhat, A.H.; Amin, S.; Reshi, B.A.; Zargar, M.A.; Masood, A.; Ganie, S.A. Elucidating Critical Proteinopathic Mechanisms and Potential Drug Targets in Neurodegeneration. Cell Mol. Neurobiol. 2020, 40, 313–345. [Google Scholar] [CrossRef]
- Rahman, A. The role of adenosine in Alzheimer’s disease. Curr. Neuropharmacol. 2009, 7, 207–216. [Google Scholar] [CrossRef]
- Ul Islam, B.; Khan, M.S.; Jabir, N.R.; Kamal, M.A.; Tabrez, S. Elucidating Treatment of Alzheimer’s Disease via Different Receptors. Curr. Top Med. Chem. 2017, 17, 1400–1407. [Google Scholar] [CrossRef]
- Dal Pra, I.; Armato, U.; Chiarini, A. Family C G-Protein-Coupled Receptors in Alzheimer’s Disease and Therapeutic Implications. Front. Pharmacol. 2019, 10, 1282. [Google Scholar] [CrossRef]
- Zanotti-Fregonara, P.; Barth, V.N.; Liow, J.S.; Zoghbi, S.S.; Clark, D.T.; Rhoads, E.; Siuda, E.; Heinz, B.A.; Nisenbaum, E.; Dressman, B.; et al. Evaluation in vitro and in animals of a new 11C-labeled PET radioligand for metabotropic glutamate receptors 1 in brain. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 245–253. [Google Scholar] [CrossRef][Green Version]
- Janssens, N.; Lesage, A.S. Glutamate receptor subunit expression in primary neuronal and secondary glial cultures. J. Neurochem. 2001, 77, 1457–1474. [Google Scholar] [CrossRef] [PubMed]
- Castillo, C.A.; Leon, D.A.; Ballesteros-Yanez, I.; Albasanz, J.L.; Martin, M. Glutamate differently modulates excitatory and inhibitory adenosine receptors in neuronal and glial cells. Neurochem. Int. 2010, 57, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Spelta, V.; Sim, J.; North, R.A.; Surprenant, A. Differential assembly of rat purinergic P2 × 7 receptor in immune cells of the brain and periphery. J. Biol. Chem. 2001, 276, 23262–23267. [Google Scholar] [CrossRef]
- Shruti, S.; Urban-Ciecko, J.; Fitzpatrick, J.A.; Brenner, R.; Bruchez, M.P.; Barth, A.L. The brain-specific Beta4 subunit downregulates BK channel cell surface expression. PLoS ONE 2012, 7, e33429. [Google Scholar] [CrossRef]
- Tsou, K.; Brown, S.; Sanudo-Pena, M.C.; Mackie, K.; Walker, J.M. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 1998, 83, 393–411. [Google Scholar] [CrossRef]
- Palmer, S.; Hughes, K.T.; Lee, D.Y.; Wakelam, M.J. Development of a novel, Ins(1,4,5)P3-specific binding assay. Its use to determine the intracellular concentration of Ins(1,4,5)P3 in unstimulated and vasopressin-stimulated rat hepatocytes. Cell Signal. 1989, 1, 147–156. [Google Scholar] [CrossRef]
- Gerwins, P. Modification of a competitive protein binding assay for determination of inositol 1,4,5-trisphosphate. Anal. Biochem. 1993, 210, 45–49. [Google Scholar] [CrossRef]
- Murphy, M.G.; Moak, C.M.; Byczko, Z.; MacDonald, W.F. Adenosine-dependent regulation of cyclic AMP accumulation in primary cultures of rat astrocytes and neurons. J. Neurosci. Res. 1991, 30, 631–640. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Aranda, P.S.; LaJoie, D.M.; Jorcyk, C.L. Bleach gel: A simple agarose gel for analyzing RNA quality. Electrophoresis 2012, 33, 366–369. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Aβ25-35 24 h | Aβ25-35 48 h | |
| Total mGluR | |||
| Bmax (pmol/mg prot) | 590 ± 31 | 952 ± 9 ** | 1178 ± 93 *** |
| KD (µM) | 3.069 ± 0.062 | 1.607 ± 0.423 ** | 1.522 ± 0.163 ** |
| A1R | |||
| Bmax (fmol/mg prot) | 149 ± 6 | 545 ± 101 ** | 423 ± 66 * |
| KD (nM) | 1.16 ± 0.16 | 7.51 ± 2.35 * | 5.51 ± 1.33 * |
| A2AR | |||
| Bmax (fmol/mg prot) | 656 ± 51 | 557 ± 56 | 1178 ± 43 *** |
| KD (nM) | 13.26 ± 0.26 | 10.33 ± 1.22 | 13.96 ± 4.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo, C.A.; Ballesteros-Yáñez, I.; León-Navarro, D.A.; Albasanz, J.L.; Martín, M. Early Effects of the Soluble Amyloid β25-35 Peptide in Rat Cortical Neurons: Modulation of Signal Transduction Mediated by Adenosine and Group I Metabotropic Glutamate Receptors. Int. J. Mol. Sci. 2021, 22, 6577. https://doi.org/10.3390/ijms22126577
Castillo CA, Ballesteros-Yáñez I, León-Navarro DA, Albasanz JL, Martín M. Early Effects of the Soluble Amyloid β25-35 Peptide in Rat Cortical Neurons: Modulation of Signal Transduction Mediated by Adenosine and Group I Metabotropic Glutamate Receptors. International Journal of Molecular Sciences. 2021; 22(12):6577. https://doi.org/10.3390/ijms22126577
Chicago/Turabian StyleCastillo, Carlos Alberto, Inmaculada Ballesteros-Yáñez, David Agustín León-Navarro, José Luis Albasanz, and Mairena Martín. 2021. "Early Effects of the Soluble Amyloid β25-35 Peptide in Rat Cortical Neurons: Modulation of Signal Transduction Mediated by Adenosine and Group I Metabotropic Glutamate Receptors" International Journal of Molecular Sciences 22, no. 12: 6577. https://doi.org/10.3390/ijms22126577
APA StyleCastillo, C. A., Ballesteros-Yáñez, I., León-Navarro, D. A., Albasanz, J. L., & Martín, M. (2021). Early Effects of the Soluble Amyloid β25-35 Peptide in Rat Cortical Neurons: Modulation of Signal Transduction Mediated by Adenosine and Group I Metabotropic Glutamate Receptors. International Journal of Molecular Sciences, 22(12), 6577. https://doi.org/10.3390/ijms22126577